

Abstract
Based on the structures of small-molecule hits targeting the HIV-1 gp41, N-(4-carboxy-3-hydroxy)phenyl-2,5-dimethylpyrrole (2, NB-2) and N-(3-carboxy-4-chloro)phenylpyrrole (A1, NB-64), 42 N-carboxyphenylpyrrole derivatives in two categories (A and B series) were designed and synthesized. We found that 11 compounds exhibited promising anti-HIV-1 activity at micromolar level and their antiviral activity was correlated with their inhibitory activity on gp41 six-helix bundle formation, suggesting that these compounds block HIV fusion and entry by disrupting gp41 core formation. The structure-activity relationship and molecular docking analysis revealed that the carboxyl group could interact with either Arg579 or Lys574 to form salt bridges and two methyl groups on the pyrrole ring were favorable for interaction with the residues in gp41 pocket. The most active compound, N-(3-carboxy-4-hydroxy)phenyl-2,5-dimethylpyrrole (A12), partially occupied the deep hydrophobic pocket, suggesting that enlarging the molecular size of A12 could improve its binding affinity and anti-HIV-1 activity for further development as a small-molecule HIV fusion and entry inhibitor.
Keywords: HIV-1 gp41, HIV entry inhibitor, six-helix bundle
Introduction
According to the estimate of UNAIDS, about 33.2 million people worldwide are living with HIV, and more than 25 million patients have died of AIDS (www.unaids.org/en/KnowledgeCentre/HIVData/EpiUpdate/EpiUpdArchive/2007/). Thus far, 28 anti-HIV drugs have been licensed by the United States Food and Drug Administration (FDA) (http://www.hivandhepatitis.com/hiv_and_aids/hiv_treat.html). Most of these drugs belong to two categories: reverse transcriptase inhibitors (RTI) and protease inhibitors (PI). Combined application of these antiretroviral drugs has shown significant synergistic effects.1 However, an increasing number of patients with HIV infection/AIDS can no longer use such drugs as a result of drug resistance and serious adverse effects.2–4 Therefore, it is essential to develop novel anti-HIV drugs targeting HIV entry.
HIV-1 envelope glycoprotein (Env) transmembrane subunit gp41 plays an important role in virus fusion and entry5 and can serve as a target for the development of HIV-1 fusion inhibitors.6, 7 The gp41 ectodomain contains a fusion peptide (FP), the N- and C-terminal heptad repeat (NHR and CHR, respectively). The peptides derived from the gp41 CHR region (designated C-peptides) are potent HIV fusion inhibitors.8–10 One of the C-peptides, T-20 (1, Enfuvirtide, a 36-amino acid synthetic peptide)11, 12 was approved by the U.S. FDA in 2003 as the first member of a new class of anti-HIV drugs – HIV fusion inhibitors for treating HIV/AIDS patients who have failed to respond to RTI and PI. Drug 1 was believed to interact with the HIV-1 gp41 NHR and block the gp41 six-helix bundle (6-HB) core formation, thereby inhibiting fusion between the viral and target cell membranes.10, 13, 14 Although 1 is very potent in inhibiting HIV infection, it has two critical limitations as a drug: lack of oral availability and high production cost.15 Therefore, it is necessary to develop orally available non-peptide small-molecule fusion inhibitors, but with a mechanism of action similar to C-peptides. By screening a drug-like chemical library using a high-throughput screening technique,16 we previously identified two small molecules, N-(4-carboxy-3-hydroxy)phenyl-2,5-dimethylpyrrole (2, NB-2) and N-(3-carboxy-4-chloro)phenylpyrrole (A1, NB-64) 17 (Fig. 1), which inhibit HIV-1 fusion and entry by interfering with the gp41 6-HB formation. These promising hits prompted us to focus on the modification of 2 and A1 to discover and develop new lead compounds with novel scaffold and higher potency. Here we report the results of our hit-to-lead process, including design, synthesis, and biological evaluation of 42 N-carboxyphenyl pyrroles and related derivatives. Their primary structure-activity relationships are discussed.
Figure 1.

Structures of hits 2 and A1.
Design
Previous studies have identified an attractive target in gp41 for small-molecule HIV fusion inhibitors, i.e., the deep hydrophobic pocket (~16 Å long, ~7 Å wide, and 5–6 Å deep) on the surface of the internal N-helix trimer that is filled by three conserved hydrophobic residues with large side chains (Ile635, Trp631, and Trp628) in the gp41 CHR region.13, 18 The combined molecular mass of these residues inside the pocket is ~600 Da, which is within the size range for binding of an orally bioavailable, small-molecule drug.19 Based on this information, we docked a known hit 2 into the gp41 hydrophobic pocket (Fig. 2). This step allowed us to image the gp41 binding “pocket” shape, the orientation and effective binding conformation of inhibitor 2, and more surface amino acids, such as Trp571, Gln575, Arg579, Leu581, Gln577, Lys574 and Ile573. The docking results indicated that 2 partially occupies the pocket. Our previous studies indicated that the carboxyl group of A1 orients to the positively charged Lys574, which is a key surface amino acid in forming a salt “bridge” with an inhibitor.20–22 On the other hand, the carboxyl group of 2 interacts with Arg579 rather than Lys574 (Fig. 2). Thus, it can be hypothesized that the acid group in the compounds can interact with either of these two positively charged residues around the pocket to form a salt bridge which stabilizes the interaction between the compound and the HIV-1 gp41. Consequently, our hit-to-lead optimization and rational design aimed at improving the shape complementation with the binding pocket in order to provide more binding points and increase affinity with the biologic target. It is anticipated that these results will, in turn, provide the basis for discovering more potent leads with a new scaffold.
Figure 2.

Molecular modeling: the docking conformation of 2 inside the hydrophobic pocket of gp41.
In the hit-to-lead optimization process, both the structural similarities and differences between 2 and A1 led us to explore the structural impact of the phenylpyrrole compounds. Two parallel series of N-phenylpyrrole and N-phenyl-2,5-dimethylpyrrole derivatives (A1-A10 and A11-A20) with a p- or m-COOH on the benzene ring were first designed and synthesized to determine which structural moieties are necessary and which position of the carboxyl group is most favorable for anti-HIV potency. Specifically, it was possible that the carboxyl group on the benzene ring could significantly affect the molecular geometry and binding orientation because it is expected to form a salt bridge with Lys574 or Arg579 on the surface of binding site. 20–22 Next, more boundary substituents were introduced on the N-phenylpyrrole skeleton to produce more potential target-inhibitor interaction points for enhancing binding affinity. This resulted in an increase of anti-HIV potency. Meanwhile, an isosteric replacement of the carboxyl group was also performed by a tetrazole moiety. Next, the strategy of isosteric replacement was then applied by using several five-member heterocycles instead of the pyrrole ring to investigate if the pyrrole ring necessary for anti-HIV potency and, hopefully, to identify new potential structural moieties. Therefore, a series of carboxyphenyl compounds with an 1,2,4-oxadiazole (B1-B11), thiadiazole (B12), maleimide (B13-B15), or rhodanine (B16-B22) ring, respectively, were designed and synthesized. In the B series of compounds, a polar functional group, or side chain on the five-member ring, could enlarge the size and change the shape of inhibitors. Both the design and modification of all new compounds were driven by bioassays for anti-HIV-1 activity against HIV-1 replication and gp41 six-helix bundle formation. Meanwhile, molecular modeling studies were performed to elucidate the structure and activity relationship for these small-molecule fusion inhibitors.
Chemistry
As shown in Scheme 1, the Paal-Knorr reaction was used to synthesize A1-A9 and A11-A20 by the condensation of anilines or benzylamines with 2,5-dimethoxytetrahydrofuran or acetonylacetone (hexane-2,5-dione),23 respectively. The Paal-Knorr reaction was performed with or without glacial acetic acid as solvent under microwave irradiation in a sealed tube at 100–150 °C for 10 min.24 Microwave irradiation greatly reduced reaction time and byproducts, resulting in yields ranging from 50 to 88%, which was higher than the 28–56% in the traditional condition. N-Benzylpyrrole A10 was prepared by treatment of 4-(bromomethyl)phenylacetic acid and pyrrole in the presence of potassium tert-butoxide (t-BuOK) under microwave irradiation with a yield of 84%. However, the several syntheses to obtain pure 2 as a positive control in our bioassays were unsuccessful, even though N-(3-carboxy-4-hydroxy)phenyl-2,5-dimethylpyrrole (A12), an isomer of 2, was successfully synthesized with a 79% yield. This appeared to be caused by the instability of 2 during work-up and purification when color changed rapidly from white to deep-red.
Scheme 1.

Synthesis of A1-A20. (i) and (ii) AcOH, microwave, 150 °C, 10 min; (iii) t-BuOK, DMSO, microwave, 193 °C, 10 min.
The 1,2,4-oxadiazole derivatives B1-B11 were synthesized by the acylation of amidoxime,24 as shown in Scheme 2. Cyanobenzoic acids or ester were treated with hydroxylamine hydrochloride in the presence of 8-hydroxyquinoline25 to produce corresponding benzamidoximes 4–6, respectively. Compound 6 reacted with either bromoacetyl bromide or chloroacetyl chloride in THF at reflux temperature to afford 5-substituted 3-aryl-1,2,4-oxadiazoles B1 or B2, respectively. 5-Chloromethyl group on the oxadiazole ring of B2 was then converted into 5-thiocyanomethyl by treatment with ammonium thiocyanate in DMF at 90 °C to produce B3. Following a basic hydrolysis of B1 in EtOH, an ester exchange product 3-(4′-ethoxycarbonyl)phenyl-5-hydroxymethyl oxadiazole (B4) was produced. However, hydrolysis of B2 in the basic condition yielded 3-(4′-carboxyl)phenyl-5-hydroxymethyl oxadiazole (B5). Otherwise, esters B2 and B3 were hydrolyzed in the presence of HCl/AcOH (1:1) to yield corresponding 3-(4′-carboxyl)phenyl-1,2,4-oxadiazoles B6 and B7 respectively. The preparation of 5-bromomethyl oxadiazole B8 have to hydrolyze B1 in HBr/AcOH (1:1), whereas the bromide in B1 could be replaced by chloride if the hydrolysis was carried out in HCl/AcOH. By treating with trifluoroacetic anhydride in pyridine, 4 and 5 afforded corresponding 5-trifluoromethyl-1,2,4-oxadiazoles B9 and B10 respectively. Compound B11 was synthesized from 4 in one step with a yield of 37%.
