

Abstract
The cerebral cortex, the neostriatum (Str), and the external segment of the globus pallidus (GPe) form a cortico-Str-GPe disynaptic connection, which is one of the major connections in the basal ganglia circuitries and a target of dopamine modulation. The aim of this study was to examine the actions of D2-like dopamine receptors (D2LRs) in this connection using rat brain slice preparations. Electrical stimulation of the frontal cortex evoked disynaptic inhibitory postsynaptic currents (IPSCs) in cesium-filled GPe neurons voltage-clamped at 0 mV. The IPSCs evoked by threshold stimulation were small, <10 pA. Bath or local applications of the D2LR agonist quinpirole to Str decreased the amplitude of the cortical stimulation-induced IPSCs. Electrical stimulation of Str evoked monosynaptic IPSCs in GPe neurons. Local application of quinpirole to GPe decreased the Str stimulation-induced IPSCs. Bath application of quinpirole decreased the frequency of large miniature IPSCs (mIPSCs) that were considered to be evoked by local collateral axons of GPe neurons. These results suggested that activation of D2LRs decrease the gain of the cortico-Str-GPe disynaptic connection, with the decrease attributed to activation of D2LRs in Str and GPe, and that both Str-GPe and GPe-GPe GABAergic inhibitions are under the control of presynaptic D2LRs.
INTRODUCTION
The cerebral cortex, the neostriatum (Str), and the external segment of the globus pallidus (GPe) form a cortico-Str-GPe disynaptic connection. This disynaptic connection might play a significant role in motor control in normal and in pathological conditions such as Parkinson's disease because GPe projects to most of basal ganglia nuclei and controls their level and pattern of firing activity (Bolam et al. 2000; Kita 1994a/b, 2007; Kita et al. 2005; Mink 1996). The connection is a target for dopamine modulation because significant changes in GPe activity take place in parkinsonian patients and in experimental parkinsonian animals (Beric et al. 1996; Filion and Tremblay 1991; Pan and Walters 1988; Sterio et al. 1994; Wichmann and DeLong 2006).
Various studies have suggested that nigral dopaminergic projections to Str and GPe modulate the activity of the cortico-Str-GPe connection in a rather complex manner. We intended to clarify some of issues regarding the actions of D2-like (mostly D2 and D4) dopamine receptors (D2LRs) in the connection. In Str, two opposing actins of D2LRs are possible, and in GPe, two types of GABAergic axons can be modulated by D2LRs, as summarized below.
Immuno-electron microscopy studies localized D2LRs on cortico-Str nerve terminals (Wang and Pickel 2002). Physiological studies reported that application of D2LR agonists decreased cortico-Str synaptic transmissions by decreasing glutamate release from the synaptic terminals (Calabresi et al. 1992; Flores-Hernandez et al. 1997; Hsu et al. 1995). It has also been well documented that Str neurons projecting only to GPe express high levels of D2LRs (Gerfen et al. 1990; Meador-Woodruff and Mansour 1991). Activation of D2LRs on Str-GPe projection neurons opened potassium channels and suppressed their firing (Freedman and Weight 1988; Greif et al. 1995). Thus activation of D2LRs on both cortical terminals and Str cells may suppress activity of the cortico-Str-GPe connection. An opposing action is also possible. Because the Str neurons projecting to GPe express D2LRs, their local collateral axons and axons in GPe are also likely to express D2LRs. In addition, fast-spiking GABAergic parvalbumin containing interneurons and cholinergic interneurons in Str and their axon terminals express D2LRs (Alcantara et al. 2003; Delle Donne et al. 1997; Lenz et al. 1994; Maurice et al. 2004; Wang et al. 2006). Activation of D2LRs on these axons would suppress GABAergic inhibitions on Str-GPe neurons and could augment activity of the cortico-Str-GPe connection. The first aim of this study was to determine how the two opposing actions of D2LR activation in Str affect the gain of disynaptic cortico-Str-GPe connection.
GPe neurons express mRNAs for D2LRs (Gerfen et al. 1990; Meador-Woodruff and Mansour 1991). Thus local collateral axons of GPe projection neurons might express D2LRs. The Str-GPe axons also express D2LRs, as mentioned above. Although sparse, GPe receives dopaminergic fibers from the substantia nigra pars compacta (Lavoie et al. 1989; Lindvall and Bjorklund 1979). Previous physiological studies have suggested that activation of D2LRs decrease GABA release (Floran et al. 1997) or GABAergic IPSCs in GPe (Cooper and Stanford 2001; Querejeta et al. 2001; Shin et al. 2003). However, it still remains uncertain whether D2LRs control only Str-GPe synapses or both Str-GPe and GPe-GPe synapses. The second aim of this study was to study this issue using whole cell patch-clamp recording from GPe neurons in rat brain slice preparations.
METHODS
Slice preparation
This study was performed in compliance with the National Institutes of Health Guide for Care and Use of Laboratory Animals. Sprague-Dawley rats (16–21 days old) were anesthetized with an intraperitoneal injection of a mixture of ketamine (85 mg/kg) and xylazine (15 mg/kg). After decapitation, the brain was rapidly removed, and blocks containing the cortex, Str, and GPe were obtained. Oblique sagittal slices (400 μm thick) were cut from the blocks on a vibrating-blade microtome (Leica VT1000S, Leica Microsystems, Nussloch, Germany) in ice-cold oxygenated artificial cerebrospinal fluid (ACSF) containing (in mM) 126 choline chloride, 3 KCl, 1.24 NaH2PO4, 26 NaHCO3, 0.5 CaCl2, 6.3 MgSO4, 0.2 thiourea, 0.2 ascorbic acid, and 20 d-glucose, pH 7.4. The slices were incubated in ACSF containing (in mM) 126 NaCl, 3 KCl, 1.24 NaH2PO4, 26 NaHCO3, 2.4 CaCl2, 1.3 MgSO4, and 10 d-glucose, equilibrated with 95% O2-5% CO2 at pH 7.4 and 32°C for 1 h before recording.
