

Abstract
Lignin-degrading peroxidases, a group of biotechnologically interesting enzymes, oxidize high redox potential aromatics via an exposed protein radical. Low temperature EPR of Pleurotus eryngii versatile peroxidase (VP) revealed, for the first time in a fungal peroxidase, the presence of a tryptophanyl radical in both the two-electron (VPI) and the one-electron (VPII) activated forms of the enzyme. Site-directed mutagenesis was used to substitute this tryptophan (Trp-164) by tyrosine and histidine residues. No changes in the crystal structure were observed, indicating that the modified behavior was due exclusively to the mutations introduced. EPR revealed the formation of tyrosyl radicals in both VPI and VPII of the W164Y variant. However, no protein radical was detected in the W164H variant, whose VPI spectrum indicated a porphyrin radical identical to that of the inactive W164S variant. Stopped-flow spectrophotometry showed that the W164Y mutation reduced 10-fold the apparent second-order rate constant for VPI reduction (k2app) by veratryl alcohol (VA), when compared with over 50-fold reduction in W164S, revealing some catalytic activity of the tyrosine radical. Its first-order rate constant (k2) was more affected than the dissociation constant (KD2). Moreover, VPII reduction by VA was impaired by the above mutations, revealing that the Trp-164 radical was involved in catalysis by both VPI and VPII. The low first-order rate constant (k3) values were similar for the W164Y, W164H, and W164S variants, indicating that the tyrosyl radical in VPII was not able to oxidize VA (in contrast with that observed for VPI). VPII self-reduction was also suppressed, revealing that Trp-164 is involved in this autocatalytic process.
Lignin degradation, a key step for carbon recycling in land ecosystems and a central issue for industrial use of lignocellulosic biomass (e.g. in paper pulp manufacture and bioethanol production), is initiated in nature by 1-electron oxidation of the benzenic rings of lignin by specialized high redox potential fungal peroxidases (1-5). The latter include lignin peroxidase (LiP;6 EC 1.11.1.14), first described in Phanerochaete chrysosporium together with manganese peroxidase (EC 1.11.1.13), and versatile peroxidase (VP; EC 1.11.1.16), described in Pleurotus and Bjerkandera species as a hybrid enzyme combining the catalytic properties of LiP and manganese peroxidase (6, 7). Oxidation of high redox potential aromatics, including non-phenolic lignin models, by LiP (8, 9) and VP (10) takes place at an exposed tryptophan by long range electron transfer (LRET) to heme in the 2-electron (Compound I) and 1-electron (Compound II) activated forms of the enzyme. This basically differs from Mn2+ oxidation by manganese peroxidase (11) and VP (12) and oxidation of different phenolic substrates and dyes by classical plant peroxidases (13) that is produced by direct electron transfer to the activated heme cofactor.
The existence of a tryptophan radical in a peroxidase (in this case neighbor to heme) has been reported in cytochrome c peroxidase, where it participates in LRET substrate oxidation (14, 15). Direct detection of a protein radical in a ligninolytic peroxidase (corresponding to the above mentioned catalytic tryptophan) was reported in Pleurotus eryngii and Bjerkandera adusta VP by low temperature EPR of its Compound I (16-18), although indirect evidence on a homologous LiP radical had been previously obtained (19-21). Based on these results, an extension of the basic catalytic cycle of VP (22) was proposed by including two forms (A and B) of both Compound I (VPI) and Compound II (VPII) (10), although no direct evidence on the existence of VPII form B (characterized by the presence of a protein radical) was reported.
Several amino acids, including tyrosine and histidine in addition to tryptophan, can form stable free radicals playing a role in enzyme catalysis as intermediates in electron or hydrogen atom transfer (23). Two putative LRET pathways initiated at exposed histidine residues were first proposed in LiP (24, 25). The homologous residues are conserved in VP isoenzymes (22, 26). However, neither of them is catalytically relevant, as shown by site-directed mutagenesis, and only the LRET pathway initiated at the conserved tryptophan residue (VP Trp-164 and LiP Trp-171) seems to be operative (8, 10, 27).
Concerning possible tyrosine radicals, it is noteworthy that ligninolytic peroxidases are completely depleted of tyrosine residues in their sequences, whereas tyrosine residues are present in peroxidases from soil basidiomycetes such as the Coprinopsis cinerea peroxidase (6). This is most probably due to the need to maintain the enzymes active in the strongly oxidative environment characterizing wood lignin degradation, where tyrosine residues could be oxidized. A single exception exists, among the more than 50 ligninolytic basidiomycete peroxidases cloned up to date, corresponding to a Trametes cervina LiP that includes in its structure a catalytic tyrosine involved in aromatic substrate oxidation (located at a different position than LiP Trp-171 and VP Trp-164) (28). However, formation of tyrosyl radicals has been suggested in other heme and non-heme proteins (29, 30) including a plant peroxidase oxidizing bulky lignin precursor by a LRET mechanism (31).
In the present study, the presence of catalytic tryptophan radicals in both P. eryngii VPI and VPII is investigated, and the operation of alternative amino acid residues substituting the catalytic Trp-164 of VP is explored. With this purpose, the resting state and peroxide-activated forms of the native enzyme and the variants obtained by Trp-164 site-directed mutagenesis were analyzed by EPR to detect the presence and nature of radicals. Moreover, their steady-state and transient state kinetics were investigated to determine whether the VPI and VPII radicals were catalytically operative. Finally, the crystal structure of a mutated variant forming a new protein radical was solved to investigate changes in the molecular structure.
MATERIALS AND METHODS
Chemicals—Dithiothreitol, ferrocyanide, hemin, isopropyl-β-d-thiogalactopyranoside, manganese sulfate, oxidized glutathione, potassium hydrogen phthalate, Reactive Black 5 (RB5), sodium tartrate, veratryl alcohol (VA), and other chemicals were purchased from Sigma-Aldrich, with the exception of hydrogen peroxide and urea, which were from Merck. Horseradish peroxidase type VI (RZ = 3) was from Sigma-Aldrich.
Site-directed Mutagenesis—The cDNA encoding the mature isoenzyme VPL (allelic variant VPL2; GenBank™ AF007222) (22) was cloned in the expression vector pFLAG1 (International Biotechnologies Inc.). Escherichia coli DH5α was selected for plasmid propagation. The plasmid pFLAG1-VPL2 was used as template for site-directed mutagenesis by PCR using the QuikChange™ kit from Stratagene.
