

Abstract
A water-soluble glycopeptide (PGY), fractionated and purified from the aqueous extract of the fruiting bodies of Ganoderma lucidum via two-step dialysis, anion-exchange and gel permeation chromatography, was constituted of two moieties of carbohydrate and peptide. Carbohydrate characterization with component analysis, methylation analysis, periodate oxidation, Smith degradation, enzymic hydrolysis, and IR and NMR experiments demonstrated that the carbohydrate moiety possessed a backbone of approximately thirty-three (1 → 3)-linked β-D-glucopyranosyl residues, and side chains, at positions 6, of single α-L-arabinofuranosyl residues for every three Glcp residues in the main chain. On the basis of the results of amino acid composition and trypsin digestion, the peptide moiety shown to consist of Arg, Ser, Ala, and Gly in a ratio of 1:1:2:2, exhibited the sequence of Ser-Arg-[(Ala)2(Gly)2], and was O-attached to the carbohydrate moiety via Ser. To contribute toward our understanding of structure-activity relationship, a series of expected derivatives generated from PGY by trypsin digestion, debranching, and NaIO4-oxidation following reduction experiments, including PTC, DB-PGY, and PPP, were obtained. All of them, as well as PGY and reference compound (BHT), were evaluated with two conventional antioxidant testing systems of DPPH and superoxide radicals scavenging, and found to have their respective antioxidant activities in a concentration-dependent manner. Comparable radical scavenging activities observed between PTC and PGY demonstrated that the removal of Ala and Gly in peptide moiety did not result in the variation of biological functions of PGY. However, it was very interesting to note that the scavenging activity of PPP was higher for DPPH radicals with an SC50 value of 116.4 ± 5.1 μg/mL, and lower for superoxide radicals with an SC50 value of 205.2 ± 14.4 μg/mL than that of PGY with corresponding SC50 values of 133.5 ± 5.5 and 140.5 ± 7.7 μg/mL, and moreover, DB-PGY displayed the weakest scavenging potency in the tested samples, indicating that the carbohydrate moiety, in particular the side chain of nonreducing end-units of Araf residues as the functional region played a vital role in the structure and antioxidant activity. In addition, compared with the SC50 value of BHT, PGY showed lower DPPH radical scavenging activity, but possessed higher superoxide radical quenching potency, suggesting that it could be presented as a possible new source of natural antioxidants.
Keywords: glycopeptide, structure, functional regions, antioxidants, relationship
Introduction
One of the most common cotranslational/posttranslational modifications of proteins in eukaryotes of plants, fungi and animals, is attributed to their glycosylation in intimate association with protein folding and intracellular trafficking.1,2 The oligosaccharide/polysaccharide moieties of glycoproteins are covalently attached to the proteins by N- or O-linked forms, and involved in many important cellular communication processes associated with cell adhension, host-pathogen interaction, development, and immune responses.3–7 Nonetheless, protein glycosylation accompanying a series of posttranslational events that are not under direct genetic control, usually generate a variety of glycoforms bound to proteins that in large part have the akin polypeptide backbone but differ in the pendant sugar chains. For this reason, of particular importance is the successful isolation and characterization of homogeneous glycopeptides and glycoproteins that are indispensable for the investigation of detailed structure-activity relationship and biomedical application of glycoproteins/glycopeptides, due to hampering of their structural micro-heterogeneity. By comparison with a glycoprotein, it is herein worthy of mention that, a glycopeptide is similar in molecular composition but has a shorter chain of amino acids. In addition, putting in place the correct glycosylation pattern is also fundamentally important for developing effective glycoprotein-based therapeutics.
The superoxide anion and hydroxyl radicals are generated in living organisms as adverse by-products through many metabolic pathways and readily react with and oxidize biomolecules, such as carbohydrates, proteins, lipids, and DNA. Several lines of evidence have demonstrated that free radicals accumulated in biologic systems contribute to oxidative damage to tissues that lead to cellular integrity and function, and also are associated with the pathogenesis of various diseases, such as arthritis, cancer, inflammation, and heart diseases, as well as aging-related health deterioration in humans.8–10 Nevertheless, due to the various risks of many artificially synthesized antioxidants, such as BHA, BHT, and TBHQ that probably result in the chronic occurrence of cancer,11 the search for appropriate antioxidant agents has recently focused on many plants and mushrooms used in traditional medicine.
To the best of our knowledge, the crude extracts and chemical components of a medicinal mushroom Ganoderma lucidum are well known to contribute to various biological and medicinal functions including hypoglycemic, antitumour, carcinostatic, anti-mutagenic, anti-inflammatory, anti-allergic, anti-androgenic and anti-viral properties.12–19 On the basis of these effects, in addition to small molecules, numerous attentions have been donated to structural studies of G. lucidum and other fungal polysaccharides/glycoconjugates. Most recently, Ye, et al.20 has reported the isolation of a glycopeptide indicative of the ability of stimulating the proliferation of mouse spleen lymphocytes from G. lucidum fruiting bodies, and structural characterization of its polysaccharide moiety utilizing chemical analysis in combination with NMR technology, but information on the peptide part is still very limited. Taken together, however, on account of the difficulties to provide more structural data, in many cases, the in vitro/in vivo effects of many polysaccharides/glycoconjugates remain unclear.