Electrophysiological recordings
The slices were transferred to a recording chamber with oxygenated ACSF continuously perfused at a flow rate of ≈2 ml/min. The temperature of the recording chamber was kept at 32 ± 1°C. Whole cell recording pipettes with a tip diameter of ∼1.5 μm were pulled from 1.5 mm, thin-wall, borosilicate glass capillaries on a horizontal electrode puller (P-97, Sutter Instruments, Navato, CA). GABAA-mediated inhibitory postsynaptic currents (IPSCs) and miniature IPSCs (mIPSCs) were recorded with pipettes filled with (in mM) 110 Cs2SO4, 5 tetraethylammonium, 0.5 CaCl2, 2 MgCl2, 5 EGTA, 5 HEPES, 5 Mg-ATP, 0.2% Neurobiotin, and 3 QX-314 with the pH adjusted to 7.2 with CsOH. Cs2SO4, tetraethylammonium, and QX-314 were used to block voltage-gated potassium currents and action potential generation. Neurobiotin was used to histologically confirm the locations of recorded GPe neurons. The resistance of these recording pipettes was 3–8 MΩ. All IPSCs and mIPSCs in this study were recorded from neurons voltage clamped at 0 mV. Neurons and recording pipettes were visualized using an infrared-differential interference contrast microscope BX50WI (Olympus, Tokyo, Japan), with a ×40 water immersion objective LUM Plan PL (Olympus) and a CCD camera (OLY-150, Olympus).
Whole cell recordings were obtained with an amplifier Axopatch-200B (Axon Instruments, Foster City, CA). The output of the amplifier was monitored with an oscilloscope HM407 (HAMEG, East Meadow, NY). Signals were filtered at 5 kHz, digitized at 10 kHz with a computer interface ITC-18 (InstruTECH, Port Washington, NY), and stored on the hard disc drive of a Macintosh G4 computer, using the data acquisition and analysis system Axopatch 4.6 (Axon Instruments).
Postsynaptic responses
To evoke cortico-Str-GPe disynaptic IPSCs, triple stimulation (a train of 3 current pulses with 3-ms interpulse intervals, each pulse 200 μs in duration and ≤200 μA) was applied through a bipolar electrode (tip distance, 0.5 mm) every 10 s. The cathode of the bipolar electrode was placed on the subcortical white matter, and the anode was on the deep layer of the cortex. To evoke Str-GPe monosynaptic IPSCs, double stimulation (at 50-ms interpulse intervals) was applied to Str using similar stimulus electrodes.
To block ionotropic glutamatergic responses, the AMPA/kainate receptor antagonist 1,2,3,4-tetrahydro-6-nitro-2,3-dioxobenzo(f)quinoxaline-7sulfonamide (NBQX; 10 μM) and the N-methyl-d-aspartate (NMDA) receptor antagonist, 3-(2-carboxypiperzin-4-yl)-propyl-1-phosponic acid (CPP, 30 μM), were applied to the bath. To block GABAA responses, gabazine (10 μM) was applied to the bath. To record action potential-independent mIPSCs, TTX (1 μM) was applied to the bath in addition to the glutamate blockers. The D2LR agonist, (−)quinpirole hydrochloride, was applied to the bath or locally to the cortex, Str, or GPe. For local application, a stainless steel tube with an inner diameter of ≈0.6 mm was used. The shaft of the tube was positioned parallel to bath flow and tilted ≈30 degree from the horizontal plain, with the orifice about 0.3 mm above the slice and 0.5 mm upstream to a desired drug application site. The quinpirole-containing (10–50 μM) ACSF was ejected (0.2–0.3 ml/min) from the tube (Fig. 3A). We assume that the ejected quinpirole was diluted with bath ACSF before reaching the effective site of the slice, with the degree of the dilution dependent on factors such as relative location from the tube orifice.
Data analysis and statistics
All group data were expressed as a means ± SD and analyzed statistically using Student's paired t-test. P < 0.05 was considered statistically significant. mIPSCs were analyzed using the Mini Analysis Program (Synaptosoft, Decatur, GA). Events were ranked by amplitude and interevent interval for the preparation of cumulative probability distributions within 1- to 3-min epochs for control and drug conditions. The cumulative probability distributions were compared with the Kolmogorov-Smirnov test in the Mini Analysis Program.
Chemicals
NBQX, CPP, (−)quinpirole hydrochloride, (S)-(−) sulpiride, TTX, and 2-[3-carboxypropyl]-3-amino-6-[4-methoxyphenyl]pyridazinium bromide (gabazine) were obtained from Sigma-Aldrich (St. Louis, MO). QX-314 was obtained from Alomone Labs (Jerusalem, Israel).