Scheme 2.

Synthesis of B1-B11. (i) Na2CO3, EtOH, reflux, 4 h; (ii) THF, reflux, 8 h; (iii) DMF, 90 °C, 3 h; (iv) 1N NaOH, EtOH, r.t., 4 h; (v) HCl or HBr/AcOH (1:1), 100 °C, 8–12 h; (vi) pyridine, reflux, 2 h.
The syntheses of B12-B22 are summarized in Scheme 3. These compounds possess a five-member heterocyclic ring moiety instead of the pyrrole ring in the A series of compounds. According to methods found in the literature,26 thiosemicarbazone (7) was prepared from p-carboxylbenzaldehyde by treating with thiosemicarbazide in ethanol followed by a cyclization in aq. ammonium ferric sulfate solution to produce 4-carboxyphenyl-1,3,4-thiadiazole (B12). Anilines reacted with maleic anhydride afforded intermediate N-aryl maleic monoamides 8 and 9, followed by dehydration and cyclization in acetic anhydride and triethylamine,26 which produced corresponding N-aryl maleimide derivatives B13 and B14, respectively. The acidic hydrolysis of B14 produced an expected compound B15, but a basic hydrolysis destroyed the maleimide ring. Subsequently, N-phenyl-substituted rhodanine derivatives B16-B22 were prepared from various anilines by treatment of bis(carboxymethyl)-trithio carbonate in water with a 28%~88% yield, respectively.27 A direct substitution between NH of rhodanine and benzene bromide failed in spite of using various bases, such as potassium tert-butoxide, sodium hydride, potassium (or sodium) hydroxide, and potassium carbonate, even though this reaction seemed simple and convenient.
Scheme 3.

Synthesis of B12-B22. (i) EtOH, reflux, 1.5 h; (ii) H2O, reflux, 10 h; (iii) EtOAc, r.t., 1 h; (iv) NEt3, 60 °C, 1 h; (v) HCl/AcOH (1:1), 100 °C, 8 h; (vi) H2O, 100 °C, 19 h.
Results and Discussion
A total of 42 newly synthesized N-arylpyrrole derivatives (20 in the A series and 22 in the B series) were tested in parallel with A1 for their inhibitory activity on HIV-1 replication (as determined by p24 production), HIV-1-mediated cell-cell fusion, and gp41 6-HB formation. The compounds A2-A10 and A11-A20 contain the same backbone structures of A1 and 2, respectively. A majority of these compounds exhibited significant inhibitory activity on HIV-1 replication and gp41 6-HB formation as determined by ELISA and native-PAGE (Table 1 and Fig. 3). The anti-HIV-1 activities of these compounds were correlated with their inhibitory activities on 6-HB formation (Fig. 4), suggesting that these compounds, like 2 and A1, may interact with the gp41 NHR region to disrupt the fusion-active core formation, resulting in blockage of gp41-mediated membrane fusion and inhibition of HIV-1 replication. Indeed, most of the active compounds displayed inhibitory activity on HIV-1-mediated cell-cell fusion (data not shown). But unlike the majority of the active compounds, a few compounds, did not have good correlation between their anti-HIV-1 activity and their inhibitory activity on 6-helix bundle formation. One of the explanations is that these active compounds may target more than one step of the HIV life cycle. For example, betulinic acid, a bifunctional anti-HIV-1 compound, inhibits HIV infection by targeting both HIV-1 entry and maturation steps.28 Therefore, it is worthwhile to further investigate the mechanism(s) of action of those compounds that do not have good correlation between their activities against HIV-1 infection and 6-helix bundle formation.
Table 1.
Anti-HIV activity data of pyrrole derivatives A1-A20 as HIV-1 gp-41 inhibitors.
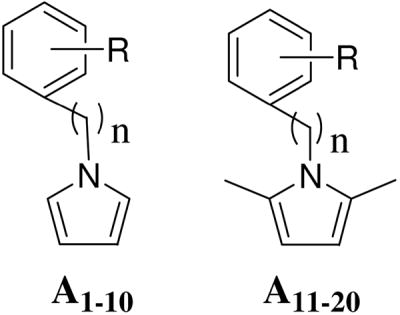 | ||||||||
|---|---|---|---|---|---|---|---|---|
| Code# | R on benzene ring |
Inhibiting p24 production EC50 (μM) | Cytotoxicity CC50 (μM) | SI* | Inhibition of 6-HB formation IC50 (μM) | |||
| p- | m- | o- | n | |||||
| A1 (NB-64) | Cl | CO2H | H | 0 | 2.39 | 335.72 | 140.47 | 58.74 |
| A2 | OH | CO2H | H | 0 | 9.66 | >492.61 | >50.99 | 48.72 |
| A3 | H | CO2H | H | 0 | 44.81 | 301.93 | 6.74 | 210.7 |
| A4 | H | tetrazolyl | H | 0 | 41.04 | 436.11 | 10.63 | 226.11 |
| A5 | CO2CH3 | OH | H | 0 | 35.44 | 379.12 | 10.70 | >460.83 |
| A6 | CO2H | H | H | 0 | 69.25 | >534.76 | >7.72 | 129.47 |
| A7 | CH2CO2H | H | H | 0 | 371.24 | >497.51 | >1.34 | >497.51 |
| A8 | CO2H | OH | H | 0 | 81.67 | 442.56 | 5.42 | 70.84 |
| A9 | CO2H | H | H | 1 | 63.93 | >497.51 | >7.78 | 156.72 |
| A10 | CH2CO2H | H | H | 1 | >465.12 | >465.12 | - | >465.12 |
| A11 | Cl | CO2H | H | 0 | 1.52 | 210.76 | 138.66 | 78.68 |
| A12 | OH | CO2H | H | 0 | 0.69 | 133.46 | 193.42 | 37.36 |
| A13 | H | CO2H | H | 0 | 11.81 | 88.42 | 7.49 | 42.23 |
| A14 | H | tetrazolyl | H | 0 | 7.7 | 249.46 | 32.40 | 25.61 |
| A15 | CO2CH3 | OH | H | 0 | 59.14 | 88.9 | 1.50 | 286.7 |
| A16 | CO2H | H | H | 0 | 173.72 | 136.28 | 0.78 | 188.74 |
| A17 | CH2CO2H | H | H | 0 | 2.1 | 203.84 | 97.07 | 225.15 |
| A18 | Cl | H | CO2H | 0 | 8.56 | >400.00 | >46.73 | 60.34 |
| A19 | CO2H | H | H | 1 | 15.55 | 298.56 | 19.20 | 160.22 |
| A20 | SO2NH2 | H | H | 0 | 99.08 | 116.8 | 1.18 | 182.44 |
SI = selectivity index, CC50/EC50
Figure 3.

Inhibition of the HIV-1 gp41 6-HB formation by small-molecule compounds (N-PAGE). N36 alone (lane 1) exhibited no band because it carries net positive charges and may migrate off the gel 35. C34 alone (lane 2) displayed a band at a lower position in the gel. The mixture of N36 and C34 (lane 3) showed a band at the upper position in gel, corresponding to that of 6-HB 35. In the presence of A3 (lane 4) and A4 (lane 5), N36 and C34 could still form the 6-HB, while addition of A1 (lane 6), A2 (lane 8), A12 (lane 9) and A14 (lane 7) resulted in disappearance of the 6-HB band and reappearance of the C34 band, suggesting that these compounds can block the 6-HB formation between N36 and C34.
Figure 4.

Correlation between the inhibitory activities of the A series compounds on HIV-1 replication and gp41 6-HB formation.
Notably, the 2,5-dimethylpyrrole compounds A11-A14, A17 and A19 were significantly more potent than the corresponding pyrrole compounds A1-A4, A7 and A9, respectively. This fact, along with previous theoretical work,29 indicated that the two methyl groups on the pyrrole ring and a near perpendicular conformation between benzene and pyrrole rings by steric crowding effect may be favorable for targeting the gp41 binding site, as shown in Fig 2. Further comparisons between A13 (EC50 11.81 μM) and A16 (EC50 173.72 μM), A2 (EC50 9.66 μM) and A8 (EC50 81.67 μM), and A3 (EC50 44.81 μM) and A6 (EC50 69.25 μM) indicated that a carboxyl group at the m-position of the benzene ring is more suitable than the p-position for enhancing anti-HIV potency. After an isosteric replacement of carboxyl, A14 with a tetrazolyl moiety at the m-position of the benzene ring was more potent (EC50 7.70 μM) than A13. More interestingly, compound A14 exhibited more potent inhibitory activity against six-helix bundle formation (IC50 25.61 μM) than A1 (IC50 58.74 μM), suggesting that tetrazolyl moiety could be a potential structural fragment for designing novel small-molecule HIV fusion inhibitors targeting gp41. By introducing an additional hydroxyl or chloride on the phenyl ring, compounds A1, A2, A11, A12, and A18 all exhibited potent anti-HIV activity with an EC50 value range of 0.69 – 9.66 μM. This suggests that more boundary substituents may greatly enhance molecular affinity with the binding site. Meanwhile, Compound A17 with more linear molecular size exhibited higher potency (EC50 2.10 μM) than its counterpart A16 (EC50 173.72 μM), thus guiding us to expand molecular size in order to enhance the affinity of inhibitor with the binding site.
Among the phenylpyrrole derivatives, A12 was the most effective in inhibiting p24 production (EC50 0.69 μM) and gp41 6-HB formation (IC50 37.36 μM). It also effectively inhibited HIV-1-mediated cytopathic effect (CPE) and cell-cell fusion (data not shown). The other two N-carboxylphenyl-2,5-dimethypyrroles, A11 and A17, also showed promising potency.