The present study was focused on a new water-soluble glycopeptide, PGY isolated from G. lucidum fruiting bodies by two-step dialysis, anion-exchange and gel-permeation chromatography, as illustrated in Figure 1. One of the objectives for this biopolymer was to characterize in detail the primary structure by virtue of chemical analysis, and NMR and IR spectroscopy together with digestion with specific enzymes. On the other hand, to monitor the functional region in PGY, it was subjected to trypsin digestion, debranching and periodate-oxidation following reduction experiments aimed at generating a series of expected derivatives, all of which, together with PGY and reference compound (BHT), were evaluated through two scavenging assays of free radicals including DPPH and superoxide. It was evident that this work may apparently provide further insight into the carbohydrate-conjugate constituents of mushrooms, and a better understanding of the relationship between structure and activity, that was also fundamental to the development of targeted medicine of the market share for human therapeutics.
Figure 1.

Scheme for the purification of glycopeptide, PGY from G. lucidum. The part highlighted with blue was indicative of the inverse procedure in the step of dialysis. The WAF was fractionated by HPGPC to allow for the present of three moieties by elution with water, in which the second one was collected for the current study.
Materials and Methods
General procedures
The determination of optical rotation was carried out using a Perkin-Elmer 141 polarimeter. IR spectrum was recorded with a Beckman Acculab 10 instrument with KBr pellets. The sample was deuterium-exchanged for hydrogen by repeated freeze-drying from D2O (5 × 5 mL), followed by 13C NMR spectrum monitored on a Bruker 500-instrument (Billerica, MA) for solutions in D2O at 37°C.
Data processing was performed using vendor-supplied software. Chemical shifts were reported in parts per million using internal acetone in D2O (δC 31.45) as reference. The NMR DEPT experiment was conducted using a polarization-transfer pulse of 135°. The HMBC spectrum was recorded on the Bruker Avance DPX-500 spectrometer using different mixing times: 50, 100, and 200 ms. Protein was determined according to the method of Sedmark and Grossberg21 with Coomassie Brilliant Blue and bovine serum albumin as the standard.
Preparation of the glycopeptide
The artificially cultivated fungus fruit bodies of our collection was purchased from a mountainous district in mainland China. Air-dried G. lucidum fruit bodies, ~1.6 kg were pulverized in a commercial grinder and successively extracted with 6 vols of 95% EtOH for three 24-hour periods to defat. After centrifugation, the pellets were air-dried and then extracted three times with distilled water for 5 h at 60 °C, and the combined aqueous extracts were concentrated into small volumes under diminished pressure, and precipitated with 3 vols of 95% EtOH following the 24-hour dialysis (Spectrapor tubes, molecular weight cutoff = 6000~10000) against distilled water for three times. The dialysate, not retentate was combined and concentrated into a small volume. Subsequent dialysis for 36 h against distilled water (Spectrapor tubes, molecular weight cutoff = 3500) was conducted, and the retentate was collected, concentrated under diminished pressure at 45°C using a Büchi Rotavapor rotary evaporator and freeze-dried in a vacuum desiccator to afford crude extracts, GCE (1.3 g, 0.08%yield), followed by elution from a column (5 × 60 cm) of fast-flow DEAE-Sepharose (Pharmacia) with (a) water (400 mL); (b) a gradient of NaCl solution (0.05 → 1 M, 500 mL). The water-eluted anthrone-positive22 fractions, WAF were combined, concentrated, and further purified by gel permeation chromatography on a high-resolution Sephacryl S-300 column (3.6 × 60 cm) employing distilled water as the eluent. Three peaks containing polysaccharide moieties were observed using a Waters 2414 refractive index detector, and the second peak was collected and designated as PGY (197.5 mg, 0.012%yield).
Sugar composition and linkage analysis
Hydrolysis of the PGY was performed with 2 M TFA at 100 °C for 1 h. After reduction with sodium borohydride and acetylation, the samples were analyzed by GLC. The absolute configurations of the sugars present in the PGY were determined essentially as described by Leontein et al.23 by GLC of their acetylated glycosides, using (+)-2-butanol.24 Alditol acetates and acetylated 2-butyl glycosides were separated on an HP-5 fused silica column (0.25 mm × 30 m; Hewlett-Packard) using a temperature program of 180 °C (1 min) and 3 °C min−1 to 210 °C. The column was fitted to a Hewlett-Packard model 5890A gas chromatograph equipped with a flame ionization detector. Methylation analysis was performed according to the procedure of Ciucanu & Kerek.25 Partially methylated alditol acetates were analyzed with a Hewlett-Packard GC 5890A, using a mass selective detector 5970B, a Durabond fused-silica capillary column (DB-225, 30 m × 0.25 mm), and a temperature program of 180–250°C at 10 °C min−1 followed by an isothermic phase. All partially methylated alditol acetates were positively identified using authentic standards.