The following primers were used to produce the different variants (direct constructions, with the changed triplets in bold and mutations underlined, are shown): (i) W164Y, 5′-CCCGTCGAGGTTGTTTACCTCCTGGCTTCGC-3′; (ii) W164H, 5′-CCCGTCGAGGTTGTTCACCTCCTGGCTTCGCACTCC-3′; (iii) W164S, 5′-CCCGTCGAGGTTGTTTCGCTCCTGGCTTCGC-3′; (iv) R257A/A260F double variant, 5′-CGAAGATTCAGAACGCTTTCGCTTTTACCATGTCGAAGATGGCTCTTCTTGGC-3′. The W164Y/R257A/A260F triple variant was obtained using the plasmid pFLAG1-VPL2 (R257A/A260F) as template and direct and reverse oligonucleotides W164Y as primers. The mutated genes were sequenced using an ABI 3730 DNA analyzer (Applied Biosystems) to ensure that only the desired mutations occurred.
PCR reactions (50 μl final volume) were carried out in a PerkinElmer Life Sciences GeneAmp PCR system 240 using 10 ng of template DNA, 500 μm each dNTP, 125 ng of direct and reverse primers, 2.5 units of PfuTurbo polymerase (Stratagene), and the manufacturer's buffer. Reaction conditions were as follows: (i) a “hot start” at 95 °C for 1 min; (ii) 18 cycles at 95 °C for 50 s, 55 °C for 50 s, and 68 °C for 10 min; and (iii) a final cycle at 68 °C for 10 min.
Enzyme Production—Native (non-mutated) recombinant VP protein and five site-directed variants were obtained by E. coli W3110 expression after transformation with the corresponding plasmids (32). Cells were grown for 3 h in Terrific Broth, induced with 1 mm isopropyl-β-d-thiogalactopyranoside, and grown for a further 4 h. The apoenzyme accumulated in inclusion bodies was recovered using 8 m urea. Subsequent in vitro folding was performed using 0.16 m urea, 5 mm Ca2+, 20 μm hemin, 0.5 mm oxidized glutathione, 0.1 mm dithiothreitol, and 0.1 mg/ml protein concentration, at pH 9.5 (32). Active enzyme was purified by Resource-Q chromatography using a 0-0.3 m NaCl gradient (2 ml/min, 20 min) in 10 mm sodium tartrate (pH 5.5) containing 1 mm CaCl2. Electronic absorption spectra were recorded at 25 °C using a Shimadzu UV-160 spectrophotometer. The concentrations of native VP and variants were calculated from the absorption at 407 nm using an 150 mm-1 · cm-1 extinction coefficient (22).
EPR Measurements—Solutions of native VP and its directed variants (final concentration ∼275 μm) were prepared in 0.1 m phthalate buffer (pH 4.5). The W164Y variant and native VP were treated with 8 eq of H2O2, whereas the W164H and W164S variants were treated with 1 eq of H2O2, and the reactions were stopped by rapid immersion of the EPR tube in liquid N2. For VPII analysis, the enzyme solutions were treated with equimolecular amounts of H2O2 and ferrocyanide.
CW-X-band (9.4 GHz) EPR measurements were carried out with a Bruker E500 Elexsys series using the Bruker ER 4122 SHQE cavity and an Oxford helium continuous flow cryostat (ESR900). Spin quantification was performed by double integration of the experimental EPR radical signal when compared with the iron signal. EPR simulations were performed using the X-Sophe package (Bruker).
Dimerization Analysis—Native VP and the mutated variants were incubated with 8 eq of H2O2 overnight at 4 °C in 10 mm sodium tartrate, pH 5.0. SDS-PAGE of the treated samples was performed in 12% polyacrylamide gels, including low molecular mass standards (Bio-Rad). Amino acid composition was determined in a Biochrom 20 autoanalyzer (Amersham Biosciences) after hydrolysis of 7 μg of the treated proteins with 6 m HCl at 110 °C under vacuum for 20 h.
Crystallization and Structure Determination—Crystals of the W164Y variant were obtained by the sitting-drop vapor diffusion method. The protein (10 mg/ml in 10 mm sodium tartrate, pH 5.0) was mixed 1:1 with a solution containing 100 mm sodium cacodylate (pH 5.0), 4% 1,3-propanediol, and 1.4 m ammonium sulfate. Crystals appeared after 14 days at 25 °C and were “frozen” in the crystallization medium plus 25% glycerol. A complete data set was collected at the beamline ID14.2 of the European Synchrotron Radiation Facility (ESRF, Grenoble, France). Data reduction was done using MOSFLM and SCALA, from the CCP4 package (33). The structure was solved by molecular replacement using MOLREP (34) and the native recombinant VP model deposited at the Protein Data Bank (PDB; entry 2BOQ) as a search probe. Consecutive cycles of refinement and manual rebuilding were done using REFMAC (35) and COOT (36). The statistics of data collection, processing, and refinement are shown in Table 1. The structure was validated using MolProbity (37). The W164Y variant crystal structure was deposited as PDB entry 2W23. Fig. 3 was produced using PyMOL (DeLano Scientific LLC).
TABLE 1.
Statistics of W164Y variant crystal structure (PDB entry 2W23)
| Data collection | |
| Space group | I41 |
| Cell dimensions (Å) | 96.74, 96.74, 98.19, 90.00, 90.00, 90.00 |
| Resolution (Å) | 1.94 (2.04) |
| No. of observations | |
| Measured | 233971 |
| Unique | 33450 |
| Completeness (%) | 99.8 (99.4)a |
| Rmean (%) | 8.9 (27.4) |
| Multiplicity | 4.1 (3.9) |
| I/sigma I | 13.6 (5.4) |
| Refinement | |
| Rcryst/Rfree (%) | 14.44 (17.75) |
| r.m.s.d.b | |
| Bonds (Å) | 0.014 |
| Angles (°) | 1.366 |
| Ramachandran plot | |
| Favored | 98.4 |
| Outliers | 0 |
Values in parenthesis are for the highest resolution shell.
Root mean square deviations.
FIGURE 3.

Electron density map of the W164Y variant (2W23) (A) and a superimposition with native VP model (2BOQ in green)(B). Shown is a representative (2Fo - Fc) electron density map, contoured at the 1.5 σ level, displaying the tyrosine residue introduced at the position of the catalytic Trp-164 and neighbor Glu-243 and Arg-257.
