

Abstract
The tripeptide glutathione is involved in cellular defense mechanisms for xenobiotics and reactive oxygen species. This study investigated glutathione-dependent mechanisms in the model organism Aspergillus nidulans. A recombinant dimeric protein of A. nidulans glutathione reductase (GR) contained FAD and reduced oxidized glutathione (GSSG) using NADPH as an electron donor. A deletion strain of the GR gene (glrA) accumulated less intracellular reduced glutathione (GSH), indicating that the fungal GR contributes to GSSG reduction in vivo. Growth of the deletion strain of glrA was temperature-sensitive, and this phenotype was suppressed by adding GSH to the medium. The strain subsequently accumulated more intracellular superoxide, and cell-free respiration activity was partly defective. Growth of the strain decreased in the presence of oxidants, which induced glrA expression 1.5-6-fold. These results indicated that the fungal glutathione system functions as an antioxidant mechanism in A. nidulans. Our findings further revealed an initial proteomic differential display on GR-depleted and wild type strains. Up-regulation of thioredoxin reductase, peroxiredoxins, catalases, and cytochrome c peroxidase in the glrA-deletion strain revealed interplay between the glutathione system and both the thioredoxin system and hydrogen peroxide defense mechanisms. We also identified a hypothetical, up-regulated protein in the GR-depleted strains as glutathione S-transferase, which is unique among Ascomycetes fungi.
Glutathione (γ-glutamyl-cysteinyl-glycine, GSH) is a ubiquitous tripeptide of which the thiol/thiolate group of its cysteine residue is reversibly oxidized to generate oxidized glutathione (GSSG). It is the most abundant intracellular redox-active sulfhydryl compound, and it acts as a major cellular redox buffer. Maintenance of the cellular redox state is the most important role of glutathione because reactive oxygen species (ROS),2 due to their powerful oxidant reactivity, cause oxidative damage to nucleotides, proteins, and lipids if they are not properly removed from cells. Glutathione functions as an electron donor for glutathione peroxidase that reduces hydrogen peroxide (H2O2) to water and contributes to ROS degradation (1). Glutaredoxins have glutathione-dependent disulfide reductase activity and reduce protein disulfide generated by oxidation of protein sulfhydryl groups by ROS, and thus, this mechanism is important for repairing oxidative damage to proteins as well as for regulating the redox state of the proteins (glutathione-glutaredoxin system) (2, 3). The mechanisms of the thioredoxin and glutathione-glutaredoxin systems are similar. Thioredoxins are small (12-13 kDa) protein disulfide reductases like glutaredoxins (4, 5), with a redox-active dithiol group, and they serve as electron donors for thioredoxin peroxidase (peroxiredoxin) that degrades H2O2. After oxidation, glutathione and thioredoxin are re-reduced by glutathione reductase (GR) and thioredoxin reductase (TR), respectively.
Glutathione reductase is a FAD-containing protein that reduces GSSG to GSH using NADPH as an electron donor. It constitutes a family of proteins with sulfide dehydrogenase, dihydrolipoamide dehydrogenase, trypanothione reductase, and TR, all of which catalyze NAD(P)H:disulfide oxidoreductase (6). The absence of the GR gene (pgr1+) causes a low respiration rate, inactivates oxidation-labile enzymes containing FeS, and arrests growth at the G1 phase in Schizosaccharomyces pombe and, thus, is essential for its survival (7). Although GLR1, the sole GR gene in Saccharomyces cerevisiae, is not required for normal growth, it is required for growth in the presence of oxidative stress (8). By contrast, GR is dispensable for the normal growth and maintenance of cellular GSH in Escherichia coli (9). These imply that the contribution of GR to cell growth differs according to species. Meanwhile, a recent report has shown that when the genes for the thioredoxins TRX1 and TRX2 are deleted, S. cerevisiae remains viable but not when the genes for both glutaredoxins (GRX1 and GRX2) are also deleted (3). The triple mutant (trx1, trx2, glr1) is also non-viable, indicating overlapping functions of thioredoxins and glutaredoxins for reducing protein disulfides (3). This seems to be true for S. pombe, in which multicopies of the thioredoxin gene (trx2+) suppress the growth deficiency of GR mutants (10). Taken together, the difference in the species requirement for GR is presumed to be caused by the extent of activity of compensation mechanisms such as the thioredoxin system. However, current comparative analyses of GR-deficient mutants from various organisms are too limited to thoroughly address this issue.