Amino acid composition
Hydrolysis of the samples with 8 mL of 6 M HCl was performed at 110 °C for 24 h under nitrogen for analysis of the amino acids, except for cysteine and tryptophan. For the analysis of cysteine, the pretreatment of samples with a performic acid solution of formic acid and hydrogen peroxide in the volume ratio of 9:1 was carried out at 0 °C for 15 h prior to the 6 M HCl hydrolysis. The hydrolysate was evaporated to dryness in a SpeedVac system, followed by the determination with an Hitachi 835–50 amino acid analyzer. The detection wavelength was 440 nm for proline and hydroxyline, and 570 nm for the other amino acids. To analyze tryptophan, the samples were incubated in 4 M barium hydroxide containing 3% thiodiethylene glycol in a sealed tube at 110 °C overnight. The tryptophan was detected by Waters 2695 HPLC with 2475 multifluorescence detector by elution with a solution of 10 mM perchloric acid in MeOH, and the eluent was monitored by measuring the fluorescence (excitation at 285 nm, emission at 348 nm).
Perioxide oxidation and Smith degradation
Periodate-oxidation, reduction, and acid hydrolysis were performed by the procedure of Johnson et al.26 A solution of PGY (40 mg) was mixed with 20 mL NaIO4 solution (45 mM) and kept at 4 °C in the dark for 6 days when the process of oxidation was complete. A portion (2 mg) of the resulting polymeric glycopeptide-polyol, PPP (23 mg) obtained by dialysis against water was completely hydrolyzed in 2 M TFA at 100 °C for 5 h, converted into the alditol acetates, and analyzed by GLC/MS (as described above).
Enzymic hydrolysis
A) A suspension of the PGY (15 mg) in a 6 mL of solution containing 50 units of exo-(1 → 3)-β-D-glucanase from Basidiomycete QM 806 in 0.05 M sodium acetate buffer (pH 4.8) was stored at 37 °C for 72 h. After 100 °C heat inactivation for 10 min, followed by the 30-minute high-speed centrifugation, the digest was concentrated into a small volume (~2.5 mL) and applied to a gel-filtration column (1.6 × 80 cm) of Sephadex G-15 (Pharmacia), eluted with distilled water. Fractions (1 mL) were collected, and the sugar content was determined by the phenol-sulfuric acid method.27 B) A solution of PGY (22 mg) in 10 mL of 0.1 M sodium acetate buffer (pH 4.5) containing 300 units of α-L-arabinofuranosidase from Bifidobacterium adolescentis was kept at 40 °C for 3 days when the hydrolytic reaction mediated by this enzyme was complete, since no α-L-arabinofuranosyl residues were detected by analysis of monosaccharide composition of polymer generated from the retentate from dialysis of enzyme digest. The reaction mixture was quenched through 15-minute 100 °C heating for inactivation, followed by the 30-minute high-speed centrifugation for the purpose of removing denatured enzymes. The digest was dialyzed against water, and the retentate was concentrated into a small volume (~2.5 mL) and purified by a Sephadex G-25 gel-filtration column (1.6 × 80 cm) (Pharmacia) by elution with distilled water.
DPPH radical-scavenging assay
The scavenging capability of samples for stable DPPH free radical was measured through the method reported by Katerere and Eloff28 with minor modifications. The different concentrations of 200 μL of tested samples were taken in different test tubes with 4 mL of a 0.006% DPPH solution in MeOH. BHT was used as standard, and the control contained all the earlier chemicals except the tested samples that were replaced by 200 μL of water. Absorbance at 517 nm was monitored after 40-minute incubation. Radical scavenging activity was expressed as the inhibition percentage, and calculated using the following formula, %Radical scavenging activity = [(A0 − A1)/A0]×100, where A0 is the absorbance of the control, and A1 is the absorbance of the samples/standard.
Superoxide radical (O2−) radical-scavenging assay
The scavenging for the superoxide radicals generated from riboflavin-light-NBT system29 resulted in the inhibition of formazan formation. The reaction mixture contained 50 mM phosphate buffer (pH 7.6), 20 mg riboflavin, 12 mM EDTA and 350 μg/mL of NBT, which was prepared in this sequence of chemicals. Reaction was initiated by illuminating the reaction mixture with different concentrations of test samples (50–250 μg/mL) for 1.5 mins. After illumination immediately, the absorbance was measured at 590 nm. The entire reaction assembly was enclosed in a box lined with aluminium foil. Identical tubes with reaction mixture were kept in the dark and served as blanks. BHT was used as standard. The percentage inhibition of superoxide anion generation was calculated using the following formula: %Inhibition = [(A0 − A1)/A0]×100; where A0 is the absorbance of the control, and A1 is the absorbance of the samples/standard.
Statistical Analysis
All of the experimental results were compared by one-way analysis of variance (ANOVA), and the data were expressed as mean±SD of three different trials. The SC50 values, which are the concentrations of samples in μg • mL−1 that scavenge 50% of the free radicals, were calculated from the dose-response curves.