RESULTS
Cortical stimulation evoked IPSCs in GPe
Single electrical stimulation of the cortex in oblique sagittal slice preparations failed to evoke IPSCs in GPe neurons. However, stimulation of the cortex with triple current pulses did evoke IPSCs in GPe neurons, although the success rate was small—less than one neuron per 10 slices. IPSCs evoked by a threshold stimulus intensity were small (<10 pA) and had long latencies (23.8 ± 4.0 ms, n = 15). When the stimulus intensity was gradually increased, the response amplitude gradually increased, and the latency gradually shortened. The response amplitude and the latency became relatively stable after reaching a certain stimulus intensity (Fig. 1, A and B). The shortest latency and the largest amplitude of IPSCs observed with ≤120-μA stimulus intensity was 16.8 ± 2.3 ms and 38 ± 5 pA (n = 15). Bath application of the ionotropic glutamate receptor antagonist mixture NBQX (10 μM) plus CPP (30 μM) or the GABAA receptor antagonist gabazine (10 μM) blocked the IPSCs (Fig. 1C).
FIG. 1.

Triple stimulation with 3-ms interpulse interval of the cortex evoked inhibitory postsynaptic currents (IPSCs) in globus pallidus (GPe) neurons. The IPSCs in this and all subsequent figures were recorded with pipettes containing Cs2SO4 (110 mM), tetraethylammonium (5 mM), and QX-314 (3 mM) to block potassium channels and from neurons voltage clamped at 0 mV. The stimulus artifacts in this and subsequent figures were truncated. A: examples of IPSCs evoked by 2 different stimulus intensities. Aa: stimulations with 63 μA evoked ∼7-pA IPSCs. Ab: increase in the stimulus intensity increased the amplitude of IPSCs and decreased the latency. B: plots of the amplitude (○) and the latency (•) of the IPSCs against the stimulus intensity. The data were obtained from the same neuron shown in A. Plots indicate that amplitudes and latencies of the IPSCs changed with the change in stimulus intensity. C: IPSCs from another GPe neuron. Ca: control in standard artificial cerebrospinal fluid (ACSF). Cb: bath application of 1,2,3,4-tetrahydro-6-nitro-2,3-dioxobenzo(f) quinoxaline-7sulfonamide (NBQX; 10 μM) and 3-(2-carboxypiperzin-4-yl)-propyl-1-phosponic acid (CPP; 30 μM) completely blocked the IPSCs. Cc: IPSCs partially recovered after a 20-min wash. Cd: subsequent application of gabazine (10 μM) also completely blocked the IPSCs.
Effects of quinpirole on cortical stimulation-evoked IPSCs in GPe
Bath application of quinpirole (10 and 20 μM) significantly decreased (∼60 and 45% of control, respectively) the amplitude of cortical stimulation-evoked IPSCs in GPe (Fig. 2, A and B). The decrease of the amplitude was associated with a 2- to 5-ms increase in the response latency. Quinpirole did not alter the input resistances of the neurons assessed by voltage pulses of 10-mV and 30-ms duration (Fig. 2C). Bath application of sulpiride (20 μM) blocked the quinpirole effects (Fig. 2E). Sulpiride itself had no significant effects on the IPSCs.
FIG. 2.

Bath application of quinpirole decreased the amplitude of the cortical stimulation-evoked IPSCs in GPe neurons. A: examples of IPSCs taken at the time points (a–d) marked in B. B: changes of the amplitude of IPSCs observed in a GPe neuron during bath application of 10 and 20 μM quinpirole for 5 min each. A partial recovery was seen after a 15-min wash. Each point in the graph represents a mean amplitude of 6 consecutive IPSCs. C: quinpirole did not alter the input resistance, monitored with 10 mV and 30-ms pulses, of 9 GPe neurons (P > 0.05, paired t-test). The 9 neurons include 4 from this evoked IPSC experiment and 5 from the miniature IPSC (mIPSC) experiment shown in Fig. 6. In this and subsequent summary figures, each line represents data from a neuron and dots with error bars represent means and SD of data from all neurons. D: a summary shows that quinpirole significantly decreased the amplitude of IPSCs (*P < 0.05, **P < 0.01, paired t-test). In this and subsequent summary figures, each data point in the graph represents a mean amplitude of 6 consecutive IPSCs obtained immediately before drug application (for control) or at the end of drug application, and the amplitudes of IPSCs are normalized by the control amplitude. The numbers of neurons are shown above the plots. E: pretreatment with sulpiride (20 μM, sulpi.) for 5 min blocked the quinpirole (quin.) effect on IPSCs. Sulpiride alone did not alter the amplitude of IPSCs.
To assess effects of D2LR activation in Str, quinpirole was applied locally to Str through a stainless steel tube (Fig. 3A). Application of 10–50 μM quinpirole to Str significantly decreased the amplitude of the cortical stimulation-evoked IPSCs in GPe (Fig. 3, B–D). Local application of quinpirole, ≤50 μM, to the cortical stimulus site did not change amplitudes of IPSCs recorded from three GPe neurons (data not shown). We were unsuccessful in the application of quinpirole locally to the GPe recording site with cortical stimulation because of technical difficulties, including the rare occurrences of stable cortical stimulation-evoked responses and the loss of recording during quinpirole application.
FIG. 3.

Local application of quinpirole to neostriatum (Str) reduced the amplitude of the cortical stimulation-evoked IPSCs in GPe neurons. A: diagram of experimental setup. Quinpirole (10–50 μM in the ACSF, 0.2–0.3 ml/min) was applied through a stainless steel tube (ID ≈ 0.6 mm) that was aimed at the middle of Str. B: examples of IPSCs recorded from a GPe neuron show that quinpirole decreased the amplitude of IPSCs [samples were taken at the time points (a–e) indicated in C]. C: decrease of IPSC amplitude observed during sequential local application of 10, 20, and 50 μM quinpirole for 5 min each. D: summary of the quinpirole effects. Quinpirole with concentrations 10–50 μM all significantly decreased the IPSC amplitudes. Only a partial recovery was observed after a 20-min wash. Paired t-test compared with control (**P < 0.01, ***P < 0.001).