Another role of GSH is mediated by glutathione S-transferases (GSTs), which comprise a group of ubiquitous small cytosolic proteins containing a redox-active sulfhydryl group. The GSTs are involved in cellular detoxification by catalyzing the conjugation of GSH with xenobiotics and endogenous products of oxidative stress (11). Fungal GSH conjugates are excreted to vacuoles and detoxified (12). Protein glutathionylation is another function of GSH through which protein function is regulated, although reports about this type of modification in fungi are sparse (13). In addition, some GSTs oxidize GSH to GSSG and acts as glutathione peroxidase and disulfide reductase (glutaredoxin) (14). The family of GSTs is very large according to their primary sequences. Early studies of GSTs from higher eukaryotes classified them as α, μ, π, κ, ω, ζ, σ, and membraneassociated proteins involved in eicosanoid and glutathione metabolism (MAPEG) types. McGoldrick et al. (15) recently investigated the phylogenetic relationships of fungal GSTs and found that most of them did not fit the above categories and, rather, clustered into distinct groups designated clusters 1 and 2, EF1Bγ, Ure2p, and MAK16. Properties of some fungal GSTs that did not fit these clusters are currently under investigation (16-20). However, fungal GSTs are so divergent that further studies are required to comprehensively understand their structures and antioxidant functions.
Aspergillus nidulans is a filamentous fungus in Ascomycota that has been used as a model organism for studying cellular events such as development, secondary metabolism, and gene expression. Thön et al. (21) recently studied the physiological functions of thioredoxin system of this fungus and found that a deletion of one of the two thioredoxin genes (trxA) resulted in slow growth and the inability to form sexual and asexual reproductive organs. They also found that adding GSH to culture medium complemented these defects and that a combination of recombinant TrxA and TR reduced GSSG in vitro (21). These results indicated that A. nidulans produces a functional thioredoxin system that interacts with the glutathione system in redox regulation like S. cerevisiae and S. pombe. Here, we investigated the glutathione system of A. nidulans using a deletion strain of the fungal GR, which was viable and depleted of most cellular reduced GSH. We further applied a proteomic approach to identify cellular proteins of which amounts were increased by GR and GSH depletion. We found that such proteins included those in the fungal thioredoxin system and catalases and peroxidases, implying functional relevance of these proteins in glutathione and thioredoxin systems and in H2O2 degradation. We also identified and characterized a novel GST exhibiting both GST and glutathione peroxidase activities. Phylogenetic analysis showed that this GST (designated GstB) constitutes a unique family of GSTs that is conserved among some filamentous fungi.
EXPERIMENTAL PROCEDURES
Strains, Culture, and Media—A. nidulans strains A26 (biA1) and A89 (biA1; argB2) were obtained from the Fungal Genetic Stock Center (University of Kansas Medical Center). Conidia (108) were transferred to 500-ml Erlenmeyer flasks containing 100 ml of MMDN (1% glucose, 10 mm NaNO3, 10 mm KH2PO4, 7 mm KCl, 2 mm MgSO4, 2 ml liter-1 Hutner's trace metals (22)) and incubated at 30 °C for 20 h at 120 rpm (preculture). Resultant mycelia (1 g wet weight) were collected by centrifugation, washed twice with 0.85% NaCl, and then inoculated into 500-ml Erlenmeyer flasks containing 100 ml of MMDN. Flasks were sealed with cotton plugs and rotated at 120 rpm for 6 h at 30 °C. Biotin (0.2 μgl-1) was added to all media. Arginine (0.2 mg l-1) was added to culture the strain under arginine auxotrophy. E. coli was cultured in Luria broth (LB) (1% Tryptone, 0.5% yeast extract, 0.5% NaCl).