To understand and interpret the bioassay results, molecular modeling studies (2D-, 3D-Quantitative Structure Activity Relationship and molecular docking) were performed for the compounds from series A.29 The docking analysis for the two active compounds A12 and A14 with the hydrophobic pocket of the gp41 N-trimer (Figures 5–6) indicates lower calculated binding free energies of ΔG, −6.9 and −5.6 kcal/mol, respectively. The following main interactions between A12 and gp41 were expected: 1) interaction of two H-bonds from the hydroxyl group of A12 with the main-chain carbonyl oxygen of Gln575 and that of the m-carboxyl group of A12 with the main-chain amine of Gln577, respectively; 2) an electrostatic interaction between the carboxyl group of A12 and Arg579; and 3) hydrophobic interactions of the pyrrole ring with Lys574 and Ile573 and that of the phenyl ring with the Trp571. The electrostatic force oriented A12 in the direction of Arg579, and the steric force of the methyl groups of A12 resulted in a T-shaped effective binding conformation between the benzene and pyrrole rings. However, A14 with a three-aromatic-ring skeleton occupied more binding site space, thus providing more hydrophobic interactions between A14 and surface amino acid residues of the gp41 binding site, i.e., the benzene ring with Ile573 and the pyrrole ring with the hydrocarbon chain of Lys574. The tetrazolyl ring with more negative charges orients A14 closer to Arg579 and serves as an H donor to form an H-bond with main-chain carbonyl oxygen of Gln572. However, the molecular modeling results indicated that, while both A12 and A14 have similar binding orientation and perpendicular conformation between aromatic rings, the three-ring angle geometric conformation of A14 might match the binding site better, thus enhancing binding affinity.
Figure 5.

Surface representation of the hydrophobic pocket of HIV-1 gp41 with the docked compound A12 (left) or A14 (right).
Figure 6.

The docking conformations of active compounds A12 (left) and A14 (right) inside the hydrophobic pocket of HIV-1 gp41and residues of the pocket surrounding ligands.
Among compounds B1-B11 with an oxadiazole ring, only a few (e.g., B6 and B10) displayed moderate inhibitory activity against HIV-1 replication and 6-HB formation with selectivity index (SI) values > 16. However, the B series of compounds with a thiadiazole B12, maleimide B13-B15, or rhodanine ring B16-B22, respectively, showed no significant inhibitory activity on HIV-1 replication and 6-HB formation (Table 2). The results suggest that these heterocyclic structures may be less effective than pyrrole in interacting with the gp41 pocket and, therefore, less effective in inhibiting HIV-1 fusion and entry.
Table 2.
Anti-HIV activity data of five-member heterocyclic compounds B1-B22 as HIV-1 gp41 inhibitors.
 | |||||||
|---|---|---|---|---|---|---|---|
| # | R | Aryl |
Inhibiting p24 production EC50 (μM) | Cytotoxicity CC50 (μM) | SI* | Inhibition of 6-HB formation IC50 (μM) | |
| Type | R′ | ||||||
| B1 | p-COOMe | CH2Br | 87.95 | 32.73 | 0.37 | >336.70 | |
| B2 | p-COOMe | CH2Cl | 41.42 | 14.23 | 0.34 | >395.26 | |
| B3 | p-COOMe | CH2SCN | 10.95 | 43.67 | 3.99 | 171.38 | |
| B4 | p-COOEt | CH2OH | 373.02 | 384.84 | 1.03 | 177.90 | |
| B5 | p-COOH | CH2OH | 152.73 | 454.55 | 2.98 | 26.45 | |
| B6 | p-COOH |
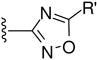
|
CH2Cl | 3.91 | 63.99 | 16.37 | >420.17 |
| B7 | p-COOH | CH2SCN | 67.97 | 383.14 | 5.64 | 177.74 | |
| B8 | p-COOH | CH2Br | 127.03 | 81.91 | 0.64 | >353.36 | |
| B9 | m-COOH | CF3 | >400 | >400 | - | >1000 | |
| B10 | p-COOH | CF3 | 3.06 | 82.25 | 26.88 | 223.64 | |
| B11 | m-COOH | CH2Cl | 57.7 | 64.9 | 1.12 | 346.2 | |
| B12 | p-COOH |
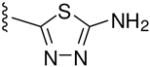
|
>400 | >400 | - | >1000 | |
| B13 |
p-COOH p-COOMe |
205.44 | >400 | >1.95 | >1000 | ||
| B14 |
m-OH p-COOH |
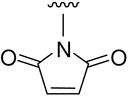
|
39.39 | 66.15 | 1.68 | 100.61 | |
| B15 | m-OH | >400 | >400 | - | >1000 | ||
| B16 | m-CF3 | 115.8 | 85.4 | 0.74 | >1000 | ||
| B17 | p-OCH3 | 63.4 | 89.6 | 1.41 | >1000 | ||
| B18 | p-OH | 35.7 | 84.9 | 2.38 | >1000 | ||
| B19 | p-COOH |
|
257.9 | 143.2 | 0.56 | >1000 | |
| B20 |
m-COOH m-COOH |
203.7 | 165.3 | 0.81 | >1000 | ||
| B21 |
p-Cl m-COOH |
179.4 | 155.3 | 0.87 | >1000 | ||
| B22 | p-OH | 165.8 | 152.0 | 0.92 | >1000 | ||
SI = selectivity index, CC50/EC50
In conclusion, we have designed and synthesized 42 N-carboxyphenylpyrrole derivatives in both the A and B categories. These novel compounds were based on the structures of small-molecule hits targeting the HIV-1 gp41, 2 and A1. We found that a majority of the compounds in the A series exhibited significant inhibitory activity on HIV-1 entry and replication as well as 6-HB formation. Their anti-HIV-1 activity is also correlated with their ability to disrupt the gp41 fusion-active core formation, suggesting that these compounds inhibit HIV-1 fusion and entry in agreement with the mechanism of action of 2 and A1 and the anti-HIV peptide C34.15, 30 Furthermore, 2,5-dimethylpyrrole compounds were generally more potent than the corresponding pyrrole compounds, suggesting that the presence of dimethyl groups resulted in a favorable T-shape conformation between the benzene and pyrrole rings. Compound A12 was the most active compound; therefore, further development is justified. In contrast, the majority of the compounds in the B series showed no significant anti-HIV-1 activity, indicating that these heterocyclic structures might be unfavorable for binding to the gp41 pocket. The docking analysis also suggests that the positively charged residues Arg579 and Lys574 are important for interaction with the compounds by forming salt bridges for stabilizing the binding of the compounds to the hydrophobic pocket. On the other hand, all compounds in the A and B series may not be large enough (< 300 Da) to fully occupy the deep hydrophobic pocket, which can accommodate a compound with a mass of ~600 Da. Therefore, current active compounds should be increased in molecular size in order to improve their efficacy in blocking the 6-HB formation and inhibiting HIV fusion and entry.
Experimental Section
Chemistry
Melting points were measured with a RY-1 melting apparatus without correction. The proton nuclear magnetic resonance (1H NMR) spectra were measured on a JNM-ECA-400 (400 MHz) spectrometer using tetramethylsilane (TMS) as internal standard. The solvent used was DMSO-D6 unless indicated. Mass spectra (MS) were measured on either a Micromass ZabSpec or an ABI Perkin-Elmer Sciex API-3000 mass spectrometer with electrospray ionization. Elementary analyses were performed with a Model-1106 analyzer. Active target compounds were analyzed for C, H, and N and gave values within ±0.4% of the theoretical values. HPLC analyses were performed using an Agilent 1100 series and an Eclipse XDB-C18 column eluting with a mixture of solvents A and B (condition 1: A/B = water/acetonitrile 30:70, flow rate 0.8 mL/min; condition 2: A/B = water/methanol 20:80, flow rate 0.6 mL/min; UV 254 nm). The microwave reactions were performed on a microwave reactor from Biotage, Inc. Thin-layer chromatography (TLC) was performed on silica gel GF254 plates. Silica gel GF254 (200–300 mesh) from Qingdao Haiyang Chemical Company was used for TLC, preparative TLC, and column chromatography. Medium-pressure column chromatography was performed using a CombiFlash® Companion™ purification system. All chemicals were obtained from Beijing Chemical Works or Sigma-Aldrich, Inc.
General procedure for the preparation of N-phenylpyrrole derivatives A1-A7, A9
To a solution of aminobenzoic acid analogs (1 eq) in 3 mL of glacial acetic acid was added 2,5-dimethoxy-tetrahydrofuran (1.1 eq). The mixture was stirred and irradiated under microwave at 150 °C for 10 min as monitored by TLC (petroleum ether/EtOAc 3:1). The mixture was poured into ice-water. The solid was filtered out and washed with water and then crude product was purified by flash column (gradual eluant: EtOAc/petroleum ether with AcOH (v/v 1:0.005) to obtain pure expected product.
N-(3-Carboxy-4-chloro)phenylpyrrole (A1, NB-64)
Yield: 82%, starting with 331 mg (1.93 mmol) of 5-amino-2-chlorobenzoic acid to afford A1, white solid, mp 125–128 °C; 1H NMR δ ppm 7.90 (1H, d, J = 2.0 Hz, ArH-2), 7.75 (1H, dd, J = 8.4 and 2.0 Hz, ArH-6), 7.60 (1H, d, J = 8.4 Hz, ArH-5), 7.43 (2H, m, PyH-2,5), 6.29 (2H, t, J = 2.2 Hz, PyH-3,4). MS m/z (%) 221 (M+, 100), 223 (M+2, 36). Anal. (C11H8ClNO2) C, H, N.
N-(3-Carboxy-4-hydroxy)phenylpyrrole (A2)
Yield: 51%, starting with 306 mg (2 mmol) of 5-aminosalicylic acid to afford A2, white solid, mp 178–180 °C; 1H NMR δ ppm 11.34 (1H, br, COOH), 7.82 (1H, d, J = 2.8 Hz, ArH-2), 7.73 (1H, dd, J = 9.2 and 2.8 Hz, ArH-6), 7.26 (2H, m, PyH-2,5), 7.07 (1H, d, J = 9.2 Hz, ArH-5), 6.24 (2H, t, J = 2.2 Hz, PyH-3,4); MS m/z (%) 203 (M+, 100). Anal. (C11H9NO3) C, H.
N-(3-Carboxy)phenylpyrrole (A3)
Yield: 57%, starting with 274 mg (2 mmol) of 3-aminobenzoic acid to afford A3, white solid, mp 175–178 °C; 1H NMR δ ppm 13.18 (1H, br, COOH), 7.98 (1H, d, J = 2.0 Hz, ArH-2), 7.80 (2H, dd, J = 8.4 and 2.0 Hz, ArH-4,6), 7.57 (1H, t, J = 8.4 Hz, ArH-5), 7.40 (2H, m, PyH-2,5), 6.26 (2H, t, J = 2.2 Hz, PyH-3,4). MS m/z (%) 187 (M+, 100). Anal. (C11H9NO2) C, H, N.