Steady-state Enzyme Kinetics—Oxidation of VA to veratraldehyde (ε310 9300M-1 · cm-1) was estimated at pH 3.0, and that of RB5 (ε59830000 m-1 · cm-1) at pH 3.5. Oxidation of Mn2+ was estimated by the formation of Mn3+-tartrate complex (ε238 6500 m-1 · cm-1) using 0.1 m sodium tartrate (pH 5). All enzymatic activities were measured as initial velocities taking linear increments (decreases in the case of RB5) at 25 °C in the presence of 0.1 mm H2O2. Mean values and standard errors for apparent affinity constant (Michaelis-Menten constant, Km) and maximal enzyme turnover (catalytic constant, kcat) were obtained by non-linear least-squares fitting to the Michaelis-Menten model. Fitting of these constants to the equation v = (kcat/Km)[S]/(1+[S]/Km) yielded the efficiency or selectivity constant (kcat/Km) with its standard error.
Transient State Enzyme Kinetics—Transient state kinetic constants were measured at 25 °C (or 10 °C when required) using a stopped-flow equipment (Bio-Logic) including a three-syringe module (SFM300) synchronized with a diode array detector (J&M Analytik AG) and Bio-Kine software. VPI formation was investigated by mixing the resting enzyme with increasing concentrations of H2O2 in 100 mm sodium tartrate (pH 3) under pseudo-first-order conditions and followed at 397 nm (the isosbestic point of VPI and VPII). To investigate VPII formation, VPI was first prepared by mixing 4 μm resting enzyme with 1 eq of H2O2 in 10 mm sodium tartrate (pH 5). After 0.6 s of aging, an excess of substrate in 100 mm (final concentration) sodium tartrate, pH 3 (VA) or pH 3.5 (RB5) was added, and VPII formation was followed at 416 nm (the isosbestic point of VPII and resting enzyme). The first step to investigate VPII reduction was production and reduction of VPI by premixing (6 s) a solution of 4 μm enzyme and 4 μm ferrocyanide with 1 eq of H2O2 in 10 mm sodium tartrate (pH 5). VPII reduction was followed at 406 nm (the Soret maximum of resting enzyme) after mixing with different concentrations of VA or RB5 in the same buffers used for VPI reduction. In all cases, the final concentration of enzyme was 1 μm. All kinetic traces exhibited single exponential character from which pseudo-first-order rate constants were calculated.
RESULTS
Site-directed Variants
Five VP variants were obtained by PCR mutagenesis and, together with native VP, investigated by EPR, crystallographic, and steady and transient state kinetic techniques. The W164Y and W164H variants included tyrosine or histidine residues potentially forming protein radicals in substitution of the catalytic Trp-164, whereas W164S was used as a control lacking activity on high redox potential substrates. The W164Y/R257A/A260F triple variant combined the W164Y mutation with two changes in the Trp-164 environment that have been previously shown (38) and increased the capability of VPI and VPII for oxidizing VA (the R257A/A260F variant was analyzed for comparison). The resting state and VPI and VPII electronic absorption spectra (the two latter obtained under stopped-flow conditions) of the different variants and native VP showed that the mutations had not caused any substantial changes in the heme environment.
EPR Analyses
Fig. 1A (top) shows the low temperature (20 K) 9-GHz EPR spectrum of the resting state of the W164Y variant. This spectrum maintained the same characteristics of the native VP resting state (Fig. 2A) with the Fe(III) in the high spin state (g⊥ = 6 and g∥ = 2.0). The resting state of the W164H and W164S variants (not shown) also had the same characteristics, confirming the information provided by the electronic absorption spectra.
FIGURE 1.

Low temperature EPR spectra of native VP and its directed variants (W164Y, W164H and W164S). A, spectra of W164Y variant resting state (RS) characterized by the Fe(III) signal and after the addition of 8 eq of H2O2 showing a protein radical signal (g = 4.3 is due to a non-heme iron impurity). B, high resolution narrow scan spectra of the protein radical in the peroxide-activated W164Y variant described above, paired with its simulation (Sim.) and with the tryptophanyl radical found in the activated native enzyme (VP). C, spectra of the W164H (dotted line) and W164S (continuous line) variants after H2O2 activation (obtained 10 s after the addition of 1 eq). EPR spectra were recorded under the following conditions: ν, 9.39 GHz; modulation amplitude, 1 mT (A), 0.2 mT (B), or 0.4 mT (C); microwave power, 2 milliwatts; modulation frequency, 100 kHz; and temperature, 20 K (A and B) or 9 K (C). The actual values used in the simulation of the W164Y variant spectrum (B, top) were the following: Ax(Hβ1) = 2.20, Ay(Hβ1) = 2.09, Az(Hβ1) = 2.16 mT and Ax(Hβ2) = 0.64, Ay(Hβ2) = 0.83, and Az(Hβ2) = 0.27 mT, whereas those of the α-protons at the phenol ring are smaller in magnitude, more anisotropic, and almost invariant for the Ax (H3,5) = 1.00, Ay (H3,5) = 0.36, Az (H3,5) = 0.75 mT, and for Ax (H2,6) = 0.18, Ay (H2,6) = 0.27, Az (H2,6) = 0.08 mT.
FIGURE 2.

Low temperature EPR spectrum of VP resting state (A) when compared with those recorded after the addition of equimolecular amounts of H2O2 and ferrocyanide to native VP (B) and its W164Y variant (C). All spectra were recorded at ν 9.39 GHz; modulation amplitude, 1 mT; microwave power, 2 milliwatts; modulation frequency, 100 kHz; and temperature, 20 K. EPR spectra were normalized to obtain a better resolution; real spin quantitation is reported under “Results.”
Adding 8 eq of H2O2 to the W164Y variant resulted in VPI formation, showing characteristic UV-visible maxima detectable by stopped-flow. In its EPR spectrum (Fig. 1A, bottom), the appearance of a radical-like signal was evident, with the concomitant disappearance of the iron signal. The new radical signal had a giso = 2.0049(1) with a radical yield of 0.07 spin/heme. Saturation measurements (see supplemental Fig. 1) showed that the radical was weakly coupled with a paramagnetic ion, suggesting the same situation described for Trp-164 radical in native VP (16, 17, 39).
Fig. 1B (top and center) shows an expansion of the well resolved EPR signal of the W164Y variant radical reported in Fig. 1A (simulated and experimental spectra) when compared with the expansion of the EPR signal assigned to a Trp-164 neutral radical of native enzyme VPI (Fig. 1B, bottom). These signals were dominated by the hyperfine couplings of the β-methylene protons, β1 and β2 at the side-chain Cβ (see supplemental Fig. 2), one of them being much larger than the other. The EPR spectrum of the W164Y radical was characterized by a doublet with well resolved sub-splittings on both components. This radical signal showed a higher giso value (2.0049) than that of native VPI (giso = 2.0027), indicative of the formation of an organic radical including oxygen or sulfur (40). The hyperfine couplings constants obtained for the β-methylene protons of the W164Y variant radical (Fig. 1) are in fair agreement with those previously reported for the native enzyme Trp-164 radical (17). As these hyperfine couplings constants are related to the dihedral angle (41), we could deduce that the phenolic side chain had the same orientation in the mutated variant like that indolic ring in the native protein VPI radical. Finally, the g tensor components (gx = 2.0087(2), gy = 2.0041(2), and gz = 2.0019(2)) were consistent with a tyrosyl radical (40, 42-44), although more accurate determination of these values might require EPR experiments at higher microwave frequencies and magnetic field strengths.