Effects of quinpirole on Str stimulation-evoked IPSCs in GPe
We examined effects of quinpirole application to GPe on Str stimulation-evoked IPSCs because these were more readily recorded than cortical stimulation-evoked IPSCs and because some loss of neurons during recording could be tolerated. The properties of Str stimulation-evoked gabazine-sensitive IPSPs in GPe have been studied previously. Low-intensity Str stimulation evoked long, ∼8 ms, latency IPSCs that were considered to be evoked mainly by Str-GPe axons. High-intensity stimulation evoked short, ∼5 ms, latency IPSCs that were composed of an early component evoked by intrapallidal collateral axons of GPe-Str projection neurons overlapped with the long latency component evoked by Str-GPe axons (Kita 2007; Ogura and Kita 2000). In this study, double stimulation of Str with 50-ms interpulse intervals was used to evoke IPSCs in GPe neurons in slices perfused with ACSF containing the AMPA/kainate receptor antagonist NBQX (10 μM) and the NMDA receptor antagonist CPP (30 μM). Long- and short-latency IPSCs were evoked by adjusting the stimulus intensity.
Quinpirole (10–50 μM) was applied to the recording sites in GPe through a stainless steel tube with the orifice placed ∼0.5 mm upstream of the bath flow similar to the Str application described above. Quinpirole significantly reduced the amplitude of IPSCs (Fig. 4, A–C). The reduction of the amplitude was accompanied by an increase in paired pulse ratios (PPRs, the ratio of the amplitude of IPSCs from the second test to the first conditioning stimulation; Fig. 4D). Similar experiments were performed with higher Str stimulus intensities to evoke large IPSCs with ∼5 ms latency. Quinpirole significantly reduced the amplitude of IPSCs (Fig. 4, E–G) without a significant increase in PPRs (Fig. 4H). Quinpirole application to the recording site ≤50 μM did not alter the input resistances of the neurons (n = 10, data not shown).
FIG. 4.

Application of quinpirole to the GPe recording site reduced the amplitude of Str stimulation-evoked IPSCs in GPe neurons. ACSF contained NBQX (10 μM) and CPP (30 μM) to block glutamatergic responses. Long- (A–D) or short-latency (E–H) IPSCs were evoked by adjusting the strength of striatal stimulation. A and E: sample traces of long (7.4 ms) latency small IPSCs (A) and short (5.2 ms) latency IPSCs (E) to double Str stimulation [samples were taken at the time points (a and b) indicated in B and F]. B and F: decrease of the amplitude of both long- and short-latency IPSCs observed during sequential application of 10, 20, and 50 μM quinpirole to GPe for 5 min each. C and G: summaries of the quinpirole effects on IPSC amplitude. Quinpirole significantly decreased the IPSC amplitudes. D and H: Quinpirole increased paired pulse ratios of only long-latency IPSCs. Paired t-test compared with control (*P < 0.05, **P < 0.01, ***P < 0.001).
Local application of quinpirole (20 and 50 μM) to the Str stimulus site also significantly reduced the amplitudes of long-latency IPSCs without altering their PPRs (Fig. 5, A and B). However, quinpirole (≤50 μM) failed to alter short-latency IPSCs (Fig. 5, C and D).
FIG. 5.

Application of quinpirole to the Str stimulus site reduced amplitudes of Str stimulation-evoked long latency IPSCs but not short-latency IPSCs in GPe neurons. A and C: summaries of the quinpirole effects on the amplitude of both long- and short-latency IPSCs observed during sequential application of 10, 20, and 50 μM quinpirole to Str for 5 min each. B and D: quinpirole did not alter paired pulse ratios of these responses. Paired t-test compared with control (*P < 0.05, **P < 0.01).
Quinpirole reduced the frequency but not the amplitude of mIPSCs
To examine whether quinpirole suppresses GABA release from synaptic boutons of intra-GPe collateral axons, mIPSCs were recorded from GPe neurons in the presence of NBQX (10 μM), CPP (30 μM), and TTX (1 μM). Only mIPSCs with amplitudes exceeding 10 pA were analyzed, and neurons exhibiting mIPSCs with frequencies exceeding 1 Hz were selected for this experiment (Fig. 6A). Gabazine (10 μM) application blocked the mIPSCs, confirming that they were mediated via GABAA receptors (data not shown). Bath application of quinpirole (5–20 μM) significantly decreased the frequency of mIPSCs (Fig. 6B) without changing their mean amplitude (Fig. 6C) or the amplitude distribution (Fig. 6D). Bath application of quinpirole ≤20 μM did not alter the input resistance of these GPe neurons (n = 5; Fig. 2C). It has been shown that activation of presynaptic serotonergic receptors decreases GABA release in GPe. To test the possibility that quinpirole activates serotonin receptors on GABAergic terminals, slices were pretreated with 10 μM methysergide, a serotonin 1/2/5/6/7 receptor antagonist that blocks receptors on GABAergic terminals in GPe (Hashimoto and Kita 2008). Methysergide did not occlude 20 μM quinpirole effects on the frequency of mIPSPs (Fig. 6B).
FIG. 6.