N-(3-(1H-tetrazol-5-yl)phenylpyrrole (A4)
Yield: 88%, starting with 161 mg (1 mmol) of 3-tetrazolylaniline to afford A4, white solid, mp 210–212 °C; 1H NMR δ ppm 8.19 (1H, d, J = 2.0 Hz, ArH-2), 7.92 (1H, d, J = 8.4 Hz, ArH-4), 7.83 (1H, dd, J = 8.4 and 2.0 Hz, ArH-6), 7.71 (1H, t, J = 8.4 Hz, ArH-5), 7.47 (2H, m, PyH-2,5), 6.35 (2H, t, J = 2.2 Hz, PyH-3,4); MS m/z (%) 211 (M+, 100). Anal. (C11H9N5) C, H, N.
N-(3-Hydroxy-4-methoxycarbonyl)phenylpyrrole (A5)
Yield: 71%, starting with 167 mg (1 mmol) of methyl 4-aminosalicylate to afford A5, white solid, mp 121–124 °C; 1H NMR δ ppm 10.75 (1H, s, OH), 7.85 (1H, d, J = 8.4 Hz, ArH-5), 7.52 (2H, m, PyH-2,5), 7.25 (1H, d, J = 8.4 Hz, ArH-6), 7.23 (1H, s, ArH-2), 6.31 (2H, t, J = 2.2 Hz, PyH-3,4), 3.91 (3H, s, OCH3). MS m/z (%) 217 (M+, 100). Anal. (C12H11NO3·1/8 H2O) C, H, N.
N-(4-Carboxy)phenylpyrrole (A6)
Yield: 65%, starting with 274 mg (2 mmol) of 4-aminobenzoic acid to afford A6, white solid, mp 199–201 °C; 1H NMR δ ppm 12.92 (1H, br, COOH), 8.01 (2H, d, J = 8.4 Hz, ArH-3,5), 7.73 (2H, d, J = 8.4 Hz, ArH-2,6), 7.50 (2H, m, PyH-2,5), 6.32 (2H, t, J = 2.2 Hz, PyH-3,4); MS m/z (%)187 (M+, 86). Anal. (C11H9NO2) C, H, N.
N-(4-Carboxymethene)phenylpyrrole (A7)
Yield: 77%, starting with 151 mg (1 mmol) of 4-aminophenylacetic acid to afford A7, white solid, mp 161–164 °C; 1H NMR δ ppm 12.38 (1H, br, COOH), 7.52 (2H, d, J = 8.4 Hz, ArH-2,6), 7.34 (2H, m, PyH-2,5), 7.32 (2H, d, J = 8.4 Hz, ArH-3,5), 6.26 (2H, t, J = 2.2 Hz, PyH-3,4), 3.59 (2H, s, CH2); MS m/z (%) 201 (M+, 64). Anal. (C12H11NO2·1/8 H2O) C, H, N.
N-(4-Carboxy-3-hydroxy)phenylpyrrole. (A8)
To a solution of A5 (538 mg, 2.48 mmol) in 10 mL methanol was added 15 mL of 40% aq. NaOH, and the mixture was stirred at room temperature for 6 h monitored by TLC (petroleum ether/EtOAc 3:1). The mixture was then poured into water and acidified with 5% aq. HCl to pH 3. The solid was collected, washed with water, and purified by Flash chromatography [eluant: EtOAc/petroleum ether with AcOH (4:0.02) 0~20%] to afford 157 mg of A8, 31% yield, white solid, mp 208–210 °C; 1H NMR δ ppm 13.94 (1H, br, COOH), 10.83 (1H, s, OH), 7.52 (1H, d, J = 8.4 Hz, ArH-5), 7.51 (2H, m, PyH-2,5), 7.25 (1H, d, J = 8.4 Hz, ArH-6), 7.22 (1H, s, ArH-2), 6.30 (2H, t, J = 2.2 Hz, PyH-3,4); MS m/z (%): 203 (M+, 100). Anal. (C11H9NO3) calcd C, H, N.
N-(4-Carboxy)benzylpyrrole (A9)
Yield: 70%, starting with 151 mg (1 mmol) of 4-aminomethylbenzoic acid to afford A9, white solid, mp 168–170 °C; 1H NMR δ ppm 12.92 (1H, br, COOH), 7.90 (2H, d, J = 8.4 Hz, ArH-3,5), 7.24 (2H, d, J = 8.4 Hz, ArH-2,6), 6.83 (2H, m, PyH-2,5), 6.64 (2H, t, J = 2.2 Hz, PyH-3,4), 5.19 (2H, s, CH2); MS m/z (%) 201 (M+, 93). Anal. (C12H11NO2) C, H, N.
N-(4-Carboxymethene)benzylpyrrole (A10)
To a solution of (4-bromomethyl)-phenylacetic acid (57 mg, 0.25 mmol) and pyrrole (0.02 mL, 0.29 mmol) in 2 mL of DMSO was added t-BuOK (70 mg, 0.63 mmol). The mixture was stirred and irradiated under microwave at 193 °C for 20 min monitored by TLC (chloroform/methanol/ammonia 6:1:0.1). The mixture was poured into ice-water and acidified with 2N HCl to pH 3. The solid was collected, washed with water, and purified with Flash column [eluant: EtOAc/petroleum ether with AcOH (4:0.02), 0~20%] to afford 45 mg of A10, 84% yield, pale yellow solid, mp 106–109 °C; 1H NMR δ ppm 12.28 (1H, br, COOH), 7.21 (2H, d, J = 8.4 Hz, ArH-3,5), 7.12 (2H, d, J = 8.4 Hz, ArH-2,6), 6.80 (2H, m, PyH-2,5), 6.01 (2H, t, J = 2.2 Hz, PyH-3,4), 5.06 (2H, s, N CH2), 3.53 (2H, s, -CH2CO); MS m/z (%) 215 (M+, 98). Anal. (C13H13NO2·1/8 H2O) C, H, N.
General procedure for the preparation of N-aryl-2,5-dimethylpyrroles A11-A20
To a solution of amino benzoic acid analogs (1 eq) in 3 mL of glacial acetic acid was added hexane-2,5-dione (1.1 eq). The reaction mixture was irradiated under microwave at 150 °C for 10 min and monitored by TLC (petroleum ether/EtOAc 3:1). The mixture was poured into ice-water and left overnight. The precipitated solid was filtered, washed with water, and purified by a Flash column [gradual eluant: EtOAc/petroleum ether with AcOH (4:0.02), 0~20%] to afford pure expected products.
N-(3-Carboxy-4-chloro)phenyl-2,5-dimethylpyrrole (A11)
Yield: 66%, starting with 343 mg (2 mmol) of 5-amino-2-chlorobenzoic acid to afford A11, white solid, mp 140–142 °C; 1H NMR δ ppm 13.63 (1H, br, COOH), 7.69 (1H, d, J = 8.4 Hz, ArH-5), 7.61 (1H, d, J = 2.0 Hz, ArH-2), 7.48 (1H, dd, J = 8.4 and 2.0 Hz, ArH-6), 5.83 (2H, s, PyH), 1.98 (6H, s, Py-CH3×2); MS m/z (%) 249 (M+, 100), 251 (M+2, 42). Anal. (C13H12ClNO2) C, H, N.
N-(3-Carboxy-4-hydroxy)phenyl-2,5-dimethylpyrrole (A12)
Yield: 79%, starting with 153 mg (1 mmol) of 5-aminosalicylic acid to afford A12, white solid, mp 169–171 °C; 1H NMR δ ppm 11.50 (1H, br, COOH), 7.54 (1H, d, J = 2.0 Hz, ArH-2), 7.43 (1H, dd, J = 8.4 and 2.0 Hz, ArH-6), 7.09 (1H, d, J = 8.4Hz, ArH-5), 5.78 (2H, s, PyH), 1.94 (6H, s, Py-CH3×2); MS m/z (%) 231 (M+, 100). Anal. (C13H13NO3·1/8 H2O) C, H, N.
N-(3-Carboxy)phenyl-2,5-dimethylpyrrole (A13)
Yield: 50%, starting with 274 mg (2 mmol) of 3-aminobenzoic acid to afford A13, white solid, mp 145–148 °C; 1H NMR δ ppm 13.18 (1H, br, COOH), 7.97 (1H, d, J = 8.4 Hz, ArH-4), 7.65 (1H, s, ArH-2), 7.62 (1H, t, J = 8.4 Hz, ArH-5), 7.52 (1H, d, J = 8.4 Hz, ArH-6), 5.78 (2H, s, PyH), 1.92 (6H, s, Py-CH3×2); MS m/z (%) 214 (M−H, 100). Anal. (C13H13NO2) C, H, N.
N-(3-(1H-tetrazol-5-yl)phenyl-2,5-dimethylpyrrole (A14)
The mixture of 3-tetrazolylaniline (161 mg, 1 mmol) and hexane-2,5-dione (0.3 mL, 2.5 mmol), without glacial acetic acid, was irradiated under microwave at 100°C for 10 min to afford 137 mg of A14, 57% yield, white solid, mp 147–148 °C; 1H NMR δ ppm 8.14 (1H, d, J = 8.4 Hz, ArH-4), 7.87 (1H, s, ArH-2), 7.76 (1H, t, J = 8.4 Hz, ArH-5), 7.53 (1H, d, J = 8.4 Hz, ArH-6), 5.85 (2H, s, PyH), 2.02 (6H, s, Py-CH3×2); MS m/z (%) 239 (M+, 76). Anal. (C13H13N5) C, H, N.
N-(3-Hydroxy-4-methoxycarbonyl)phenyl-2,5-dimethylpyrrole (A15)
Yield: 62%, starting with 378 mg (2.26 mmol) of methyl 4-aminosalicylate to afford A15, yellow solid, mp 58–61 °C. 1H NMR (CDCl3) δ ppm 10.92 (1H, s, OH), 7.94 (1H, d, J = 8.4 Hz, ArH-5), 6.86 (1H, d, J = 2.0 Hz, ArH-2), 6.76 (1H, dd, J = 8.4 and 2.0 Hz, ArH-6), 5.91 (2H, s, PyH), 3.99 (3H, s, OCH3), 2.08 (6H, s, Py-CH3×2); MS m/z (%) 245 (M+, 100); HPLC purity 98.6 %.