In Fig. 1C, the EPR spectra of the W164H (dotted line) and W164S (continuous line) variants after the addition of 1 eq of H2O2 (resulting in VPI formation) are superimposed. Both VPI EPR spectra were very similar in shape, characterized by the same giso value (2.00(1)) (with a peak to peak amplitude equal to 0.22 mT), and resembled the [Fe(IV) = O Por+·] intermediate spectrum reported in other heme proteins (45-47). Unlike the organic radical signals of native VP and W164Y VPI, which were well resolved also at T = 70 K, the EPR spectra of the W164H and W164S VPI showed a trend with the variation of temperature similar to that obtained for the porphyrinyl radical of horseradish peroxidase Compound I (see supplemental Fig. 3), confirming the above identification.
With the aim to investigate differences at the VPII state, ferrocyanide was used to reduce VPI to VPII before EPR analysis. In Fig. 2B, the low temperature EPR spectrum of native VP after the addition of 1 eq of H2O2 and 1 eq of ferrocyanide is presented. The EPR spectrum of the W164Y variant, obtained under the same conditions, is shown in Fig. 2C. The two spectra are compared with the EPR spectrum of the VP resting state (Fig. 2A). These spectra were normalized to the more intense peak. Double integration was performed to calculate the spin quantitation of radical yield with respect to the resting state Fe(III). The yield of VPIIB in the native enzyme was ∼0.025 spin/heme with 0.14 spin/heme of resting state Fe(III). The corresponding EPR spectrum of the W164Y variant showed an iron signal and a radical signal with the same intensity giving a yield for VPIIB equal to 0.026 spin/heme. No Fe(III) from resting state was present.
Crystallographic Studies
The VP crystal structure has recently been determined at 1.33 Å resolution (PDB entry 2BOQ), and some mutated variants at the Trp-164 environment (PDB entry 2VKA) and the Mn2+ binding site (12) have been solved at different resolutions. The W164Y variant was also crystallized, and its structure was determined at 1.94 Å (Fig. 3A) and deposited as PDB entry 2W23. The W164Y model includes residues 1-315, the heme group, two calcium ions, one sulfate ion, one glycerol molecule, and 387 water molecules. The phenolic ring of the tyrosine was in the same plane as the indole group of the tryptophan residue in the native structure (Fig. 3B), in agreement with the EPR results. The tyrosine residue was stabilized by a hydrogen bond with Glu-243 and by stacking with the aliphatic side chain of Arg-257. This arginine residue was in a double conformation in the structure of native VP. However, its interaction with the tyrosine residue seemed to stabilize only one of the conformations in the W164Y variant, this being the main difference found in the new structure. Other small variations, due to the different conditions required to crystallize the mutated variant, concerned: (i) residues 288-291 appearing slightly shifted due to the presence of a glycerol molecule; and (ii) the presence of a sulfate ion affecting the conformation of the side chains of residues 136 and 140.
Steady-state Kinetics of the VP Variants
Table 2 shows the steady-state kinetic constants for oxidation of VA, RB5, and Mn2+ by the five variants analyzed, when compared with native VP. Substitution of Trp-164 by a histidine, serine, or tyrosine residues caused a complete loss of activity on VA and RB5. This fact showed that Trp-164 is involved in oxidation of these substrates, and demonstrated that a tyrosine or histidine residue cannot play “the same role” of Trp-164 in VP catalysis (under steady-state conditions). Changes in the kinetic constants for oxidation of Mn2+ were always small, confirming that this cation is oxidized at a different site.
TABLE 2.
Steady-state kinetic constants of native VP and directed variants
Shown are Km (μm), kcat (s−1) and kcat/Km (mm−1 · s−1) for oxidation of VA, RB5 and Mn2+. Reactions at 25 °C in 0.1 m tartrate, pH 3 for VA, pH 3.5 for RB5, and pH 5 for Mn2+. Means and 95% confidence limits.
| Constants | VP | W164S | W164H | W164Y | W164Y/R257A/A260F | R257A/A260F | |
|---|---|---|---|---|---|---|---|
| VA | Km | 4130 ± 320 | −a | − | − | − | 13500 ± 1200 |
| kcat | 9.5 ± 0.2 | 0 | 0 | 0 | 0 | 17.9 ± 0.7 | |
| kcat/Km | 2.3 ± 0.1 | 0 | 0 | 0 | 0 | 1.3 ± 0.1 | |
| RB5 | Km | 3.4 ± 0.3 | − | − | − | − | 4.9 ± 0.5 |
| kcat | 5.5 ± 0.3 | 0 | 0 | 0 | 0 | 9.1 ± 0.4 | |
| kcat/Km | 1610 ± 90 | 0 | 0 | 0 | 0 | 1900 ± 100 | |
| Mn2+ | Km | 181 ± 10 | 110 | 133 ± 6 | 78 ± 4 | 76 ± 4 | 150 ± 8 |
| kcat | 275 ± 4 | 207 ± 3 | 328 ± 4 | 164 ± 2 | 216 ± 3 | 207 ± 3 | |
| kcat/Km | 1520 ± 70 | 1900 ± 100 | 2470 ± 100 | 2110 ± 90 | 2850 ± 140 | 1380 ± 60 |
−, Not determined because of lack of activity.
We considered that tyrosyl radicals in two W164Y molecules could interact forming dityrosine cross-links that would inactivate the enzyme (the VP sequence has no tyrosine residues). No dimerization was observed either by SDS-PAGE or by amino acid analysis of the W164Y variant treated with H2O2 in the absence of reducing substrates (and the same result was obtained with the other variants), confirming that the lack of activity was not due to inactivation by intermolecular coupling.
Transient State Kinetics
VPI Formation—The kinetic constants for formation of VPI (Reaction 1) in the five site-directed variants were estimated at 397 nm and compared with those of native VP (see supplemental Table 1). The similar k1app values (all of them in the range 3-4 × 106 m-1 · s-1) showed that the mutations introduced did not affect the formation of VPI.