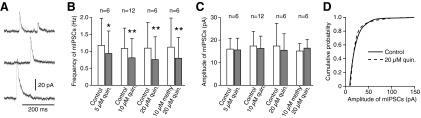
Bath application of quinpirole reduced the frequency but not the amplitude of mIPSCs. mIPSCs with amplitudes exceeding 10 pA were recorded from GPe neurons with ACSF containing NBQX (10 μM), CPP (30 μM), and TTX (1 μM). A: sample traces of mIPSCs. B and C: frequencies and amplitudes of mIPSCs before and during application of 5–20 μM quinpirole. Quinpirole significantly diminished the frequencies but not the mean amplitudes of mIPSCs. The pretreatment of slices with 10 μM methysergide, a serotonin receptor antagonist, did not occlude 20 μM quinpirole effects on the frequency of mIPSCs (B). Paired t-test compared with control (*P < 0.05, **P < 0.01). D: cumulative amplitude distributions of mIPSCs recorded from a GPe neuron show no amplitude shift with 20 μM quinpirole (P > 0.05, Kolmogorov-Smirnov test).
DISCUSSION
The primary aim of this study was to clarify the effects of D2LR activation in the cortico-Str-GPe disynaptic connection. The results showed that D2LRs control the gain of the disynaptic connection in both Str and GPe through presynaptic mechanisms.
Cortical stimulation evoked IPSCs in GPe
Stimulation of the cortex in oblique sagittal slices cut from rat brains could evoke IPSCs in GPe neurons. There are two possible connections that could induce IPSCs: cortico-Str-GPe and cortico-GPe-GPe (Naito and Kita 1994). The following data suggest that the IPSCs were mediated by the cortico-Str-GPe disynaptic connection. The IPSCs were sensitive to bath application of either glutamate receptor or GABAA receptor antagonists. The latency of the IPSCs shortened with an increase in the stimulus intensity, and the latency matched those expected from the conduction time of the cortico-Str-GPe connection. The IPSCs evoked by threshold stimulation were small (<10 pA). This suggests that Str projection neurons evoke small IPSCs in GPe neurons, because their local collateral axons evoke small IPSCs in neighboring Str neurons (Czubayko and Plenz 2002; Jaeger et al. 1994; Koos et al. 2004) and that co-activation of a large number of Str neurons is required to inhibit autonomously active GPe neurons.
The yield of this experiment was low for two main reasons: only small numbers of the disynaptic connection survived in slice preparations, and Str neurons in slice preparations had very negative resting potential and required large excitatory postsynaptic potentials (EPSPs) to reach their spike threshold. The fact that we could observe IPSCs with triple but not with single stimulation is consistent with the second reason.
Effects of quinpirole on cortical stimulation-evoked IPSCs in GPe
Bath application of quinpirole decreased IPSCs, whereas sulpiride application antagonized the quinpirole effect. This observation suggested that the overall effect of D2LR activation was a decrease in the gain of cortico-Str-GPe connection. Activation of D2LRs in Str can inhibit Str projection neurons by presynaptically decreasing cortico-Str excitation (Calabresi et al. 1992; Flores-Hernandez et al. 1997; Hsu et al. 1995; Wang et al. 2006) and postsynaptically opening potassium channels of Str projection neurons (Freedman and Weight 1988; Waszczak et al. 1998). Activation of D2LRs in Str can also disinhibit Str projection neurons by inhibiting fast firing GABAergic interneurons (Trevitt et al. 2005) and by decreasing GABA release from local collateral axons of striato-GPe projection neurons (Delgado et al. 2000). This study showed that the effects of D2LR activation that inhibit are stronger than those that disinhibit Str-GPe neurons.
Local application of quinpirole to the cortical stimulation site had no effect on the cortical stimulation-evoked IPSCs in GPe. This result may not be caused by a lack of quinpirole effects on cortico-Str neurons. Instead, we speculate that the result was caused by the placement of the stimulation electrodes. The cathode of the bipolar electrodes was located on the subcortical white matter and the anode on the deep layer of the cortex. This arrangement was suited for stimulating cortico-Str axons but not the somata or dendrites of cortical neurons.
Effects of quinpirole on Str stimulation-evoked IPSCs in GPe
Because of technical difficulties, we could not examine the effects of locally applied quinpirole to GPe recording sites with cortical stimulation-induced IPSCs. Instead, we examined the effects of local quinpirole application on Str stimulation-evoked IPSCs. Str stimulation at low intensity evoked ∼8-ms-latency IPSCs with <10-pA amplitude, and stimulation at high-intensity evoked ∼5-ms-latency IPSCs with large amplitude in GPe neurons. Both IPSCs had a constant latency, followed double shock, and were completely blocked by application of 10 μM gabazine. We believe that the long-latency IPSCs were evoked mainly by Str-GPe axons and that the short-latency IPSCs were composed of an early component evoked by intrapallidal collateral axons of GPe-Str projection neurons (Kita 2007; Kita and Kitai 1994; Ogura and Kita 2000). Application of quinpirole to the GPe recording site decreased the amplitude of long-latency IPSCs and increased PPRs, suggesting that activation of D2LRs on Str-GPe axons decreased the probability of GABA release. Quinpirole also significantly decreased the amplitude of short-latency IPSCs with no significant change in their PPRs, suggesting that activation of D2LRs on intra-GPe collateral axons also suppressed GABA release. To confirm these possibilities, we examined effects of bath application of quinpirole on mIPSCs with the amplitude exceeding 10 pA. We confirmed that quinpirole decreased the frequency but not the amplitude or the amplitude distribution of mIPSCs. The serotonin antagonist methysergide, which blocked the presynaptic serotonin effects in rat GPe in vitro (Hashimoto and Kita 2008), did not occlude the quinpirole effect, suggesting quinpirole did not activate presynaptic serotonin receptors. The amplitude of IPSCs evoked by threshold cortical stimulation and threshold Str stimulation were <10 pA (also see Kita 2007). Thus most of the mIPSCs recorded in the recent study were likely to be evoked by intra-GPe collateral axons. These results indicated that activation of D2LRs suppresses GABA release from synaptic boutons of both Str-GPe and local collateral axons of GPe neurons projecting to Str.