5′-Rapid Amplification of cDNA Ends—Complementary DNA was synthesized using the 5′-rapid amplification of cDNA end system, Version 2.0 (Invitrogen) according to the manufacturer's instructions. Total RNA was prepared from strain A26 cultured in MMDN medium for 24 h as above. The gene-specific primers for 5′-rapid amplification of cDNA ends were GSP1 (5′-TTAGGATGTGCGGCGCAGTGTA-3′) and GSP2 (5′-GCACGAGGTCAATTCCTTCGTT-3′), and the products were cloned into pGEM-T easy (Promega, Madison, WI).
Production of GlrA-Green Fluorescence Protein (GFP) Fusion Protein in A. nidulans—The plasmid pGRgfp1 and pGRgfp1-M86A were constructed using the MultiSite Gateway Three-Fragment Vector Construction kit (Invitrogen) according to the manufacturer's instructions. The gene promoters of gpdA encoding glyceraldehyde-3-phosphate dehydrogenase (Pgpd) and of the argB gene were amplified by PCR using the primers 5′-GGGGACAACTTTGTATAGAAAAGTTGCGTTGACCTAGCTGATTCTG-3′ and 5′-GGGGACTGCTTTTTTGTACAAACTTGTGTGATGTCTGCTCAAGCG-3′ and primers 5′-GGGACAGCTTTCTTGTACAAAGTGGGAACGCATGCAATAATTGCAGC-3′ and 5′-GGGGACAACTTTGTATAATAAAGTTGGTCGACCTACAGCCATTG-3′ (attB recombination sites are underlined) and cloned into pDONRP4-P1R and pDONRP2R-P3, respectively. Fragments of DNA encoding enhanced GFP, and glrA were amplified using the primers G1 (GGAAGAGCTTGTTACTTTGCGTATGGTGAGCAAGGGCGAGGA) and G2 (GGGGACCACTTTGTACAAGAAAGCTGGGTTTACTTGTACAGCTCGTCCATG) and A1 (GGGGACAAGTTTGTACAAAAAAGCAGGCTATGCTCTCTCGCTCCTCGCTT) and A2 (TCCTCGCCCTTGCTCACCATACGCAAAGTAACAAGCTCTTCC), respectively. The resulting DNA fragments were fused by a second PCR using the primers A1 and G2 and cloned into pDONR221. These three plasmids and pDESTR4-R3 were used to generate the plasmid pGRgfp1 containing PgpdA::glrA::gfp fusion and argB. The glrA::gfp fusion was replaced by that mutated at the Met86 codon to an Ala codon to construct pGRgfp1-M86A as described above. The mutated DNA fragment was generated by PCR using pGRgfp1 as a template and the primers A1 and 5′-CACCGGCGGCGCGTTGCTCGAG-3′ and G2 and 5′-CTCGAGCAACGCGCCGCCGGTG-3′ and fused by the second PCR using primers A1 and G2. Both pGRgfp1 and pGRgfp1-M86A were introduced into A. nidulans A89.
Fluorescence Microscopy—Conidia of the transformants harboring pGRgfp1 and pGRgfp1-M86A were incubated on glass coverslips in MMDN medium at 37 °C for 10 h followed by MitoTracker Red CMXRos (Invitrogen) for 10 min and then analyzed using the fluorescence microscope DMLB (Leica, Heidelberg, Germany) with a BP 450-490 filter for fluorescence excitation of GFP and a BP 515-560 filter for excitation of MitoTracker Red.