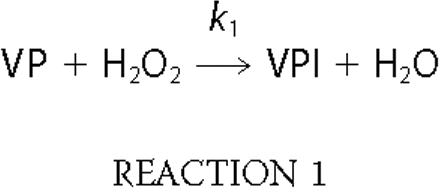
VPI Reduction—The single electron reduction of VPI by VA and RB5 (Reaction 2) was examined at 416 nm, and the pseudo-first-order rate constants (k2obs) were calculated. Plots of k2obs versus VA concentration showed saturation kinetics in the case of native VP and the W164Y variant (Fig. 4A), and first-order rate constants (k2) and equilibrium dissociation constants (KD2) for VPI reduction could be calculated (Table 3). By contrast, reduction of VPI of the other variants exhibited a linear dependence of the VA concentration, from which the apparent second-order rate constants (k2app) were calculated.
FIGURE 4.

Kinetics of reduction of VPI and VPII of native VP and its directed variants by VA. Stopped-flow reactions were carried out at 25 °C using 1 μm enzyme, 0.1 m tartrate (pH 3), and varying concentrations of VA. The insets show kinetic curves at a smaller vertical scale. Means and 95% confidence limits of replicate assays are shown. The symbols are as follows: native VP (•), W164Y (▪), W164H (♦), W164S (▴), W164Y/R257A/A260F (□), and R257A/A260F (○).
TABLE 3.
Transient state kinetic constants for reduction of VPI of native enzyme and directed variants by VA and RB5
Shown are first-order rate constants (k2), equilibrium dissociation constants (Kd2) and apparent second-order rate constants (k2app). Hill equation used for RB5 k2 and Kd2 estimation due to sigmoidal kinetic curves (supplemental Fig. 4B). Means and 95% confidence limits.
| k2 | Kd2 (0.5) | k2app | |
|---|---|---|---|
| s−1 | m | m−1 · s−1 | |
| VPI reduction by VA | |||
| VP | 168 ± 13 | (60 ± 7) × 10−3 | (2.8 ± 0.1) × 103 |
| W164Y | 36 ± 5 | (114 ± 20) × 10−3 | (3.2 ± 0.1) × 102 |
| W164H | -a | - | (2.9 ± 0.1) × 102 |
| W164S | - | - | (5.6 ± 0.6) × 101 |
| R257A/A260F | - | - | (6.2 ± 0.1) × 104 |
| W164Y/R257A/A260F | - | - | (2.3 ± 0.1) × 102 |
| VPI reduction by RB5 | |||
| VP | - | - | (3.9 ± 0.1) × 106 |
| W164Y | 5.2 ± 0.1 | (21.3 ± 0.7) × 10−6 | 2.5 × 105 |
| W164H | 21.5 ± 2.2 | (41.5 ± 5.1) × 10−6 | 5.2 × 105 |
| W164S | 6.8 ± 1.2 | (29.1 ± 0.7) × 10−6 | 2.3 × 105 |
| R257A/A260F | - | - | ∼3.0 × 107b |
| W164Y/R257A/A260F | 23.7 ± 2.7 | (29.4 ± 3.5) × 10−6 | 8.1 × 105 |
-, Not determined because of non-saturation kinetics.
Measured at 10 °C.
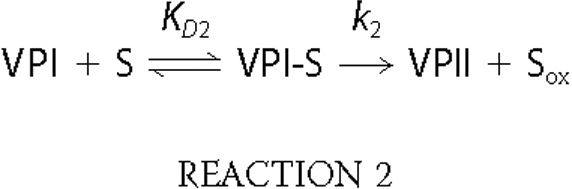
VPI reduction by VA was impaired in all the variants harboring Trp-164 mutations, although in different degrees. The k2app values for the W164Y, W164Y/R257A/A260F, and W164H variants were an order of magnitude higher than calculated for the W164S variant. These constants suggested participation of the tyrosine and histidine residues introduced in VPI reduction by VA. It was found that the difference between native VP and the W164Y variant was due first to a decrease in VA oxidation by the mutated VPI (4.7-fold lower k2) and second to a decrease in the stability of enzyme-bound VA (1.9-fold higher KD2).
Plots of k2obs versus RB5 concentration were linear for native VP (and the R257A/A260F variant) (supplemental Fig. 4A), and the apparent second-order rate constant for VPI reduction (k2app) was determined (Table 3). All variants harboring mutations in Trp-164 exhibited sigmoidal kinetics (supplemental Fig. 4A, inset), and the corresponding kinetics constants (Table 3) were significantly lower than those for native VPI reduction by RB5. However, no important differences between the different variants were observed. This showed that VPI Trp-164 is involved in oxidation of RB5 and that its substitution by a tyrosine (or other residue) resulted in mutated variants whose VPI is not able to oxidize RB5 at this site.
VPII Reduction—The single electron reduction of VPII by VA and RB5 (Reaction 3) was examined at 406 nm.
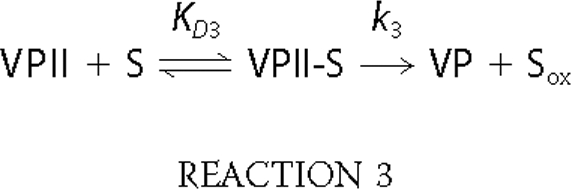
Native VP and the R257A/A260F, W164H, and W164Y/R257A/A260F variants exhibited saturation kinetics when VA was used as reducing substrate (Fig. 4B), and the corresponding kinetic constants were calculated (Table 4). The k3 and KD3 values for the two latter variants were dramatically affected, and the resulting k3app decreased by 2 orders of magnitude. The W164S and W164Y variants maintained the same residual activity levels (kobs) at all VA concentrations tested, and only a rough estimation of k3 could be obtained, the value being similar to that of the W164H variant.
TABLE 4.