N-(4-Carboxy)phenyl-2,5-dimethylpyrrole (A16)
Yield: 83%, starting with 274 mg (2 mmol) of 4-aminobenzoic acid to afford A16, white solid, mp 177–179 °C; 1H NMR δ ppm 13.07 (1H, br, COOH), 8.06 (2H, d, J = 8.4 Hz, ArH-3,5), 7.40 (2H, d, J = 8.4 Hz, ArH-2,6), 5.83 (2H, s, PyH), 1.99 (6H, s, Py-CH3×2); MS m/z (%): 215 (M+, 100). Anal. (C13H13NO2) C, H, N.
N-(4-Carboxymethene)phenyl-2,5-dimethylpyrrole (A17)
Yield 42%, starting with 521 mg (3.45 mmol) of 4-aminophenylacetic acid to afford A17, white solid, mp 110–112 °C; 1H NMR δ ppm 7.40 (2H, d, J = 8.4 Hz, ArH-2,6), 7.20 (2H, d, J = 8.4 Hz, ArH-3,5), 5.78 (2H, s, PyH), 3.65 (2H, s, -CH2), 1.93 (6H, s, Py-CH3×2); MS m/z (%): 229 (M+, 100); Anal. (C14H15NO2) C, H, N.
N-(4-Chloro-2-carboxy)phenyl-2,5-dimethylpyrrole (A18)
The mixture of 172 mg (1 mmol) of 2-amino-5-chlorobenzoic acid and 0.3 mL of hexane-2,5-dione (2.5 mmol), without acetic acid, was irradiated under microwave at 100°C for 10 min to produce 109 mg of A18, 44% yield, pink solid, mp 124–127 °C; 1H NMR δ ppm 13.16 (1H, br, COOH), 7.86 (1H, d, J = 2.0 Hz, ArH-3), 7.75 (1H, dd, J = 8.4 and 2.0 Hz, ArH-5), 7.33 (1H, d, J = 8.4 Hz, ArH-6), 5.74 (2H, s, PyH), 1.86 (6H, s, Py-CH3×2); MS m/z (%) 248 (M−H, 100), 251 (M+2, 47). Anal. (C13H12ClNO2·1/8 H2O) C, H, N.
N-(4-Carboxy)benzyl-2,5-dimethylpyrrole (A19)
Yield: 77%, starting with 302 mg (2 mmol) of 4-aminomethylbenzoic acid to afford A19, white solid, mp 171–175 °C; 1H NMR (CDCl3) δ ppm 8.04 (2H, d, J = 8.4 Hz, ArH-3,5), 6.98 (2H, d, J = 8.4 Hz, ArH-2,6), 5.88 (2H, s, PyH), 5.07 (2H, s, CH2), 2.13 (6H, s, Py-CH3×2); MS m/z (%) 229 (M+, 100). Anal. (C14H15NO2) C, H, N.
N-(4-Aminosulfonyl)phenyl-2,5-dimethylpyrrole (A20)
The mixture of sulfanilamide (172 mg, 1 mmol) and hexane-2,5-dione (0.3 mL, 2.5 mmol), without glacial acetic acid, was irradiated under microwave at 180°C for 15 min to afford 188 mg of A20, purified with Flash chromatography [eluant: EtOAc/petroleum ether with Et3N (4:0.02), 0~20%], 75% yield, white solid, mp 158–159 °C; 1H NMR (CDCl3) δ ppm 8.05 (2H, d, J = 8.4 Hz, ArH-2,6), 7.39 (2H, d, J = 8.4 Hz, ArH-3,5), 5.94 (2H, s, PyH), 4.95 (2H, s, -SO2NH2), 2.06 (6H, s, Py-CH3×2); MS m/z (%) 250 (M+, 100). Anal. (C12H14N2O2S) C, H, N.
(Z)-(3-Carboxyl)benzamidoxime (4)
To a solution of 3-cyanobenzoic acid (588 mg, 4 mmol) in 30 mL ethanol was added hydroxylamine hydrochloric acid (595 mg, 8.56 mmol) in water (3 mL) and sodium carbonate (687 mg, 6.48 mmol) in water (6 mL), successively, in the presence of 8-hydroxyquinoline (2 mg, 0.013 mmol). The mixture was heated to reflux for 4 h monitored by TLC (petroleum ether/EtOAc/AcOH 1:1:0.03). After removal of ethanol solvent under reduced pressure, the residue was diluted with 30 mL water, and the water solution was slowly acidified with 10% HCl to pH 3.2. The white precipitate was filtrated, washed with water to neutral pH, and dried to afford 668 mg of 4, 93% yield, white solid, mp 197–199 °C; 1H NMR δ ppm 13.03 (1H, br, COOH), 9.77 (1H, s, OH), 8.27 (1H, s, ArH-2), 7.95 (2H, dd, J = 8.4 Hz, ArH-4,6), 7.53 (1H, t, J = 8.4 Hz, ArH-5), 5.95 (2H, br, NH2).
(Z)-(4-Carboxyl)benzamidoxime (5)
The preparation was the same as the synthesis of 4. Starting with 735 mg (5 mmol) of 4-cyanobenzoic acid to afford 611 mg of 5, 68% yield, white solid, mp 228 °C dec; 1H NMR δ ppm 13.02 (1H, br, COOH), 9.89 (1H, s, OH), 7.94 (2H, d, J = 8.4 Hz, ArH-3,5), 7.80 (2H, d, J = 8.4 Hz, ArH-2,6), 5.94 (2H, s, NH2); MS m/z (%) 179 (M−H, 100).
(Z)-(4-Methoxycarbonyl)benzamidoxime (6)
The preparation was the same as that of 4. Starting with methyl 4-cyanobenzoate (3) (806 mg, 5 mmol) to afford 901 mg of 6, white solid, 93% yield, mp 164–166 °C; 1H NMR δ ppm 9.91 (1H, s, OH), 7.96 (2H, d, J = 8.4 Hz, ArH-3,5), 7.83 (2H, d, J = 8.4 Hz, ArH-2,6), 5.95 (2H, br, NH2), 3.86 (3H, s, -OCH3); MS m/z (%) 195 (M+H, 100).
5-Bromomethyl-3-(4-methoxycarbonyl)phenyl-1,2,4-oxadiazole (B1)
The mixture of 6 (388 mg, 2 mmol) and bromoacetyl bromide (0.19 mL, 2.2 mmol) in THF (10 mL) was heated to reflux for 8 h and monitored by TLC (petroleum ether/EtOAc 4:1). After solvent removal, residue was purified by a silica gel column (petroleum ether/EtOAc 4:1) to afford 378 mg of B1, yield 64%, white solid, 100–102 °C; 1H NMR δ ppm 8.19 (4H, dd, J = 8.4 Hz, ArH), 5.03 (2H, s, CH2), 3.90 (3H, s, -OCH3); MS m/z (%): 296 (M+, 48), 298 (M+2, 44). Anal. (C11H9BrN2O3) C, H, N.
5-Chloromethyl-3-(4-methoxycarbonyl)phenyl-1,2,4-oxadiazole (B2)
Preparation was the same as that described for B1. Chloroacetyl chloride (0.18 mL, 2.2 mmol) was reacted with 6 (388 mg, 2 mmol) to afford B2:367 mg, 73% yield, white solid, mp 57–58 °C; 1H NMR δ ppm 8.19 (4H, dd, J = 8.4 Hz, ArH), 5.22 (2H, s, CH2), 3.90 (3H, s, OCH3); MS m/z (%) 252 (M+, 37), 254 (M+2, 13). Anal. (C11H9ClN2O3·½ H2O) C, H, N.
3-(4-Methoxycarbonyl)phenyl-5-(thiocyanatomethyl)-1,2,4-oxadiazole (B3)
A mixture of B2 (184 mg, 0.73 mmol) and ammonium thiocyanate (228 mg, 3 mmol) in 5 mL of DMF was heated at 90 °C for 3 h and monitored by TLC (petroleum ether/EtOAc 4:1). The solution was poured into ice-water and a precipitated yellow solid was filtered, washed with water, and purified by a silica gel column (petroleum ether/EtOAc 4:1) to obtain B3:89 mg, 44% yield, pale yellow solid, mp 124–126 °C; 1H NMR δ ppm 8.20 (4H, dd, J = 8.4 Hz, ArH), 4.88 (2H, s, CH2), 3.91 (3H, s, OCH3); MS m/z (%) 275 (M+, 46); HPLC purity 98.7 %.
3-(4-Ethoxycarbonyl)phenyl-5-hydroxymethyl-1,2,4-oxadiazole (B4)
A solution of B1 (90 mg, 0.3 mmol) in ethanol (3 mL) in the presence of 1 mL of aq. NaOH (1N) was stirred at room temperature for 4 h. The mixture was poured into water, acidified with 5% HCl to pH 5, and then extracted with EtOAc three times. Then organic solvent was removed in vacuo, and residue was purified by a silica gel column (petroleum ether/EtOAc 4:1) to afford B4:52 mg, 70% yield, white solid, mp 145–148 °C; 1H NMR (CDCl3) δ ppm 9.05 (1H, s, OH), 8.17 (2H, d, J = 8.4 Hz, ArH-3,5), 7.85 (2H, d, J = 8.4 Hz, ArH-2,6), 4.49 (2H, s, -CH2OH), 4.42 (2H, q, -OCH2CH3), 1.44 (3H, t, -CH2CH3); ESI-MS m/z (%) (C12H12N2O4) 248 (M+, 100); HPLC purity 97.1%.
3-(4-Carboxy)phenyl-5-hydroxymethyl-1,2,4-oxadiazole (B5)
A mixture of B2 (30 mg, 0.12 mmol) in ethanol (3 mL) and 1 mL of aq. NaOH (1N) was stirred at room temperature for 4 h. After removal of ethanol in vacuo, more water (ca. 8 mL) was added, washed with EtOAc (2×5 mL), and acidified with 5% HCl to pH 2. Solid was filtered out, washed with water to neutral pH, and dried to afford B5:18 mg, 68% yield, white solid, mp 240–242 °C; 1H NMR δ ppm 13.27 (1H, br, COOH), 11.53 (1H, s, OH), 8.03 (2H, d, J = 8.4 Hz, ArH-3,5), 7.88 (2H, d, J = 8.4 Hz, ArH-2,6), 4.41 (2H, s, CH2); MS m/z (%) 219 (M−H, 100); HPLC purity 99.6%.