Production of Recombinant Proteins—Plasmid pETglrA was constructed to produce truncated recombinant glutathione reductase with an amino-terminal Met86 as follows. We prepared glrA cDNA by PCR using the cDNA of A. nidulans (see below) and the respective 5′- and 3′-PCR oligonucleotide primers: 5′-GCCATATGCCGCCGGTGGAGACAAA-3′ and 5′-CCTAGCGGCCGCACGCAAAGTAACAAGCTCTTCC-3′ (restriction sites are underlined). The PCR products were purified, digested by NdeI and NotI, then ligated to the plasmid vector pET21a (Novagen, Darmstadt, Germany) that had been digested with the same restriction enzymes. Plasmids for producing recombinant thioredoxin (rTrxA) and the gene product of AN6024.3 were constructed essentially as described above. The respective cDNAs were amplified using the primers 5′-GGGAATTCCATATGGGTGCCTCTGAACACGT-3 and 5′-CCCAAGCTTAGCAAGCAGAGCCTTGATAC-3′ (for trxA) and 5′-CCCATATGTCCCAGCCAGTCTACCA-3′, and 5′-CCCTCGAGCTTATGCTCCGCAATATACTC-3′ (for AN6024.3). We used pET22b and pET21a to construct respective plasmids to generate pETtrxA and pETgstB. Standard DNA manipulation techniques proceeded according to Sambrook et al. (23).
The plasmids pETtrxA and pETgstB were introduced into E. coli Rosetta (DE3) pLysS (Novagen) and cultured in LB containing 50 μgml-1 ampicillin sodium salt for 12 h, and then a portion (3 ml) was agitated on a rotary shaker at 120 rpm in 200 ml of LB containing 50 μgml-1 ampicillin sodium salt in 500-ml flasks at 30 °C. After the optical density reached 1.0, we added 0.1 mm isopropyl 1-thio-β-d-galactoside (final concentration) to the medium and further incubated the flask for 12 h at 120 rpm at 30 °C. The E. coli cells were harvested by centrifugation, suspended in 50 ml of 20 mm potassium phosphate (pH 7.5), and disrupted by sonication. The sonicate was centrifuged at 6000 × g for 15 min to remove cellular debris and unbroken cells, and then the resulting cell-free extract was centrifuged at 100,000 × g for 60 min to obtain supernatant (soluble) fractions. These fractions were passed through columns containing nickel-nitrilotriacetic acid-agarose (inner diameter, 0.5 × 2 cm, Qiagen) equilibrated with 20 mm potassium phosphate (pH 7.5). The columns were washed with 10 ml of 20 mm potassium phosphate (pH 7.5) containing 50 mm imidazole, and proteins were eluted with the same buffer containing 100 mm imidazole. All manipulations proceeded at temperatures below 4 °C.
Gene Disruption of A. nidulans glrA—A 720-bp DNA fragment encoding 5′-region of glrA fused with appropriate restriction sites was amplified using the primers 5′-CCGCGGCCGCACATTCCTTCGTCAGGG-3′ and 5′-CCGGATCCTTCTGGTTATACTGAAATTG-3′ (restriction sites are underlined). After restriction digestion with the enzymes NotI and BamHI, the ends were ligated with pBSarg1 that was spliced beforehand with the same restriction enzymes. The 3′-region of glrA was amplified using the primers 5′-GGCTGCAGGTGTGTTGCTATTCATCC-3′ and 5′-GGCTCGAGAGTGGTTACACCGAAC-3′, cut with PstI and XhoI, and inserted into the same restriction sites of the resulting plasmid to generate pDGLR1. The plasmid pBSarg1 was generated by inserting a 1.8-kilobase BamHI-SphI fragment containing the argB of A. nidulans into SmaI-digested pBluescript KS+ and transformed into A. nidulans. Total fungal DNAs prepared as described by Takasaki et al. (24) were Southern-blotted using a DIG DNA labeling and detection kit (Roche Applied Science) according to the manufacturer's instructions.
Enzyme Assays—Fungal cells were collected by filtration, washed twice with 0.7% NaCl, suspended in buffer A (20 mm potassium phosphate (pH 7.2), 10% glycerol, 0.3 mm N-tosyl-l-phenylalanine, 0.3 mm phenylmethylsulfonyl fluoride), and homogenized as described (24). Cellular debris was sedimented by centrifugation at 1500 × g for 10 min, and then the supernatant was further separated by centrifugation at 10,000 × g for 1 h. The supernatant was further separated by centrifugation at 100,000 × g for 60 min to obtain cell-free extracts.