Other observations
GPe neurons express D4 dopamine receptors (Ariano et al. 1997; Mauger et al. 1998), which increase potassium conductance and postsynaptically reduce GABAergic IPSCs in GPe neurons in mice (Shin et al. 2003). This study excluded the effects of D4 receptor activation because intracellular Cs ions and tetraethylammonium should largely block potassium currents of the neurons. Indeed, quinpirole application to bath or to GPe recording sites did not alter the input resistance of recorded neurons.
Washing slices for 15–20 min resulted in only partial recovery of the quinpirole effects. The slow recovery was probably caused by slow washout of quinpirole from the inside of the tissues because of the use of relatively high concentrations of quinpirole, relatively thick (400 μm) slices, and a slow flow rate (≈2 ml/min) of bath medium. Although the washout was slow, the sulpiride sensitivity suggested that the quinpirole effects observed in this study was D2 receptor mediated. A previous in vitro GPe study reported that the inhibition of evoked IPSCs or mIPSCs by 3 μM dopamine was only partially reversible, and the inhibition by >3 μM was irreversible (Cooper and Stanford 2001). They suggested the possibility that D2 receptor activation could evoke long-term changes downstream of receptor binding in GPe (Cooper and Stanford 2001).
Functional considerations
An increase in GPe firing activity was shown in some in vivo studies with systemic (Carlson et al. 1987; Hooper et al. 1997) or local (Bergstrom and Walters 1984; Querejeta et al. 2001) dopamine agonist application to GPe. The results were often interpreted as a presynaptic suppression of cortico-Str excitation or suppression of Str-GPe inhibition. The results of this study suggest that D2LR activation can suppress the gain of cortico-Str-GPe connection by suppressing both cortico-Str excitation and Str-GPe inhibition. Another role of D2LR activation in GPe may be feedback control of GPe activity through pre- and postsynaptic suppression of GPe-GPe inhibition.
The firing activity of GPe neurons in Parkinson's disease patients and experimental parkinsonian animals increases in irregularity and bursting compared with controls (Beric et al. 1996; Filion and Tremblay 1991; Sterio et al. 1994; Wichmann and DeLong 2006). Increases in sensory responses such as the movement of multiple joints in GPe were also reported (Tremblay et al. 1989). These observations suggest that the gain of cortico-Str-GPe inputs is augmented in Parkinson's disease subjects (Albin et al. 1989; Alexander and Crutcher 1990; DeLong 1990). The results of this study suggest that D2LRs control this disynaptic connection at both Str and GPe.
GRANTS
This work was supported by National Institute of Neurological Disorders and Stroke Grants NS-47085 and NS-57236.
Acknowledgments
We thank R. Kita for editing the manuscript.
The costs of publication of this article were defrayed in part by the payment of page charges. The article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
REFERENCES
- Albin et al. 1989.Albin RL, Young AB, Penney JB. The functional anatomy of basal ganglia disorders. Trends Neurosci 12: 366–375, 1989. [DOI] [PubMed] [Google Scholar]
- Alcantara et al. 2003.Alcantara AA, Chen V, Herring BE, Mendenhall JM, Berlanga ML. Localization of dopamine D2 receptors on cholinergic interneurons of the dorsal striatum and nucleus accumbens of the rat. Brain Res 986: 22–29, 2003. [DOI] [PubMed] [Google Scholar]
- Alexander and Crutcher 1990.Alexander GE, Crutcher MD. Functional architecture of basal ganglia circuits: neural substrates of parallel processing. Trends Neurosci 13: 266–271, 1990. [DOI] [PubMed] [Google Scholar]
- Ariano et al. 1997.Ariano MA, Wang J, Noblett KL, Larson ER, Sibley DR. Cellular distribution of the rat D4 dopamine receptor protein in the CNS using anti-receptor antisera. Brain Res 752: 26–34, 1997. [DOI] [PubMed] [Google Scholar]
- Bergstrom and Walters 1984.Bergstrom DA, Walters JR. Dopamine attenuates the effects of GABA on single unit activity in the globus pallidus. Brain Res 310: 23–33, 1984. [DOI] [PubMed] [Google Scholar]
- Beric et al. 1996.Beric A, Sterio D, Dogali M, Fazzini E, Eidelberg D, Kolodny E. Characteristics of pallidal neuronal discharges in Parkinson's disease patients. Adv Neurol 69: 123–128, 1996. [PubMed] [Google Scholar]