3-(4-Carboxy)phenyl-5-chloromethyl-1,2,4-oxadiazole (B6)
To a solution of B2 (30 mg, 0.12 mmol) in glacial acetic acid (1 mL) was dropped 1 mL of hydrochloric acid (36~38%). The mixture was heated at 100 °C for 8 h. After cooling to room temperature, precipitated solid was filtered out, washed with water to neutral pH, and dried to afford B6:16 mg, 55% yield, white solid, mp 210–212 °C; 1H NMR δ ppm 13.35 (1H, br, COOH), 8.17 (4H, dd, J = 8.4 Hz, ArH), 5.22 (2H, s, CH2);MS m/z (%): 237 (M−H, 94), 239 (M−H+2, 28); HPLC purity 99.2%.
3-(4-Carboxy)phenyl-5-thiocyanatomethyl-1,2,4-oxadiazole (B7)
Preparation was the same as that described for B6. Starting with 27 mg of B3 to afford B7, 38% yield, white solid, mp 240–244 °C; 1H NMR δ ppm 13.26 (1H, br, COOH), 8.08 (4H, dd, J = 8.4 Hz, ArH), 4.53 (2H, s, CH2); MS m/z (%) 274 (M−CN+K, 100), HPLC purity 96.2%.
5-Bromomethyl-3-(4-carboxy)phenyl-1,2,4-oxadiazole (B8)
A mixture of B1 (40 mg, 0.13 mmol) in 1 mL of glacial acetic acid and 1 mL hydrobromic acid (40%) was heated at 100 °C for 12 h to afford 25 mg of B8, white solid, 65% yield, mp 209–211 °C; 1H NMR δ ppm 13.31 (1H, br, COOH), 8.16 (4H, dd, J = 8.4 Hz, ArH), 5.02 (2H, s, CH2); MS m/z (%) 283 (M+, 69), 281 (M-2, 100); HPLC purity 99.8 %.
3-(3-Carboxyphenyl)-5-(trifluoromethyl)-1,2,4-oxadiazole (B9)
A mixture of 4 (180 mg, 1 mmol) and trifluoroacetic anhydride (0.42 mL, 3 mmol) in anhydrous pyridine (3 mL) was heated to reflux for 3 h and monitored by TLC (petroleum ether/EtOAc/AcOH 3:1:0.03). The mixture was poured into ice-water and adjusted with HCl (10%) to pH 4, then extracted with EtOAc three times. After solvent was removed, the residue was purified with Flash silica column [eluant: EtOAc/petroleum ether with AcOH (4:0.03) 3~20%] to afford B9:107 mg, 42% yield, white solid, mp 114–117 °C; 1H NMR δ ppm 13.48 (1H, s, COOH), 8.58 (1H, s, ArH-2), 8.32 (2H, dd, J = 8.4 Hz, ArH-4,6), 7.80 (1H, t, J = 8.4 Hz, ArH-5); MS m/z (%) 257 (M−H, 65); HPLC purity 97.1%.
3-(4-Carboxyphenyl)-5-(trifluoromethyl)-1,2,4-oxadiazole (B10)
The preparation was the same as that of B9. Starting with 5 (180 mg, 1 mmol) to afford 136 mg of B10, white solid, 53% yield, mp 169–171°C; 1H NMR δ ppm 13.44 (1H, br, COOH), 8.21 (2H, d, J = 8.4 Hz, ArH-3,5), 8.15 (2H, d, J = 8.4 Hz, ArH-2,6); MS m/z (%) 258 (M+, 100); HPLC purity 97.1%.
3-(3-Carboxy)phenyl-5-chloromethyl-1,2,4-oxadiazole (B11)
As described for the preparation of B2, a mixture of 4 (270 mg, 1.5 mmol) and chloroacetyl chloride (0.16 mL, 1.9 mmol) in 8 mL anhydrous THF was refluxed for 4 h. The mixture was poured into ice-water, and precipitated solid was collected, washed with water to neutral pH, and dried in vacuo to afford B11:132 mg, 37% yield, white solid, mp 128–132 °C; 1H NMR δ ppm 13.39 (1H, s, COOH), 8.56 (1H, s, ArH-2), 8.28 (1H, d, J = 8.4 Hz, ArH-4), 8.18 (1H, d, J = 8.4 Hz, ArH-6), 7.76 (1H, t, J = 8.4 Hz, ArH-5); MS m/z (%) 237 (M−H, 62), 239 (M−H+2, 36); HPLC purity 96.9%.
4-Carboxylbenzaldehyde thiosemicarbazone (7)
A solution of 4-carboxybenzaldehyde (450 mg, 3 mmol) and thiosemicarbazide (300 mg, 3.3 mmol) in ethanol (6 mL) was heated to reflux for 1.5 h and monitored by TLC (CHCl3/CH3OH/AcOH 3:1:0.05). After reaction mixture was cooled, the solid was filtered out, washed with ethanol, and dried to give 606 mg of 7, 91% yield, yellow solid, mp 250 °C dec; 1H-NMR δ ppm 13.04 (1H, br, COOH), 11.56 (1H, s, -CH=), 8.29 (1H, br, NH), 8.11 (2H, br, NH2), 7.93 (4H, m, ArH); MS m/z (%) 222 (M−H, 100).
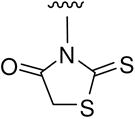
5-Amino-3-(4-carboxy)phenyl-1,3,4-thiadiazole (B12)
Thiosemicarbazone 7 (370 mg, 1.66 mmol) and ammonium ferric sulfate (3.2 g, 6.64 mmol) were soluble in H2O (15 mL), and the mixture was heated to reflux for 10 h. The mixture was then poured into ice-water, basified with 10% NaOH aq to pH 5, and extracted with EtOAc three times. Next, organic solvent was removed in vacuo, and residue was purified by a silica gel column (CHCl3/CH3OH/AcOH 9:1:0.02) to obtain 171 mg of B12, 47% yield, yellow solid, mp 240 °C dec; 1H NMR δ ppm 8.00 (2H, d, J = 8.4 Hz, ArH-3,5), 7.84 (2H, d, J = 8.4 Hz, ArH-2,6), 7.57 (2H, br, NH2); MS m/z (%): 222 (M+H, 100); HPLC purity 98.9%.
(Z)-4-[(4-Carboxy)phenylamino]-4-oxobut-2-enoic acid (8)
A solution of maleic anhydride (108 mg, 1.1 mmol) and 4-aminobenzoic acid (137 mg, 1 mmol) in EtOAc (7 mL) was stirred for 1 h at r.t., resulting in production of abundant crystal solid. The solid was filtered and washed with EtOAc to afford 223 mg of 8, 95% yield, yellow solid, mp 218–220 °C; 1H NMR δ ppm 12.82 (1H, br, COOH), 10.60 (1H, s, NH), 7.92 (2H, d, J = 8.4 Hz, ArH-3,5), 7.74 (2H, d, J = 8.4 Hz, ArH-2,6), 6.51 (1H, d, J = 12.0 Hz, =CH-2), 6.34 (1H, d, J = 12.0 Hz, =CH-3); MS m/z (%) 234 (M−H, 100).
(Z)-4-[(3-Hydroxy-4-methoxycarbonyl)phenylamino]-4-oxobut-2-enoic acid (9)
The method of synthesis was the same as that of 8. Starting with 334 mg (2 mmol) of methyl 4-aminosalicylate, 234 mg, 88% yield, yellow solid, mp 204–207 °C; 1H-NMR δ ppm 12.87 (1H, br, COOH), 10.61 (1H, s, OH), 10.56 (1H, s, NH), 7.76 (1H, d, J = 8.4 Hz, ArH-5), 7.41 (1H, s, ArH-2), 7.13 (1H, d, J = 8.4 Hz, ArH-6), 6.49 (1H, d, J = 12.0 Hz, =CH-3), 6.34 (1H, d, J = 12.0 Hz, =CH-2), 3.87 (3H, s, OCH3); MS m/z (%) 264 (M−H, 100).
N-(4-Carboxy)phenylmaleimide (B13)
A mixture of 8 (223 mg, 0.95 mmol) and triethylamine (0.06 mL, 0.475 mmol) in 3 mL acetic anhydride was stirred and heated at 60 °C for 1 h. The mixture was poured into ice-water, and the solid isolated by filtration was washed with water to neutral pH, and dried in vacuo to afford B13:98 mg, 47% yield, white solid, mp 160–163 °C; 1H NMR δ ppm 12.91 (1H, br, COOH), 8.05 (2H, d, J = 8.4 Hz, ArH-3,5), 7.51 (2H, d, J = 8.4 Hz, ArH-2,6), 7.23 (2H, s, Maleimide-H); MS m/z (%) 217 (M+, 100); HPLC purity 98.5%.
N-(3-Hydroxy-4-methoxycarbonyl)phenylmaleimide (B14)
Preparation was the same as that of B13. Solid 9 (389 mg, 1.47 mmol) was reacted with triethylamine (0.1 mL, 0.73 mmol) in 5 mL acetic anhydride to afford 132 mg of B14, 27% yield, white solid, mp 121–123 °C; 1H NMR δ ppm 10.62 (1H, s, OH), 7.88 (1H, d, J = 8.4 Hz, ArH-5), 7.22 (2H, s, Maleimide-H), 7.04 (1H, d, J = 2.0 Hz, ArH-2), 7.00 (1H, dd, J = 8.4 and 2.0 Hz, ArH-6), 3.90 (3H, s, OCH3); MS m/z (%) 247 (M+, 54). Anal. (C12H9NO5) C, H, N.
N-(4-Carboxy-3-hydroxy)phenylmaleimide (B15)
Compound B14 (40 mg, 0.16 mmol) was hydrolyzed in the same condition as B6 to afford B15, yield 79%, white solid, mp 141–144 °C; 1H NMR δ ppm 12.81 (1H, br, COOH), 11.42 (1H, s, OH), 7.43 (1H, d, J = 8.4 Hz, ArH-5), 6.09 (1H, dd, J = 8.4 and 2.0 Hz, ArH-6), 6.06 (2H, s, Maleimide-H), 5.96 (1H, d, J = 2.0 Hz, ArH-2); MS m/z (%) 232 (M−H, 11); HPLC purity 97.5%.