Results and discussion
The procedure attributable to the isolation and purification for PGY from the fruiting bodies of G. lucidum was depicted in Figure 1. The water-eluted anthrone-positive fractions, WAF resulting from the fractionation of GCE by fast flow DEAE-Sepharose anion-exchange chromatography, were detected by HPGPC equipped with a Waters 2414 refractive index detector, and led to the observation of a total of three peaks (Figure 2), in which the second peak corresponding to PGY with 15.5 min of retention time (RT) that was acquired by further isolation of a gel-permeation column of Sephacryl S-300. It had a specific rotation of [α]25D +42.9° (c0.1, water) and appeared as a single symmetrical peak on HPGPC with 15.1 min of RT (Figure 3, PD=Mw/Mn=1.03) similar to that in Figure 2, indicative of the identical carbohydrate-conjugates and homogeneity. Its average molecular weight was estimated to be 7102 Da by reference to standard dextrans, and the protein content was calculated by the calibration curve (Figure S1), and was found to be ~7%. Sugar analysis by GLC of the alditol acetates obtained after full acid hydrolysis of PGY revealed the presence of glucose/arabinose in the relative proportions 3:1. Determination of the absolute configurations of the monosaccharide by GLC as acetylated (+)-2-butyl glycosides indicated the presence of D-Glc and L-Ara. Methylation analysis by GLC-MS of the partially methylated alditol acetates of PGY resulted in the identification of 2,3,5-tri-O-methylarabinose 2,4,6-tri-O-methylglucose, and 2,4-di-O-methylglucose in the ratio of 1.00:2.02:1.07 (Table 1), on account of which, the polysaccharide moiety was tentatively thought to constitute a backbone of (1 → 3)-linked D-glucopyranosyl residues, and side chains, at positions 6, of terminal L-arabinofuranosyl groups.
Figure 2.

Elution profile of WAF in HPGPC, water-eluted Anthrone-positive fractions obtained by fast-flow DEAE-Sepharose (Pharmacia) ion-exchange chromatography, using a Waters 2414 refractive index detector. Three polysaccharide conjugates were clearly observed, and the second symmetrical peak with 15.5 min of retention time was collected and designated as PGY.
Figure 3.

Elution profile of PGY in HPGPC, using a Waters 2414 refractive index detector. The proteoglycan fraction was obtained by fast-flow DEAE-Sepharose (Pharmacia) ion-exchange chromatography.
Table 1.
Partially O-Methylalditol Acetates Formed on Methylation Analysis of Carbohydrate Moiety of PGY
| alditol acetate | approximate molar ratioa | fragments (diagnostic ions, m/z) | linkage indicated |
|---|---|---|---|
| 2,3,5-tri-O-methylarabitol | 1.00 | 87, 101, 102, 118, 129, 161 | 1-Araf |
| 2,4,6-di-O-methylgluctitol | 2.02 | 87,113,117,129,131,161,233 | 1,3-Glcp |
| 2,4-tri-O-methylgluctitol | 1.07 | 87,117,129,189 | 1,3,6-Glcp |
Relative to 2,3,5-tri-O-methylarabitol
To test our hypothesis and detect the branches, several lines of experiments were performed in this work. PGY was subjected to the oxidation of 45 mM NaIO4 for six days at 4 °C in the dark, followed by the consumption of a total of ~0.22 mol of NaIO4 and no liberation of formic acid per mole of glycosyl residues on the basis of the average molar mass (159) of component residues. Smith degradation of the polymeric glycopeptide-polyol (PPP) resulting from metaperiodate oxidation showed glucose attributable to 1,3-/1,3,6-linked Glcp residues, as well as glycerol generated from terminal Araf residues. These results were in approximate agreement with the expected results based on linkage analysis of PGY (NaIO4 consumption: 0.24; formic acid production: 0). The anomeric configurations of sugar residues were determined by oxidation of the acetylated PGY with chromium trioxide.30 In comparison with the survival of ~94.3% L-Araf, D-Glcp was completely destroyed, suggesting that D-Glcp was β-configurations as well as α-configurations of L-Araf, which were in agreement with the presence of IR bands at 893 (for β-anomers) and 841cm−1 (for α-anomers). To examine the positions in main chain linked to nonreducing-end units of Araf, PGY was incubated with exo-(1 → 3)-β-D-glucanase from Basidiomycete QM 806. The first few hours of reaction gave rise to glucose and several oligomers by paper chromatography,31 indicative of the presence of (1 → 3)-linkages in the backbone. The proportion of oligomers decreased with the increase in incubation time by observation in the elution profile of the digest on the gel-filtration column of Sephadex G-15, indicating the presence of a majority of β-(1 → 3)-linked glucosyl residues in the polysaccharide moiety. Eventually, an oligomer, GU remained to inhibit the enzyme to further degrade this molecule into small fragments, and was converted into its alditol acetates.25 Two peaks corresponding to 2,3,5-tri-O-methylarabinose and 1,2,3,4-tetra-O-methylglucose in a molar ratio of 1.00:1.09 were observed by GLC of the alditol acetates from the partially methylated GU, demonstrating that the structure of GU was a disaccharide repeat unit of Araf-(1 → 6)-Glcp, and shown in Figure 4. Evidence from this experiment supported our hypothesis that terminal α-L-Araf residues as the branches were tied into C-6 (not C-3) of the (1 → 3)-Glcp backbone. The digest resulting from the 3-day incubation of PGY with α-L-arabinofuranosidase, passed through a Sephadex G-25 gel-filtration column to give debranched PGY (DB-PGY), monosaccharide composition of which only showed the presence of glucose with the disappearance of α-L-arabinofuranose, indicative of completeness of the specific enzymatic reaction for eliminating Araf residues.