- Bolam et al. 2000.Bolam JP, Hanley JJ, Booth PA, Bevan MD. Synaptic organisation of the basal ganglia. J Anat 196: 527–542, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabresi et al. 1992.Calabresi P, Maj R, Pisani A, Mercuri NB, Bernardi G. Long-term synaptic depression in the striatum: physiological and pharmacological characterization. J Neurosci 12: 4224–4233, 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson et al. 1987.Carlson JH, Bergstrom DA, Walters JR. Stimulation of both D1 and D2 dopamine receptors appears necessary for full expression of postsynaptic effects of dopamine agonists: a neurophysiological study. Brain Res 400: 205–218, 1987. [DOI] [PubMed] [Google Scholar]
- Cooper and Stanford 2001.Cooper AJ, Stanford IM. Dopamine D2 receptor mediated presynaptic inhibition of striatopallidal GABA(A) IPSCs in vitro. Neuropharmacology 41: 62–71, 2001. [DOI] [PubMed] [Google Scholar]
- Czubayko and Plenz 2002.Czubayko U, Plenz D. Fast synaptic transmission between striatal spiny projection neurons. Proc Natl Acad Sci USA 99: 15764–15769, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delgado et al. 2000.Delgado A, Sierra A, Querejeta E, Valdiosera RF, Aceves J. Inhibitory control of the GABAergic transmission in the rat neostriatum by D2 dopamine receptors. Neuroscience 95: 1043–1048, 2000. [DOI] [PubMed] [Google Scholar]
- Delle Donne et al. 1997.Delle Donne KT, Sesack SR, Pickel VM. Ultrastructural immunocytochemical localization of the dopamine D2 receptor within GABAergic neurons of the rat striatum. Brain Res 746: 239–255, 1997. [DOI] [PubMed] [Google Scholar]
- DeLong 1990.DeLong MR Primate models of movement disorders of basal ganglia origin. Trends Neurosci 13: 281–285, 1990. [DOI] [PubMed] [Google Scholar]
- Filion and Tremblay 1991.Filion M, Tremblay L. Abnormal spontaneous activity of globus pallidus neurons in monkeys with MPTP-induced parkinsonism. Brain Res 547: 142–151, 1991. [PubMed] [Google Scholar]
- Floran et al. 1997.Floran B, Floran L, Sierra A, Aceves J. D2 receptor-mediated inhibition of GABA release by endogenous dopamine in the rat globus pallidus. Neurosci Lett 237: 1–4, 1997. [DOI] [PubMed] [Google Scholar]
- Flores-Hernandez et al. 1997.Flores-Hernandez J, Galarraga E, Bargas J. Dopamine selects glutamatergic inputs to neostriatal neurons. Synapse 25: 185–195, 1997. [DOI] [PubMed] [Google Scholar]
- Freedman and Weight 1988.Freedman JE, Weight FF. Single K+ channels activated by D2 dopamine receptors in acutely dissociated neurons from rat corpus striatum. Proc Natl Acad Sci USA 85: 3618–3622, 1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerfen et al. 1990.Gerfen CR, Engber TM, Mahan LC, Susel Z, Chase TN, Monsma FJ Jr, Sibley DR. D1 and D2 dopamine receptor-regulated gene expression of striatonigral and striatopallidal neurons. Science 250: 1429–1432, 1990. [DOI] [PubMed] [Google Scholar]
- Greif et al. 1995.Greif GJ, Lin YJ, Liu JC, Freedman JE. Dopamine-modulated potassium channels on rat striatal neurons: specific activation and cellular expression. J Neurosci 15: 4533–4544, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto and Kita 2008.Hashimoto K, Kita H. Serotonin activates presynaptic and postsynaptic receptors in rat globus pallidus. J Neurophysiol 99: 1723–1732, 2008. [DOI] [PubMed] [Google Scholar]
- Hooper et al. 1997.Hooper KC, Banks DA, Stordahl LJ, White IM, Rebec GV. Quinpirole inhibits striatal and excites pallidal neurons in freely moving rats. Neurosci Lett 237: 69–72, 1997. [DOI] [PubMed] [Google Scholar]
- Hsu et al. 1995.Hsu KS, Huang CC, Yang CH, Gean PW. Presynaptic D2 dopaminergic receptors mediate inhibition of excitatory synaptic transmission in rat neostriatum. Brain Res 690: 264–268, 1995. [DOI] [PubMed] [Google Scholar]
- Jaeger et al. 1994.Jaeger D, Kita H, Wilson CJ. Surround inhibition among projection neurons is weak or nonexistent in the rat neostriatum. J Neurophysiol 72: 2555–2558, 1994. [DOI] [PubMed] [Google Scholar]
- Kita 1994a.Kita H Parvalbumin-immunopositive neurons in rat globus pallidus: a light and electron microscopic study. Brain Res 657: 31–41, 1994a. [DOI] [PubMed] [Google Scholar]
- Kita 1994b.Kita H Physiology of two disynaptic pathways from the sensorimotor cortex to the basal ganglia output nuclei., In: The Basal Ganglia IV. New Ideas and Data on Structure and Function, edited by Percheron G, Mckenzie JS, Feger J. New York: Plenum Press, 1994b, p. 263–276.