General procedure for the preparation of N-phenylrhodanine derivatives B16-B22
A suspension of aniline analogs (1 eq) in 4 mL water was heated to 95 °C until the aniline was fully dissolved. Then bis(carboxymethyl) trithiocarbonate (1.1 eq) was added. The resulting solution was heated at 100 °C for 19 h and was cooled to room temperature. The precipitated solid was filtrated out and washed with water. The dried solid was purified by silica gel column (petroleum ether/acetone 3:1) or Flash silica column [eluant: EtOAc/petroleum ether with HOAc (3:0.03), 20~30%] to obtain pure product.
N-(3-(Trifluoromethyl)phenyl)rhodanine (B16)
Yield: 54%, starting with 2 mL (16 mmol) of 3-(trifluoromethyl)aniline to afford B16, yellow solid, mp 150–152 °C. 1H NMR (CDCl3) δ ppm 7.77 (1H, d, J = 8.4 Hz, ArH-4), 7.70 (1H, t, J = 8.4 Hz, ArH-5), 7.51 (1H, s, ArH-2), 7.43 (1H, d, J = 8.4 Hz, ArH-6), 4.23 (2H, s, CH2). MS m/z (%) 278 (M+H, 17); Anal. (C10H6F3NOS2) C, H, N.
N-(4-Methoxyphenyl)rhodanine (B17)
Yield: 87%, starting with 492 mg (4 mmol) of 4-methoxyaniline to afford B17, pale yellow solid, mp 124–127 °C. 1H-NMR (CDCl3) δ ppm 7.13 (2H, d, J = 8.4 Hz, ArH-2,6), 7.05 (2H, d, J = 8.4 Hz, ArH-3,5), 4.18 (2H, s, CH2), 3.85 (3H, s, OCH3); MS m/z (%) 240 (M+H, 14); Anal. (C10H9NO2S2) C, H, N.
N-(4-Hydroxyphenyl)rhodanine (B18)
Yield: 88%, starting with 436 mg (4 mmol) of 4-aminophenol to afford B18, pale yellow solid, mp 180–182 °C. 1H NMR (CDCl3) δ ppm 9.12 (1H, s, OH), 7.00 (4H, dd, J = 8.4 Hz, ArH-2,3,5,6), 4.17 (2H, s, CH2); MS m/z (%) 224 (M−H, 52); HPLC purity 99.6%.
N-(4-Carboxyphenyl)rhodanine (B19)
Yield: 77%, starting with 822 mg (6 mmol) of 4-aminobenzoic acid to afford B19, pale white solid, mp 188–190 °C; 1H NMR δ ppm 13.23 (1H, br, COOH), 8.09 (2H, d, J = 8.4 Hz, ArH-3,5), 7.43 (2H, d, J = 8.4 Hz, ArH-2,6), 4.40 (2H, s, CH2); MS m/z (%) 252 (M−H, 84); HPLC purity 99.9%.
N-(3-Carboxyphenyl)rhodanine (B20)
Yield: 86%, starting with 418 mg (3 mmol) of 3-aminobenzoic acid to afford B20, white solid, mp 185–187 °C; 1H NMR δ ppm 13.27 (1H, br, COOH), 8.05 (1H, d, J = 8.4 Hz, ArH-6), 7.87 (1H, s, ArH-2), 7.69 (1H, t, J = 8.4 Hz, ArH-5), 7.55 (1H, d, J = 8.4 Hz, ArH-4), 4.37 (2H, s, CH2); MS m/z (%) 252 (M−H, 100); HPLC purity 99.8%.
N-(3-Carboxy-4-chlorophenyl)rhodanine (B21)
Yield: 81 %, starting with 343 mg (2 mmol) of 5-amino-2-chlorobenzoic acid to afford B21, white solid, mp 184–186 °C; 1H NMR δ ppm 13.71 (1H, br, COOH), 7.78 (1H, d, J = 2.4 Hz, ArH-6), 7.75 (1H, d, J = 8.4 Hz, ArH-3), 7.50 (1H, dd, J = 8.4 and 2.4 Hz, ArH-4), 4.35 (2H, s, CH2); MS m/z (%) 286 (M−H, 100), 288 (M−H+2, 44); HPLC purity 99.5%.
N-(3-Carboxy-4-hydroxyphenyl)rhodanine (B22)
Yield: 28%, starting with 306 mg (2 mmol) of 5-aminosalicylic acid to afford B22, yellow solid, mp 174–176 °C; 1H-NMR δ ppm: 13.09 (1H, br, COOH), 11.52 (1H, s, OH), 7.72 (1H, d, J = 2.4 Hz, ArH-6), 7.41 (1H, dd, J = 8.4 and 2.4 Hz, ArH-4), 7.10 (1H, d, J = 8.4 Hz, ArH-3), 4.33 (2H, s, CH2); MS m/z (%) 269 (M+, 20), 268 (M−H, 100); HPLC purity 99.7%.
Determination of the inhibitory activity of the compounds on HIV-1 replication
The inhibitory activity of compounds on HIV-1 IIIB replication in MT-2 cells was determined as previously described.1 In brief, 1 × 104 MT-2 cells were infected with an HIV-1 strain (100 TCID50) in 200 μL RPMI 1640 medium containing 10% FBS in the presence or absence of a test compound at graded concentrations overnight. Then, the culture supernatants were removed and fresh media containing no test compounds were added. On the fourth day post-infection, 100 μL of culture supernatants were collected from each well, mixed with equal volumes of 5% Triton X-100 and assayed for p24 antigen, which was quantitated by ELISA. Briefly, wells of polystyrene plates (Immulon 1B, Dynex Technology, Chantilly, VA) were coated with HIV immunoglobulin (HIVIG), which was prepared from plasma of HIV-seropositive donors with high neutralizing titers against HIV-1IIIB, in 0.085 M carbonate-bicarbonate buffer (pH 9.6) at 4 °C overnight, followed by washes with washing buffer (0.01M PBS containing 0.05% Tween-20) and blocking with PBS containing 1% dry fat-free milk (Bio-Rad, Inc., Hercules, CA). Virus lysates were added to the wells and incubated at 37 °C for 1 h. After extensive washes, anti-p24 mAb (183-12H-5C), biotin-labeled anti-mouse IgG1 (Santa Cruz Biotech., Santa Cruz, CA), streptavidin-labeled horseradish peroxidase (Zymed, S. San Francisco, CA), and the substrate 3,3′,5,5′-tetramethylbenzidine (Sigma Chemical Co., St. Louis, MO) were added sequentially. Reactions were terminated by addition of 1N H2SO4. Absorbance at 450 nm was recorded in an ELISA reader (Ultra 386, TECAN, Research Triangle Park, NC). Recombinant protein p24 purchased from US Biological Swampscott, MA) was included for establishing standard dose response curves. Each sample was tested in triplicate. The percentage of inhibition of p24 production was calculated as previously described.31 The effective concentrations for 50% inhibition (EC50) were calculated using a computer program, designated CalcuSyn,32 kindly provided by Dr. T. C. Chou (Sloan-Kettering Cancer Center, New York, New York).
Assessment of in vitro cytotoxicity
The in vitro cytotoxicity of compounds on MT-2 cells was measured by XTT assay.33 Briefly, 100 μL of the test compound at graded concentrations were added to equal volumes of cells (5 × 105/mL) in wells of 96-well plates. After incubation at 37 °C for 4 days, 50 μL of XTT solution (1 mg/mL) containing 0.02 μM of phenazine methosulphate (PMS) was added. After 4 h, the absorbance at 450 nm was measured with an ELISA reader. The CC50 (concentration for 50% cytotoxicity) values were calculated using the CalcuSyn program.32
Sandwich ELISA and native polyacrylamide gel electrophoresis for detecting the gp41 6-HB formation
A sandwich ELISA as previously described34 was used to test the inhibitory activity of the compounds on gp41 six-helix bundle formation. Briefly, peptide N36 (2 μM) was pre-incubated with the compound at graded concentrations at 37 °C for 30 min, followed by addition of C34 (2 μM). In the control experiments, N36 was pre-incubated with C34 or PBS at 37 °C for 30 min, in the absence of the compounds tested. After incubation at 37 °C for 30 min, the mixture was added to wells of a 96-well polystyrene plate which were precoated with IgG (10 μg/ml) purified from rabbit antisera directed against the gp41 six-helix bundle. Then, the mAb NC-1, biotin-labeled goat-anti-mouse IgG, SA-HRP and TMB were added sequentially. Absorbance at 450 nm was read, and the percentage of inhibition by the compounds was calculated as previously described.34 All the samples were tested in triplicate. In addition, a native polyacrylamide gel electrophoresis (N-PAGE)35 was used to confirm the inhibitory activity of the compounds on the gp41 6-HB formation. In brief, the N-peptide N36 (100 μM) was mixed with an equimolar concentration of the C-peptide C34 in the presence or absence of the compounds tested. The mixtures were incubated at 37 °C for 30 min, followed by the addition of Tris-glycine native sample buffer (Invitrogen, Carlsbad, CA). The samples (20 μl) were then loaded onto Tris-glycine gels (18%; Invitrogen, Carlsbad, CA), which were run under 125 V constant voltage at room temperature for 2 hrs. The gels were stained with Coomassie Blue and then visualized with the FluorChem 8800 Imaging System (Alpha Innotech Corp., San Leandro, CA).
Supplementary Material
Elemental analysis and HPLC purity data for final compounds. This material is available free of charge via the Internet at http://pubs.acs.org
Acknowledgments
This investigation was supported by grants 2006AA02Z319 and 2006DFA33560 from the Ministry of Science and Technology in China and 2007G06 from Beijing Municipal Science & Technology Commission to L. Xie, grants 2005Z2-E4041 from Guangzhou Science and Technology Key Project and RO1 AI46221 from the U.S. National Institutes of Health to S. Jiang, and the PhD fellowship SFRH/BD/22190/2005 from Fundação para a Ciência e a Tecnologia (Portugal) to Cátia Teixeira.