Figure 4.
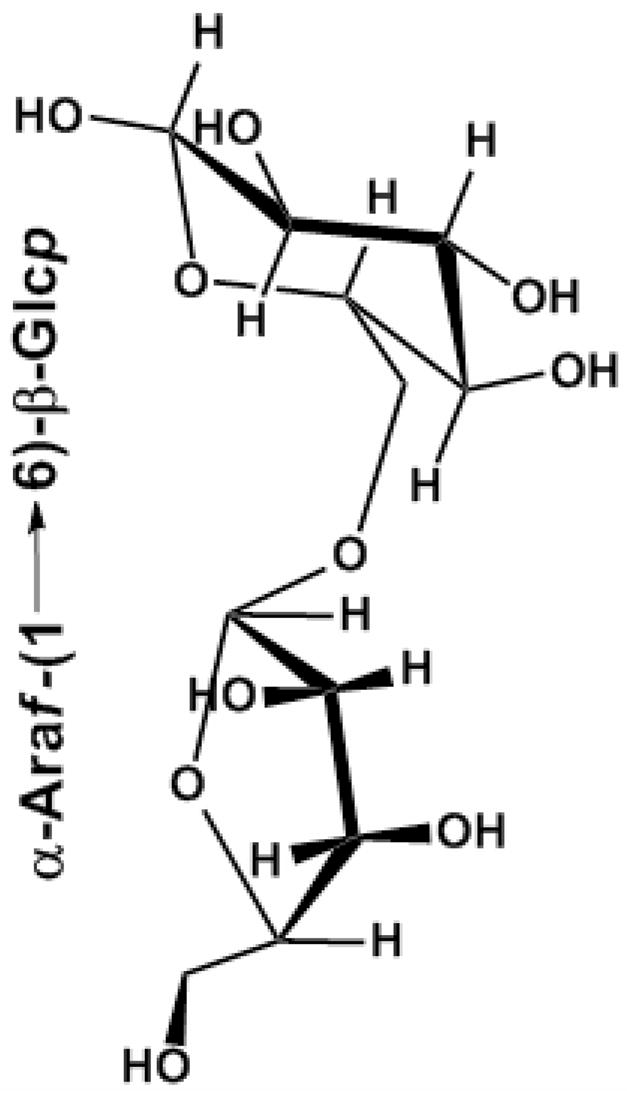
Proposed structure of GU generated by passing through Sephadex G-15 column of exo-(1 → 3)-β-D-glucanase-treated PGY.
The broad-band decoupled 13C NMR spectrum (Figure 5) showed the carbohydrate moiety of PGY to be a structurally heterogeneous polysaccharide since three signals were observed in the anomeric region. The sugar residues were donated A–C from decreasing 13C chemical shifts of the resonances of their respective anomeric carbons. Two downfield signals at δ 104.5 and 103.8 in anomeric region were respectively assigned to C-1 of B and C, however, the remaining more downfield signal at δ 107.7, indicative of furanosidic ring forms, was exclusively attributed to C-1 of A. All of the resonances were resolved31,32 and recorded in Table 2. For residue A a large downfield chemical shift displacement Δδc of 7.5 was observed for C-1. Thus, A is 1-substituted, and the residue is consequently α-L-Araf-(1 →. Residue B gave two large downfield chemical shift displacements Δδc of 7.7 and 7.1 for C-1 and C-3, respectively. Thus, B is 1,3-disubstituted and the residue is consequently →3)-β-D-Glcp-(1→. Three signals at δ 103.8, 84.1 and 66.8 present in residue C, showing large downfield chemical shift displacement Δδc of 7.8, 7.0 and 5.0 for C-1, C-3 and C-6 respectively, indicated that C was 1,3,6-trisubstituted and the residue was →3,6)-β-D-Glcp-(1→. The 13C distortionless enhancement by polarization transfer (DEPT) 135 NMR spectrum of PGY (data not shown) showed that both of signals at δ 61.6 and 61.1 were seperately assigned to C-5 of non-reducing Araf residues and C-6 of six-unsubstituted Glcp residues. Nevertheless, the relatively upfield signal at δ 66.9 resulted from C-6 of six-substituted Glcp residues. The mode of linkage for sugar residues present in the carbohydrate moiety of PGY was further corroborated by the HMBC spectrum (Figure 6) that gave the correlations between proton and carbon, viz., C-1 in A to H-6 in C, and C-1 in B to H-3 in B or C. Thus, it demonstrated that this polymeric carbohydrate moiety had a backbone of 1,3-linked β-D-Glcp residues with a side-chain of single Araf residues at O-6.
Figure 5.

13C NMR spectrum of PGY obtained from G. lucidum fruiting bodies in D2O. A, B, and C stand for repeating sugar-residue A, B, and C present in this polysaccharide-conjugate, see Table 2 for details.