Transient state kinetic constants for reduction of VPII of native VP and directed variants by VA and RB5
Shown are first-order rate constants (k3), equilibrium dissociation constants (KD3) and apparent second-order rate constants (k3app). Means and 95% confidence limits.
| k3 | KD3 | k3app | |
|---|---|---|---|
| s−1 | m | m−1 · s−1 | |
| VPII reduction by VA | |||
| VP | 6.5 ± 0.2 | (4.9 ± 0.4) × 10−3 | (1.3 ± 0.1) × 103 |
| W164Y | 0.1 ± 0.0 | -a | - |
| W164H | 0.4 ± 0.1 | (3.3 ± 0.9) × 10−2 | 1.3 × 101 |
| W164S | 0.2 ± 0.1 | - | - |
| R257A/A260F | 45.9 ± 4.5 | (6.7 ± 2.2) × 10−4 | (6.8 ± 1.7) × 104 |
| W164Y/R257A/A260F | 0.5 ± 0.1 | (1.2 ± 0.4) × 10−2 | (4.0 ± 0.0) × 101 |
| VPII reduction by RB5 | |||
| VP | 7.3 ± 0.3 | (6.4 ± 0.7) × 10−6 | (1.2 ± 0.1) × 106 |
| W164Y | 0.8 ± 0.1 | (1.3 ± 0.2) × 10−4 | 5.9 × 103 |
| W164H | 0.3 ± 0.0 | (7.9 ± 1.21) × 10−5 | 4.3 × 103 |
| W164S | 1.4 ± 0.2 | (2.2 ± 0.4) × 10−4 | 6.4 × 103 |
| R257A/A260F | 26.8 ± 0.3 | (7.9 ± 0.1) × 10−6 | (3.4 ± 0.0) × 106 |
| W164Y/R257A/A260F | 0.8 ± 0.2 | (1.0 ± 0.4) × 10−4 | 7.9 × 103 |
-, Not determined because of very low activity.
VPII reduction by RB5 was observed for native VP (Table 4 and supplemental Fig. 4B) and the R257A/A260F variant, which showed saturation and sigmoidal kinetics, respectively. The rest of the mutated variants incorporating a tyrosine, histidine, or serine residue in the position of Trp-164 exhibited very low reduction rates, with k3app values 3 orders of magnitude lower than native VP. These results suggested that both a tyrosine and a histidine residue occupying this position in VP are not catalytically active, and that an alternative low efficient oxidation site for RB5, different to the catalytic Trp-164, exits in these VP variants. VPII self-reduction was also affected by the above mutations, that of the W164Y variant being much lower than found for the native VP (0.0061 s-1 versus 0.105 s-1).
DISCUSSION
The catalytic cycle of heme peroxidases includes activation of the heme cofactor by peroxide, this oxidizer attaining the heme central pocket using an access channel (48). However, despite some plasticity degree (49), the channel in ligninolytic peroxidases is too narrow to allow lignin and other aromatic substrates to directly interact with the peroxide-activated heme (6). To overcome this limitation, LiP and VP have developed an alternative catalytic mechanism for oxidation of high redox potential aromatics at the protein surface followed by electron transfer to the activated cofactor via a LRET pathway. This pathway initiates at an exposed tryptophan residue conserved in P. chrysosporium LiP and P. eryngii VP that gives rise to a catalytic radical (8, 10). This radical was directly detected in P. eryngii VPI using low temperature EPR, in agreement with previous studies (10) that also showed that the tryptophanyl radical was in the neutral form (17). Due to VP self-reduction, some excess of hydrogen peroxide was required in the EPR experiments. The radical yield after hydrogen peroxide addition and rapid freezing of VP was estimated as 0.25 spin/heme. Substitution of Trp-164 by a residue that was unable to form a stable free radical (W164S mutation) resulted in the complete loss of the protein radical signal, and a porphyrinyl radical was identified by the low giso value (2.00(1)), similar to that described in horseradish peroxidase (50). The ability of VP to oxidize VA to veratraldehyde under steady-state conditions was simultaneously lost, and VA reduction of both VPI and VPII was strongly impaired (k2app being in the self-reduction range). The above results showed that the tryptophanyl radical was involved in catalysis by both VPI and VPII.
When Trp-164 of VP was substituted by histidine or tyrosine residues and the variants treated with hydrogen peroxide were analyzed by EPR after rapid freezing, a protein radical was detected only in the W164Y variant VPI. W164Y EPR gave a well resolved spectrum, even at high temperature (T = 70 K). The EPR signal was assigned to a tyrosyl radical whose yield was lower (Tyr· = 0.07 spin/heme) than that found for the native enzyme VPI radical (Trp· = 0.25 spin/heme). In contrast, the W164H VPI yielded a signal similar to that detected in the W164S variant that was assigned to a porphyrinyl radical. Interestingly, the W164Y VPI was able to oxidize VA, albeit with lower k2 and higher KD2 than native VP, resulting in an order of magnitude lower apparent second-order rate constant (k2app). The tyrosyl radical was involved because the k2app value was an order of magnitude higher than that of the inactive W164S variant. The W164H variant exhibited the same kinetic properties than the W164Y variant, suggesting involvement of a histidyl radical in catalysis, although it was not detected by EPR under the present conditions.
VPII was obtained by ferrocyanide reduction of VPI using equimolar amounts of VP, hydrogen peroxide, and ferrocyanide (12, 38). The objective was the EPR detection of the protein radical that should characterize the VPIIB state (see the extended VP catalytic cycle in supplemental Fig. 5) predicted by site-directed mutagenesis (10) but still to be directly detected. The existence of Compound IIB had been demonstrated in cytochrome c peroxidase, representing 10% of total Compound II at pH 5 (51). VPIIB was definitively found in native VP with a yield of around 0.025 spin/heme. The yield of Fe(III) in the same sample was 0.14 spin/heme, suggesting a certain percentage (around 11%) of enzyme resting state formed from VPII self-reduction. The latter was in agreement with kinetic data showing the spontaneous decay of the peroxide-activated VP to its resting state by a protein intramolecular reductant (10).
VPII protein radical was detected not only in native VP but also in its W164Y variant. The EPR spectrum of the peroxide-activated variant presented approximately the same yield of VPIIB (around 0.026 spin/heme) reported in wild type, but no Fe(III) from the resting state was present, indicating that the W164Y mutation blocked the VPII self-reduction. Even if the percentage of radical was more or less the same for both enzymes, substrate oxidation and self-reduction was extremely slow in the W164Y variant, and the system did not cycle back to the resting state like in the native VP. The lack of activity of the W164Y variant was not due to enzyme inactivation by intermolecular cross-linking from tyrosyl radical coupling, as observed in some heme proteins (52), but to the absence of an active radical or to steric hindrances for its interaction with the reducing substrate.