- Kita 2007.Kita H Globus pallidus external segment. Prog Brain Res 160: 111–133, 2007. [DOI] [PubMed] [Google Scholar]
- Kita and Kitai 1994.Kita H, Kitai ST. The morphology of globus pallidus projection neurons in the rat: an intracellular staining study. Brain Res 636: 308–319, 1994. [DOI] [PubMed] [Google Scholar]
- Kita et al. 2005.Kita H, Tachibana Y, Nambu A, Chiken S. Balance of monosynaptic excitatory and disynaptic inhibitory responses of the globus pallidus induced after stimulation of the subthalamic nucleus in the monkey. J Neurosci 25: 8611–8619, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koos et al. 2004.Koos T, Tepper JM, Wilson CJ. Comparison of IPSCs evoked by spiny and fast-spiking neurons in the neostriatum. J Neurosci 24: 7916–7922, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavoie et al. 1989.Lavoie B, Smith Y, Parent A. Dopaminergic innervation of the basal ganglia in the squirrel monkey as revealed by tyrosine hydroxylase immunohistochemistry. J Comp Neurol 289: 36–52, 1989. [DOI] [PubMed] [Google Scholar]
- Lenz et al. 1994.Lenz S, Perney TM, Qin Y, Robbins E, Chesselet MF. GABA-ergic interneurons of the striatum express the Shaw-like potassium channel Kv3.1. Synapse 18: 55–66, 1994. [DOI] [PubMed] [Google Scholar]
- Lindvall and Bjorklund 1979.Lindvall O, Bjorklund A. Dopaminergic innervation of the globus pallidus by collaterals from the nigrostriatal pathway. Brain Res 172: 169–173, 1979. [DOI] [PubMed] [Google Scholar]
- Mauger et al. 1998.Mauger C, Sivan B, Brockhaus M, Fuchs S, Civelli O, Monsma F Jr. Development and characterization of antibodies directed against the mouse D4 dopamine receptor. Eur J Neurosci 10: 529–537, 1998. [DOI] [PubMed] [Google Scholar]
- Maurice et al. 2004.Maurice N, Mercer J, Chan CS, Hernandez-Lopez S, Held J, Tkatch T, Surmeier DJ. D2 dopamine receptor-mediated modulation of voltage-dependent Na+ channels reduces autonomous activity in striatal cholinergic interneurons. J Neurosci 24: 10289–10301, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meador-Woodruff et al. 1991.Meador-Woodruff JH, Mansour A. A. E. Bennett Award paper. Expression of the dopamine D2 receptor gene in brain. Biol Psychiatry 30: 985–1007, 1991. [DOI] [PubMed] [Google Scholar]
- Mink 1996.Mink JW The basal ganglia: focused selection and inhibition of competing motor programs. Prog Neurobiol 50: 381–425, 1996. [DOI] [PubMed] [Google Scholar]
- Naito and Kita 1994.Naito A, Kita H. The cortico-pallidal projection in the rat: an anterograde tracing study with biotinylated dextran amine. Brain Res 653: 251–257, 1994. [DOI] [PubMed] [Google Scholar]
- Ogura and Kita 2000.Ogura M, Kita H. Dynorphin exerts both postsynaptic and presynaptic effects in the Globus pallidus of the rat. J Neurophysiol 83: 3366–3376, 2000. [DOI] [PubMed] [Google Scholar]
- Pan and Walters 1988.Pan HS, Walters JR. Unilateral lesion of the nigrostriatal pathway decreases the firing rate and alters the firing pattern of globus pallidus neurons in the rat. Synapse 2: 650–656, 1988. [DOI] [PubMed] [Google Scholar]
- Querejeta et al. 2001.Querejeta E, Delgado A, Valdiosera R, Erlij D, Aceves J. Intrapallidal D2 dopamine receptors control globus pallidus neuron activity in the rat. Neurosci Lett 300: 79–82, 2001. [DOI] [PubMed] [Google Scholar]
- Shin et al. 2003.Shin RM, Masuda M, Miura M, Sano H, Shirasawa T, Song WJ, Kobayashi K, Aosaki T. Dopamine D4 receptor-induced postsynaptic inhibition of GABAergic currents in mouse globus pallidus neurons. J Neurosci 23: 11662–11672, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sterio et al. 1994.Sterio D, Beric A, Dogali M, Fazzini E, Alfaro G, Devinsky O. Neurophysiological properties of pallidal neurons in Parkinson's disease. Ann Neurol 35: 586–591, 1994. [DOI] [PubMed] [Google Scholar]
- Tremblay et al. 1989.Tremblay L, Filion M, Bedard PJ. Responses of pallidal neurons to striatal stimulation in monkeys with MPTP-induced parkinsonism. Brain Res 498: 17–33, 1989. [DOI] [PubMed] [Google Scholar]
- Trevitt et al. 2005.Trevitt JT, Morrow J, Marshall JF. Dopamine manipulation alters immediate-early gene response of striatal parvalbumin interneurons to cortical stimulation. Brain Res 1035: 41–50, 2005. [DOI] [PubMed] [Google Scholar]
- Wang and Pickel 2002.Wang H, Pickel VM. Dopamine D2 receptors are present in prefrontal cortical afferents and their targets in patches of the rat caudate-putamen nucleus. J Comp Neurol 442: 392–404, 2002. [DOI] [PubMed] [Google Scholar]
- Wang et al. 2006.Wang Z, Kai L, Day M, Ronesi J, Yin HH, Ding J, Tkatch T, Lovinger DM, Surmeier DJ. Dopaminergic control of corticostriatal long-term synaptic depression in medium spiny neurons is mediated by cholinergic interneurons. Neuron 50: 443–452, 2006. [DOI] [PubMed] [Google Scholar]
- Waszczak et al. 1998.Waszczak BL, Martin LP, Greif GJ, Freedman JE. Expression of a dopamine D2 receptor-activated K+ channel on identified striatopallidal and striatonigral neurons. Proc Natl Acad Sci USA 95: 11440–11444, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wichmann and DeLong 2006.Wichmann T, DeLong MR. Basal ganglia discharge abnormalities in Parkinson's disease. J Neural Transm Suppl 21–25, 2006. [DOI] [PubMed]