Table 2.
Chemical Shifts (ppm) of the Signals in the 13C NMR Spectrum of PGY from G. lucidum fruiting bodies a
| Sugar residues | 13C/Chemical shifts | |||||
|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | |
| α-L-Araf-(1 → A | 107.7 (7.5) | 82.6 (0.4) | 77.8 (0.3) | 85.7 (0.9) | 61.4 (−0.9) | |
| →3)-β-D-Glcp-(1→ B | 104.5 (7.7) | 74.7 (−0.4) | 84.2 (7.1) | 71.5 (1.0) | 80.7 (1.9) | 61.0 (−0.8) |
| →3,6)-β-D-Glcp-(1→ C | 103.8 (7.8) | 73.7 (−1.4) | 84.1 (7.0) | 69.8 (−0.7) | 77.0 (0.4) | 66.8 (5.0) |
Chemical shifts displacements (Δδ) are reported in parentheses, compared to those of the corresponding monosaccharides.
Figure 6.

Part of the 1H, 13C HMBC spectrum of PGY
To detect the composition of amino acids, the acid hydrolysate of PGY was subjected to HPAEC that revealed its protein moiety to be constituted of Arg, Ser, Ala, and Gly present in the ratio of 1.00:0.96:2.03:2.06, which was very close to the molecular weight of the peptide moiety (~497 Da) calculated on the basis of the estimated percentage (~7%) of protein in the glycopeptide. Subsequent trypsin digestion of PGY generated a mixture that was fractionated by fast-flow DEAE-Sepharose anion-exchange chromatography to afford one main polymeric carbohydrate-conjugate component (PTC) detected by approach of analysis using the anthrone-sulfuric acid reagent.22 HPAEC of the mixture resulting from 6 M HCl hydrolysis of PTC allowed for the presence of Ser and Arg in the approximate ratio of 1:1. It should be herein mentioned that protein amino acid O-linked glycosylation was supposed to occur in either Thr or Ser residues, whereas Asn was attributed to N-linked glycosylation,33 and on the other hand, trypsin cleaved exclusively C-terminal to Arg and Lys residues. Therefore, it could be concluded that the oligopeptide was attached to the carbohydrate moiety via Ser, and consequently Arg was deduced to bind to Ser through the peptide bond. However, the sequence of two molecules of Ala and Gly was so far unable to be identified.
On the basis of all of the foregoing results, 1, 2, 3, and 4, shown in Figure 7, were proposed to be the structures of PGY, PPP, PTC, and DB-PGY, respectively, and provided the prerequisite for the exploitation of structure-functional properties relationship. To probe to this, we subjected PGY and three derivatives as well as BHT used as positive control to the DPPH and superoxide (O2−) radical scavenging tests aimed at monitoring the active regions of this glycopeptide. The DPPH radical scavenging capacity, based on the reduction of the stable radical DPPH to yellow-colored diphenylpicrylhydrazine in the presence of a hydrogen donor, had been widely employed to evaluate antioxidant activity due to its advantage of rapid and simple measure. The scavenging effects on the DPPH free radical were investigated as shown in Figure 8, all of these tested samples were found to possess the dose-dependent DPPH radical quenching activity, which grew as their concentrations increased to a certain extent. According to the values of SC50 corresponding to the scavenging potency of DPPH radicals, BHT exhibited the highest scavenging activity with an SC50 value of 100.7±3.1 μg/mL, whereas the compound with the weakest scavenging potency was DB-PGY (SC50 > 250 μg/mL), which had significantly lower activities than the other four tested. The SC50 of PGY was determined to be 133.5±5.5 μg/mL similar to that of PTC with an SC50 value of 129.2±8.0 μg/mL, suggesting that no significant difference was found between them. However, in comparison with PGY, a slight increase of scavenging activity was observed in PPP corresponding to an SC50 value of 116.4±5.1 μg/mL, probably due to small structural difference resulting from the NaIO4 oxidation led to the opening of terminal Araf glycosidic rings, and sodium borohydride reduction, that was considered to be, at least in part, responsible for the enhancement of its hydrogen-donating ability.
Figure 7.

Proposed structures for PGY (1) and a series of its derivatives, namely PPP (2), PTC (3), and DB-PGY (4).
Figure 8.

Scavenging effects of PGY, PPP, DB-PGY, PTC and BHT on DPPH radicals. Each sample was assayed in triplicate for each concentration. Experimental results are mean±SD of three parallel measurements. The SC50 (μg/mL) values were calculated from the dose–response curves.
On the other hand, in the case of the superoxide radical assay, all of tested compounds also quenched superoxide radicals generated from riboflavin-NBT-light system in a concentration-dependent manner. As it could be seen in Figure 9, when the SC50 values (expressed as micrograms per milliliter) were compared for the five compounds tested, PGY and PTC, with corresponding SC50 values of 140.5±7.7 and 146.2±3.4 μg/mL, possessed the highest scavenging activities with no significant difference between them, however, it was interesting to note that antioxidant activities of them were weaker than that of BHT at the dosage range of 50–130 μg/mL, but with continually increasing concentration, the scavenging effect rapidly catched up with and exceeded BHT. The superoxide radical scavenging activity of PPP, with an SC50 value of 205.2±14.4 μg/mL, showed significantly weaker than that of PGY and PTC. Likewise, compared with the experimental result of DPPH assay, the compound with the weakest activity for superoxide radical scavenging was also DB-PGY (SC50 > 250 μg/mL).