The classical peroxidase catalytic cycle is that where after the formation of Compound I (containing Fe4+-oxo and porphyrin cation radical), its reduction in two 1-electron reactions results in the intermediate Compound II (containing Fe4+-oxo after porphyrin reduction) and then the resting form of the enzyme. Because in the case of VPI and VPII about 25 and 3% of molecules, respectively, delocalize one electron equivalent on Trp-164 (being 7 and 3%, respectively, in the W164Y variant), the cycle back to the resting state for these molecules is different, and the whole catalytic cycle has to be modified, as suggested previously (10). The resulting extended cycle (supplemental Fig. 5) includes VPIB (containing Fe4+ -oxo and tryptophan radical) and VPIIB (containing Fe3+ and tryptophan radical) involved in the oxidation of VA and other substrates. VPIB and VPIIB would be in equilibrium with VPIA and VPIIA, respectively. This is the first time that catalytically relevant protein radicals (tryptophanyl radical in native VP and tyrosyl radical in its W164Y variant) are detected in Compound II of a fungal peroxidase. This finding represents an important confirmation for the extended catalytic cycle of VP (and LiP) and can be considered as a reference for all those heme proteins delocalizing electron equivalents on the protein structure. Formation of exposed catalytic radicals has high biological significance because this is the mechanism developed by high redox potential fungal peroxidase to oxidize the recalcitrant bulky polymer of lignin as the first step for carbon recycling in land ecosystems.
CONCLUSIONS
The following can be concluded concerning the presence and role of protein radicals in the catalytic cycle of native and mutated VP: 1) a tryptophanyl radical, detected by low temperature EPR, is present in both the 1-electron and the 2-electron oxidized forms of the enzyme, characterizing the so-called VPIB and VPIIB that represented, respectively, ∼25 and ∼3% of total VPI and VPII under the present conditions; 2) the radical-forming residue corresponds to exposed Trp-164, being the only site for oxidation of high redox potential VA by both VPI and VPII; 3) a tyrosyl radical, detected by EPR, is present in both VPIB and VPIIB of the W164Y variant, representing around ∼7 and ∼3% of total VPI and VPII, respectively; 4) the tyrosyl radical in W164Y VPIB is able to oxidize VA, but this is not the case with VPIIB and, therefore, steady-state oxidation of VA is blocked in this variant; 5) VPII self-reduction is also blocked in the W164Y variant, as shown by the absence of resting state Fe(III) by EPR and stopped-flow spectra.
Supplementary Material
The atomic coordinates and structure factors (code 2W23) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
This work was supported by the Spanish projects BIO2005-03569 and BIO2008-01533, Italian Miur PRIN 2007, and the BIORENEW project of the European Union (Contract NMP2-CT-2006-026456). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains five supplemental figures and a supplemental table.
Footnotes
The abbreviations used are: LiP, lignin peroxidase; LRET, long range electron transfer; RB5, Reactive Black 5; SDS-PAGE, sodium dodecylsulfate-polyacrylamide gel electrophoresis; VA, veratryl alcohol; VP, versatile peroxidase; VPI, VP Compound I; VPII, VP Compound II; mT, milliteslas; PDB, Protein Data Bank.
References
- 1.Kirk, T. K., and Farrell, R. L. (1987) Annu. Rev. Microbiol. 41 465-505 [DOI] [PubMed] [Google Scholar]
- 2.Schoemaker, H. E. (1990) Recl. Trav. Chim. Pays-Bas 109 255-272 [Google Scholar]
- 3.Martínez, A. T., Speranza, M., Ruiz-Duenñas, F. J., Ferreira, P., Camarero, S., Guillén, F., Martínez, M. J., Gutiérrez, A., and del Río, J. C. (2005) Int. Microbiol. 8 195-204 [PubMed] [Google Scholar]
- 4.Kersten, P., and Cullen, D. (2007) Fungal Genet. Biol. 44 77-87 [DOI] [PubMed] [Google Scholar]
- 5.Ruiz-Duenñas, F. J., and Martínez, A. T. (January 13, 2009) Microbial Biotechnol. 10.1111/j.1751-7915.2008.00078.x [DOI]
- 6.Martínez, A. T. (2002) Enzyme Microb. Technol. 30 425-444 [Google Scholar]
- 7.Hammel, K. E., and Cullen, D. (2008) Curr. Opin. Plant Biol. 11 349-355 [DOI] [PubMed] [Google Scholar]
- 8.Doyle, W. A., Blodig, W., Veitch, N. C., Piontek, K., and Smith, A. T. (1998) Biochemistry 37 15097-15105 [DOI] [PubMed] [Google Scholar]
- 9.Mester, T., Ambert-Balay, K., Ciofi-Baffoni, S., Banci, L., Jones, A. D., and Tien, M. (2001) J. Biol. Chem. 276 22985-22990 [DOI] [PubMed] [Google Scholar]
- 10.Pérez-Boada, M., Ruiz-Duenñas, F. J., Pogni, R., Basosi, R., Choinowski, T., Martínez, M. J., Piontek, K., and Martínez, A. T. (2005) J. Mol. Biol. 354 385-402 [DOI] [PubMed] [Google Scholar]
- 11.Gold, M. H., Youngs, H. L., and Gelpke, M. D. (2000) Met. Ions Biol. Syst. 37 559-586 [PubMed] [Google Scholar]
- 12.Ruiz-Duenñas, F. J., Morales, M., Pérez-Boada, M., Choinowski, T., Martínez, M. J., Piontek, K., and Martínez, A. T. (2007) Biochemistry 46 66-77 [DOI] [PubMed] [Google Scholar]
- 13.Smith, A. T., and Veitch, N. C. (1998) Curr. Opin. Chem. Biol. 2 269-278 [DOI] [PubMed] [Google Scholar]
- 14.Beratan, D. N., Onuchic, J. N., Winkler, J. R., and Gray, H. B. (1992) Science 258 1740-1741 [DOI] [PubMed] [Google Scholar]
- 15.Pelletier, H., and Kraut, J. (1992) Science 258 1748-1755 [DOI] [PubMed] [Google Scholar]