References
- 1.De Clercq E. Antiviral drugs in current clinical use. J Clin Virol. 2004;30:115–133. doi: 10.1016/j.jcv.2004.02.009. [DOI] [PubMed] [Google Scholar]
- 2.Johnson VA, Brun-Vezinet F, Clotet B, Conway B, D’Aquila RT, Demeter LM, Kuritzkes DR, Pillay D, Schapiro JM, Telenti A, Richman DD. Drug resistance mutations in HIV-1. Top HIV Med. 2003;11:215–221. [PubMed] [Google Scholar]
- 3.Richman DD, Morton SC, Wrin T, Hellmann N, Berry S, Shapiro MF, Bozzette SA. The prevalence of antiretroviral drug resistance in the United States. AIDS. 2004;18:1393–1401. doi: 10.1097/01.aids.0000131310.52526.c7. [DOI] [PubMed] [Google Scholar]
- 4.Carr A, Cooper DA. Adverse effects of antiretroviral therapy. Lancet. 2000;356:1423–1430. doi: 10.1016/S0140-6736(00)02854-3. [DOI] [PubMed] [Google Scholar]
- 5.Moore JP, Jameson BA, Weiss RA, Sattentau QJ. The HIV-cell fusion reaction. In: Bentz J, editor. Viral Fusion Mechanisms. CRC Press; Boca Raton: 1993. pp. 233–289. [Google Scholar]
- 6.Chan DC, Kim PS. HIV entry and its inhibition. Cell. 1998;93:681–684. doi: 10.1016/s0092-8674(00)81430-0. [DOI] [PubMed] [Google Scholar]
- 7.Jiang S, Zhao Q, Debnath AK. Peptide and non-peptide HIV fusion inhibitors. Curr Pharm Des. 2002;8:563–580. doi: 10.2174/1381612024607180. [DOI] [PubMed] [Google Scholar]
- 8.Jiang S, Lin K, Strick N, Neurath AR. HIV-1 inhibition by a peptide. Nature. 1993;365:113. doi: 10.1038/365113a0. [DOI] [PubMed] [Google Scholar]
- 9.Wild CT, Shugars DC, Greenwell TK, McDanal CB, Matthews TJ. Peptides corresponding to a predictive alpha-helical domain of human immunodeficiency virus type 1 gp41 are potent inhibitors of virus infection. Proc Natl Acad Sci USA. 1994;91:9770–9774. doi: 10.1073/pnas.91.21.9770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lu M, Blacklow SC, Kim PS. A trimeric structural domain of the HIV-1 transmembrane glycoprotein. Nat Struct Biol. 1995;2:1075–1082. doi: 10.1038/nsb1295-1075. [DOI] [PubMed] [Google Scholar]
- 11.Kilby JM, Eron JJ. Novel therapies based on mechanisms of HIV-1 cell entry. N Engl J Med. 2003;348:2228–2238. doi: 10.1056/NEJMra022812. [DOI] [PubMed] [Google Scholar]
- 12.Lalezari JP, Henry K, O’Hearn M, Montaner JS, Piliero PJ, Trottier B, Walmsley S, Cohen C, Kuritzkes DR, Eron JJ, Jr, Chung J, DeMasi R, Donatacci L, Drobnes C, Delehanty J, Salgo M. Enfuvirtide, an HIV-1 fusion inhibitor, for drug-resistant HIV infection in north and south America. N Engl J Med. 2003;348:2175–2185. doi: 10.1056/NEJMoa035026. [DOI] [PubMed] [Google Scholar]
- 13.Chan DC, Fass D, Berger JM, Kim PS. Core structure of gp41 from the HIV envelope glycoprotein. Cell. 1997;89:263–273. doi: 10.1016/s0092-8674(00)80205-6. [DOI] [PubMed] [Google Scholar]
- 14.Weissenhorn W, Dessen A, Harrison SC, Skehel JJ, Wiley DC. Atomic Structure of the Ectodomain from HIV-1 gp41. Nature. 1997;387:426–428. doi: 10.1038/387426a0. [DOI] [PubMed] [Google Scholar]
- 15.Liu S, Wu S, Jiang S. HIV entry inhibitors targeting gp41: from polypeptides to small-molecule compounds. Curr Pharm Des. 2007;13:143–162. doi: 10.2174/138161207779313722. [DOI] [PubMed] [Google Scholar]
- 16.Liu S, Boyer-Chatenet L, Lu H, Jiang S. Rapid and automated fluorescence-linked immunosorbent assay for high throughput screening of HIV-1 fusion inhibitors targeting gp41. J Biomol Screening. 2003;8:685–693. doi: 10.1177/1087057103259155. [DOI] [PubMed] [Google Scholar]
- 17.Jiang S, Lu H, Liu S, Zhao Q, He Y, Debnath AK. N-substituted pyrrole derivatives as novel human immunodeficiency virus type 1 entry inhibitors that interfere with the gp41 six-helix bundle formation and block virus fusion. Antimicrob Agents Chemother. 2004;48:4349–4359. doi: 10.1128/AAC.48.11.4349-4359.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chan DC, Chutkowski CT, Kim PS. Evidence that a prominent cavity in the coiled coil of HIV type 1 gp41 is an attractive drug target. Proc Natl Acad Sci USA. 1998;95:15613–15617. doi: 10.1073/pnas.95.26.15613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Malashkevich VN, Chan DC, Chutkowski CT, Kim PS. Crystal structure of the simian immunodeficiency virus (SIV) gp41 core: Conserved helical interactions underlie the broad inhibitory activity of gp41 peptides. Proc Natl Acad Sci USA. 1998;95:9134–9139. doi: 10.1073/pnas.95.16.9134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jiang S, Debnath AK. A salt bridge between an N-terminal coiled coil of gp41 and an antiviral agent targeted to the gp41 core is important for anti-HIV-1 activity. Biochem Biophys Res Commun. 2000;270:153–157. doi: 10.1006/bbrc.2000.2411. [DOI] [PubMed] [Google Scholar]
- 21.He Y, Liu S, Jing W, Lu H, Cai D, Chin DJ, Debnath AK, Kirchhoff F, Jiang S. Conserved residue Lys574 in the cavity of HIV-1 gp41 coiled-coil domain is critical for six-helix bundle stability and virus entry. J Biol Chem. 2007;282:25631–25639. doi: 10.1074/jbc.M703781200. [DOI] [PubMed] [Google Scholar]
- 22.He Y, Liu S, Li J, Lu H, Qi Z, Liu Z, Debnath AK, Jiang S. Conserved salt-bridge between the N- and C-terminal heptad repeat regions of HIV-1 gp41 core structure is critical for virus entry and inhibition. J Virol. 2008 Sep 3; doi: 10.1128/JVI.01060-08. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chiu PK, Sammes MP. The synthesis and chemistry of azolenines. Part 18. Preparation of 3-ethoxycarbonyl-3h-pyrroles via the paal-knorr reaction, and sigmatropic rearrangements involving competitive ester migrations to c-2, c-4 and n. Tetrahedron. 1990;46:3439–3456. [Google Scholar]
- 24.Santilli AA, Morris RL. Synthesis of 3-arylsulfonylmethyl-1,2,4-oxadiazole- 5-carboxylic acid derivatives. J Heterocycl Chem. 1979;16:1197–1200. [Google Scholar]
- 25.Cottrell DM, Capers J, Salem MM, DeLuca-Fradley K, Croft SL, Werbovetz KA. Antikinetoplastid activity of 3-aryl-5-thiocyanatomethyl-1,2,4-oxadiazoles. Bioorg Med Chem. 2004;12:2815–2824. doi: 10.1016/j.bmc.2004.03.054. [DOI] [PubMed] [Google Scholar]
- 26.Yu CY, Li CX, Wang C, Wang ZH. Preparation and performance evaluation of maleimide type bactericides. Industrial water treatment. 2004;24:36–41. [Google Scholar]
- 27.Powers JP, Piper DE, Li Y, Mayorga V, Anzola J, Chen JM, Jaen JC, Lee G, Liu J, Peterson MG, Tonn GR, Ye Q, Walker NP, Wang Z. SAR and mode of action of novel non-nucleoside inhibitors of hepatitis C NS5b RNA polymerase. J Med Chem. 2006;49:1034–1046. doi: 10.1021/jm050859x. [DOI] [PubMed] [Google Scholar]
- 28.Huang L, Yuan X, Aiken C, Chen CH. Bifunctional anti-human immunodeficiency virus type 1 small molecules with two novel mechanisms of action. Antimicrob Agents Chemother. 2004;48:663–665. doi: 10.1128/AAC.48.2.663-665.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Teixeira C, Barbault F, Rebehmed J, Liu K, Xie L, Lu H, Jiang S, Fan B, Maurel F. Molecular modeling studies of N-substituted pyrrole derivatives--potential HIV-1 gp41 inhibitors. Bioorg Med Chem. 2008;16:3039–3048. doi: 10.1016/j.bmc.2007.12.034. [DOI] [PubMed] [Google Scholar]
- 30.Liu S, Lu H, Xu Y, Wu S, Jiang S. Different from the HIV fusion inhibitor C34, the anti-HIV drug Fuzeon (T-20) inhibits HIV-1 entry by targeting multiple sites in gp41 and gp120. J Biol Chem. 2005;280:11259–11273. doi: 10.1074/jbc.M411141200. [DOI] [PubMed] [Google Scholar]
- 31.Zhao Q, Ma L, Jiang S, Lu H, Liu S, He Y, Strick N, Neamati N, Debnath AK. Identification of N-phenyl-N’-(2,2,6,6-tetramethyl-piperidin-4-yl)-oxalamides as a new class of HIV-1 entry inhibitors that prevent gp120 binding to CD4. Virology. 2005;339:213–225. doi: 10.1016/j.virol.2005.06.008. [DOI] [PubMed] [Google Scholar]
- 32.Chou TC, Hayball MP. CalcuSyn: Windows software for dose effect analysis. BIOSOFT; Ferguson, MO 63135, USA: 1991. [Google Scholar]
- 33.Debnath AK, Radigan L, Jiang S. Structure-based identification of small molecule antiviral compounds targeted to the gp41 core structure of the human immunodecifiency virus type 1. J Med Chem. 1999;42:3203–3209. doi: 10.1021/jm990154t. [DOI] [PubMed] [Google Scholar]
- 34.Jiang S, Lin K, Zhang L, Debnath AK. A screening assay for antiviral compounds targeted to the HIV-1 gp41 core structure using a conformation-specific monoclonal antibody. J Virol Methods. 1999;80:85–96. doi: 10.1016/s0166-0934(99)00041-5. [DOI] [PubMed] [Google Scholar]
- 35.Liu S, Zhao Q, Jiang S. Determination of the HIV-1 gp41 postfusion conformation modeled by synthetic peptides: applicable for identification of the HIV-1 fusion inhibitors. Peptide. 2003;24:1303–1313. doi: 10.1016/j.peptides.2003.07.013. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Elemental analysis and HPLC purity data for final compounds. This material is available free of charge via the Internet at http://pubs.acs.org