Figure 9.

Scavenging effects of PGY, PPP, DB-PGY, PTC and BHT on superoxide radicals (O2□). Each sample was assayed in triplicate for each concentration. Experimental results were mean±SD of three parallel measurements. The SC50 (μg/mL) values were calculated from the dose–response curves.
From the results of two in vitro antiradical tests, it implies that the scavenging effects against DPPH and superoxide radicals represent a direct correlation on terminal α-L-Araf residues as the side chain in the carbohydrate moiety of PGY, the removal of which results in the significant decrease of antioxidative properties. Moreover, opening, oxidation, and reduction of Araf glycosidic rings attributed to the formation of polyalcohol conjugate, also influence the antiradical potency of PGY, that argues for the important role of side-chain arabinofuranosides. In contrast, in the peptide moiety, when Ala and Gly are removed from PGY, the derivative shows no significant differences on the free-radical scavenging effects, suggesting that the peptide moiety seems not to be the major functional region of antioxidant activity, at least, the absence of Ala and Gly residues has no affect on the free-radical scavenging capability of PGY.
Conclusion
Generally, glycopeptide is constituted of a oligopeptide chain to which one or more carbohydrate moieties are covalently attached. The carbohydrate moieties are usually connected through N- (for Asn) or O- (for Thr and Ser) linkages. Glycopeptides have a wide variety of biological functions, which may be dependent on the oligopeptide part, the carbohydrate part, or on both. Understanding of their structure-function relationship is usually challenging. In this publication, we introduced the isolation and characterization of a low-molecular-weight glycopeptide from the fruiting bodies of Ganoderma lucidum, and designed a series of structural variants to explore their structure-activity relationship. Evidence from the current experiments demonstrated that the carbohydrate moiety of PGY was shown to possess a backbone of (1 → 3)-β-glucan with (1 → 6)-linked Araf branches (For every three D-glucopyranosyl residues in the main chain, there is one L-arabinofuranosyl group), and the peptide moiety (SRA2G2) is coupled to the carbohydrate moiety by O-linkage via Ser. The carbohydrate moiety, especially the side chains of terminal α-L-Araf residues, is essential for the preservation of antioxidant activity of this class of glycopeptides. In contrast, the peptide moiety appears to be nonessential for their antioxidant activity. Finally, it is worth mentioning that PGY provides higher levels of antioxidant property against superoxide radicals than that of the selected antioxidant (BHT). PGY and its derivatives may represent a new class of natural products with sugar moiety-dependent antioxidant activities.
Supplementary Material
Additional information as noted in the text is available: the calibration curve of protein content. This information is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
The authors are indebted to Mr. Baoxin Wu (Ankang Medicine Co, Ltd., Yanji, Jilin Province, People’s Republic of China) for the donation of G. lucidum fruit bodies and valuable discussions. The work at Stanford University is partially sponsored by NIH grant U01CA128416 to D. Wang.
Abbreviations
- TFA
trifluroacetic acid
- GLC
gas-liquid chromatography
- HPGPC
high-pressure gel permeation chromatography
- HPAEC
high performance anion exchange chromatography
- DPPH
1,1-diphenyl-2-picrylhydrazyl
- BHT
butylated hydroxytoluene
References
- 1.Petrescu AJ, Wormald MR, Dwek RA. Curr Opin Struct Biol. 2006;16:600–607. doi: 10.1016/j.sbi.2006.08.007. [DOI] [PubMed] [Google Scholar]
- 2.Helenius A, Aebi M. Science. 2001;291:2364–2369. doi: 10.1126/science.291.5512.2364. [DOI] [PubMed] [Google Scholar]
- 3.Varki A. Glycobiology. 1993;3:97–130. doi: 10.1093/glycob/3.2.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dwek RA. Chem Rev. 1996;96:683–720. doi: 10.1021/cr940283b. [DOI] [PubMed] [Google Scholar]