- 16.Pogni, R., Baratto, M. C., Giansanti, S., Teutloff, C., Verdin, J., Valderrama, B., Lendzian, F., Lubitz, W., Vázquez-Duhalt, R., and Basosi, R. (2005) Biochemistry 44 4267-4274 [DOI] [PubMed] [Google Scholar]
- 17.Pogni, R., Baratto, M. C., Teutloff, C., Giansanti, S., Ruiz-Duenñas, F. J., Choinowski, T., Piontek, K., Martínez, A. T., Lendzian, F., and Basosi, R. (2006) J. Biol. Chem. 281 9517-9526 [DOI] [PubMed] [Google Scholar]
- 18.Pogni, R., Teutloff, C., Lendzian, F., and Basosi, R. (2007) Appl. Magn. Reson. 31 509-526 [Google Scholar]
- 19.Blodig, W., Smith, A. T., Winterhalter, K., and Piontek, K. (1999) Arch. Biochem. Biophys. 370 86-92 [DOI] [PubMed] [Google Scholar]
- 20.Choinowski, T., Blodig, W., Winterhalter, K., and Piontek, K. (1999) J. Mol. Biol. 286 809-827 [DOI] [PubMed] [Google Scholar]
- 21.Blodig, W., Smith, A. T., Doyle, W. A., and Piontek, K. (2001) J. Mol. Biol. 305 851-861 [DOI] [PubMed] [Google Scholar]
- 22.Ruiz-Duenñas, F. J., Martínez, M. J., and Martínez, A. T. (1999) Mol. Microbiol. 31 223-236 [DOI] [PubMed] [Google Scholar]
- 23.Stubbe, J., and Der Donk, W. A. (1998) Chem. Rev. 98 705-762 [DOI] [PubMed] [Google Scholar]
- 24.Schoemaker, H. E., Lundell, T. K., Floris, R., Glumoff, T., Winterhalter, K. H., and Piontek, K. (1994) Bioorg. Med. Chem. 2 509-519 [DOI] [PubMed] [Google Scholar]
- 25.Johjima, T., Itoh, H., Kabuto, M., Tokimura, F., Nakagawa, T., Wariishi, H., and Tanaka, H. (1999) Proc. Natl. Acad. Sci. U. S. A. 96 1989-1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Camarero, S., Sarkar, S., Ruiz-Duenñas, F. J., Martínez, M. J., and Martínez, A. T. (1999) J. Biol. Chem. 274 10324-10330 [DOI] [PubMed] [Google Scholar]
- 27.Gelpke, M. D. S., Lee, J., and Gold, M. H. (2002) Biochemistry 41 3498-3506 [DOI] [PubMed] [Google Scholar]
- 28.Miki, Y., Tanaka, H., Nakamura, M., and Wariishi, H. (2006) J. Fac. Agr. Kyushu. Univ. 51 99-104 [Google Scholar]
- 29.Ivancich, A., Jakopitsch, C., Auer, M., Un, S., and Obinger, C. (2003) J. Am. Chem. Soc. 125 14093-14102 [DOI] [PubMed] [Google Scholar]
- 30.Aubert, C., Mathis, P., Eker, A. P., and Brettel, K. (1999) Proc. Natl. Acad. Sci. U. S. A. 96 5423-5427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sasaki, S., Nonaka, D., Wariishi, H., Tsutsumi, Y., and Kondo, R. (2008) Phytochemistry 69 348-355 [DOI] [PubMed] [Google Scholar]
- 32.Pérez-Boada, M., Doyle, W. A., Ruiz-Duenñas, F. J., Martínez, M. J., Martínez, A. T., and Smith, A. T. (2002) Enzyme Microb. Technol. 30 518-524 [Google Scholar]
- 33.Collaborative Computational Project Number 4 (1994) Acta Crystallogr. Sect. D Biol. Crystallogr. 50 760-76315299374 [Google Scholar]
- 34.Vagin, A., and Teplyakov, A. (1997) J. Appl. Crystallogr. 30 1022-1025 [Google Scholar]
- 35.Murshudov, G. N., Vagin, A. A., and Dodson, D. J. (1997) Acta Crystallogr. Sect. D Biol. Crystallogr. 53 255. [DOI] [PubMed] [Google Scholar]
- 36.Emsley, P., and Cowton, K. (2004) Acta Crystallogr. Sect. D Biol. Crystallogr. 60 2132. [DOI] [PubMed] [Google Scholar]
- 37.Davis, I. W., Leaver-Fay, A., Chen, V. B., Block, J. N., Kapral, G. J., Wang, X., Murray, L. W., Arendall, W. B., III, Snoeyink, J., Richardson, J. S., and Richardson, D. C. (2007) Nucleic Acids Res. 35 W375-W385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ruiz-Duenñas, F. J., Morales, M., Mate, M. J., Romero, A., Martínez, M. J., Smith, A. T., and Martínez, A. T. (2008) Biochemistry 47 1685-1695 [DOI] [PubMed] [Google Scholar]
- 39.Schunemann, V., Lendzian, F., Jung, C., Contzen, J., Barra, A. L., Sligar, S. G., and Trautwein, A. X. (2004) J. Biol. Chem. 279 10919-10930 [DOI] [PubMed] [Google Scholar]
- 40.Bleifuss, G., Kolberg, M., Potsch, S., Hofbauer, W., Bittl, R., Lubitz, W., Graslund, A., Lassmann, G., and Lendzian, F. (2001) Biochemistry 40 15362-15368 [DOI] [PubMed] [Google Scholar]
- 41.McConnell, H. M., and Robertson, R. E. (1957) J. Phys. Chem. A 61 1018 [Google Scholar]
- 42.Lendzian, F. (2005) Biochim. Biophys. Acta 1707 67-90 [DOI] [PubMed] [Google Scholar]
- 43.Un, S., Gerez, C., Elleingand, E., and Fontecave, M. (2001) J. Am. Chem. Soc. 123 3048-3054 [DOI] [PubMed] [Google Scholar]
- 44.Un, S., Atta, M., Fontecave, M., and Rutherford, A. W. (1995) J. Am. Chem. Soc. 117 10713-10719 [Google Scholar]
- 45.Ivancich, A., Mazza, G., and Desbois, A. (2001) Biochemistry 40 6860-6866 [DOI] [PubMed] [Google Scholar]
- 46.Benecky, M. J., Frew, J. E., Scowen, N., Jones, P., and Hoffman, B. M. (1993) Biochemistry 32 11929-11933 [DOI] [PubMed] [Google Scholar]
- 47.Ivancich, A., Jouve, H. M., Sartor, B., and Gaillard, J. (1997) Biochemistry 36 9356-9364 [DOI] [PubMed] [Google Scholar]
- 48.Dunford, H. B. (1999) Heme Peroxidases, Wiley-VCH, New York
- 49.Gerini, M. F., Roccatano, D., Baciocchi, E., and Di Nola, A. (2003) Biophys. J. 84 3883-3893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schulz, C. E., Devaney, P. W., Winkler, H., Debrunner, P. G., Doan, N., Chiang, R., Rutter, R., and Hager, L. P. (1979) FEBS Lett. 103 102-105 [DOI] [PubMed] [Google Scholar]
- 51.Ho, P. S., Hoffman, B. M., Kang, C. H., and Margoliash, E. (1983) J. Biol. Chem. 258 4356-4363 [PubMed] [Google Scholar]
- 52.Detweiler, C. D., Lardinois, O. M., Deterding, L. J., De Montellano, P. R., Tomer, K. B., and Mason, R. P. (2005) Free Radic. Biol. Med. 38 969-976 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.