- 5.Dwek RA, Butters TD, Platt FM, Zitzmann N. Nat Rev Drug Discov. 2002;1:65–75. doi: 10.1038/nrd708. [DOI] [PubMed] [Google Scholar]
- 6.Haltiwanger RS, Lowe JB. Annu Rev Biochem. 2004;73:491–537. doi: 10.1146/annurev.biochem.73.011303.074043. [DOI] [PubMed] [Google Scholar]
- 7.Dube DH, Bertozzi CR. Nat Rev Drug Discov. 2005;4:477–488. doi: 10.1038/nrd1751. [DOI] [PubMed] [Google Scholar]
- 8.Abe J, Berk BC. Trends Cardiovasc Med. 1998;8:59–64. doi: 10.1016/S1050-1738(97)00133-3. [DOI] [PubMed] [Google Scholar]
- 9.Busciglio J, Yankner BA. Nature. 1995;378:776–779. doi: 10.1038/378776a0. [DOI] [PubMed] [Google Scholar]
- 10.Meerson FZ, Kagan VE, Kozlov YP, Belkina LM, Arkhipenko YV. Basic Res Cardiol. 1982;77:465–485. doi: 10.1007/BF01907940. [DOI] [PubMed] [Google Scholar]
- 11.Botterweck AA, Verhagen H, Goldbohm RA, Kleinjans J, Brandt PA. Food Chem Toxicol. 2000;38:599–605. doi: 10.1016/s0278-6915(00)00042-9. [DOI] [PubMed] [Google Scholar]
- 12.Mizuno T, Wang GY, Zhang J, Kawagihi H, Nishitoba T, Li JX. Food Rev Int. 1995;11:151–166. [Google Scholar]
- 13.Lin ZB, Zhang HN. Acta Pharmacol Sin. 2004;25:387–395. [PubMed] [Google Scholar]
- 14.Kim HS, Kacew S, Lee MB. Carcinogenesis. 1999;20:1637–1640. doi: 10.1093/carcin/20.8.1637. [DOI] [PubMed] [Google Scholar]
- 15.Sliva D. Integr Cancer Ther. 2003;4:358–364. doi: 10.1177/1534735403259066. [DOI] [PubMed] [Google Scholar]
- 16.Laksmi B, Ajith TA, Jose N, Janardhanan KK. J Ethnopharmacol. 2006;107:297–303. doi: 10.1016/j.jep.2006.03.027. [DOI] [PubMed] [Google Scholar]
- 17.Akihisa T, Nakamura Y, Tagata M, Tokuda H, Yasukawa K, Uchiyama E, Suzuki T, Kimura Y. Chem Biodivers. 2007;4:224–231. doi: 10.1002/cbdv.200790027. [DOI] [PubMed] [Google Scholar]
- 18.Fujita R, Liu J, Shimizu K, Konishi F, Noda K, Kumamoto S, Ueda C, Tajiri H, Kaneko S, Suimi Y, Kondo R. J Ethnopharmacol. 2005;102:107–112. doi: 10.1016/j.jep.2005.05.041. [DOI] [PubMed] [Google Scholar]
- 19.Li YQ, Wang SF. Biotechnol Lett. 2006;11:837–841. doi: 10.1007/s10529-006-9007-9. [DOI] [PubMed] [Google Scholar]
- 20.Ye LB, Zhang JS, Ye XJ, Tang QJ, Liu YF, Gong CY, Du XJ, Pan YJ. Carbohydr Res. 2008;343:746–752. doi: 10.1016/j.carres.2007.12.004. [DOI] [PubMed] [Google Scholar]
- 21.Sedmark JJ, Grossberg SE. Anal Biochem. 1977;79:544–552. doi: 10.1016/0003-2697(77)90428-6. [DOI] [PubMed] [Google Scholar]
- 22.Morris DL. Science. 1948;107:254–255. doi: 10.1126/science.107.2775.254. [DOI] [PubMed] [Google Scholar]
- 23.Leontein K, Lingberg B, Lönngren J. Carbohyd Res. 1978;62:359–362. [Google Scholar]
- 24.Gerwig GJ, Kamerling JP, Vliegenthart JF. Carbohydr Res. 1979;77:1–7. doi: 10.1016/s0008-6215(00)83788-x. [DOI] [PubMed] [Google Scholar]
- 25.Ciucanu I, Kerek F. Carbohydr Res. 1984;131:209–217. [Google Scholar]
- 26.Johnson J, Kirkwood S, Misaki A, Nelson TE, Scaletti JV, Smith F. Chem Ind-London. 1963:820–822. [Google Scholar]
- 27.Dubois M, Gilles KA, Hamilton JK, Rebers PA, Smith F. Anal Chem. 1956;28:350–356. [Google Scholar]
- 28.Katerere DR, Eloff JN. Phytother Res. 2005;19:779–781. doi: 10.1002/ptr.1719. [DOI] [PubMed] [Google Scholar]
- 29.Martinello F, Soares SM, Franco JJ, Santos AC, Sugohara A, Garcia SB, Curti C, Uyemura SA. Food Chem Toxicol. 2006;44:810–818. doi: 10.1016/j.fct.2005.10.011. [DOI] [PubMed] [Google Scholar]
- 30.Wu YL, Hu N, Pan YJ, Zhou LJ, Zhou XX. Carbohydr Res. 2007;342:870–875. doi: 10.1016/j.carres.2007.01.005. [DOI] [PubMed] [Google Scholar]
- 31.Wu YL, Pan YJ, Sun CR. Int J Biol Macromol. 2005;36:241–245. [Google Scholar]
- 32.Wu YL, Wang DN. J Nat Prod. 2008;71:241–245. doi: 10.1021/np070323+. [DOI] [PubMed] [Google Scholar]
- 33.Geyer H, Geyer R. Biochim Biophys Acta. 2006;1764:1853–1869. doi: 10.1016/j.bbapap.2006.10.007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional information as noted in the text is available: the calibration curve of protein content. This information is available free of charge via the Internet at http://pubs.acs.org.