Glutathione reductase was assayed in a reaction mixture containing 50 mm potassium phosphate (pH 7.5), 1 μm FAD, 0.1 mm NADPH, and 1 mm GSSG. The reaction was initiated by adding rGlrA or cell-free extract, and then the absorbance at 340 nm was followed at 25 °C using a Beckman DU-7500 spectrophotometer. Apparent Km values were determined by fitting each dataset to the equation v/e = kcat [A]/(KA + [A]). Thioredoxin reductase was assayed in a mixture comprising crude extract, 100 mm potassium phosphate (pH 7.5), 0.1 mm NADPH, 0.17 mm recombinant human insulin solubilized by the method of Holmgren et al. (4), and 10 μm recombinant TrxA. The reaction was started by adding recombinant TrxA, and the decrease in NADPH at absorbance 340 nm was measured. Catalase and cytochrome c peroxidase were assayed as described (25, 26). Glutathione S-transferase was assayed in a buffer (100 mm Tris HCl (pH 6.5), 1 mm GSH, 1 mm each GSH accepter) by measuring the increase in absorption at 340 nm (for 1-chloro-2,4-dinitrobenzene and 1,2-dichloro-4-nitrobenzene as accepters) and at 270 nm (for ethacrynic acid). The molecular coefficients of the GSH-conjugants were set at 9.6-, 8.5-, and 5.0-mm-1 cm-1 (27). Glutathione peroxidase activity was measured in a buffer (100 mm Tris-HCl (pH 6.5), 1 mm GSH, 1 mm cumene hydroperoxide, 1 unit of GR (Oriental Yeast Co., Tokyo), 0.2 mm NADPH), and the reaction was started by adding 15 μg of rGstB. The amount of generated GSSG was measured as a change in absorbance at 340 nm caused by the GR-dependent oxidation of NADPH coupled to GSSG reduction. All reactions proceeded at 25 °C.
Aconitase (28), sulfite reductase (29), glucose-6-phosphate dehydrogenase (30), and cytochrome c oxidase (31) activities were assayed as described. The protein concentration was determined using Protein Assay kits (Bio-Rad).
Glutathione Determination—Conidia (108) were transferred to 500-ml Erlenmeyer flasks containing 100 ml of MMDN and precultured at 30 °C to the logarithmic growth phase (20 h for A26 and 28 h for DGR1) at 120 rpm. The resultant mycelia were collected by centrifugation, washed twice with 0.85% NaCl, and then 0.2 g (wet weight) was transferred to 30-ml test tubes containing 10 ml of MMDN. After incubating at 120 rpm for 6 h at 30 °C, the mycelia were collected by centrifugation at 10,000 × g for 10 min, vigorously mixed with ice-cold 5% 5-sulfosalicylic acid, and incubated on ice for 30 min. After centrifugation at 10,000 × g for 10 min, the supernatants were neutralized with triethanolamine on ice, and then the GSH and GSSG concentrations were determined as described (32).
Analytical Methods—Absorption spectra were measured using a DU7500 spectrophotometer, and FAD was identified and quantified as described by Faeder and Siegel (33) using ε at 450 nm = 11.5 mm-1 cm-1 after denaturation to rGlrA by heating at 75 °C for 15 min. To measure superoxide anions, fungal mycelia (0.1 g wet weight) were incubated in 100 mm potassium phosphate buffer (pH 7.2) containing 1 mm nitro blue tetrazolium at 30 °C for 10 min, collected by filtration, and homogenized using liquid N2. Homogenates were suspended with dimethyl sulfoxide and centrifuged at 15,000 × g, and then the absorption of the resulting supernatants at 540 nm was measured. Hydrogen peroxide was determined as follows. Mycelia (0.5 g wet weight) were homogenized with 5 ml of 0.2 m perchloric acid. After 15 min of centrifugation at 13,000 × g at 4 °C, the resulting supernatant was neutralized to pH 7.5 with 4 m KOH and immediately used to spectrophotometrically determine H2O2 at 590 nm using a peroxidase-based assay (34). The reaction mixture contained 12 mm 3-dimethylaminobenzoic acid in 0.375 m phosphate buffer (pH 6.5), 1.3 mm 3-methyl-2-benzothiazolidone hydrazone, and 0.25 units of horseradish peroxidase (Sigma).
Nucleotide sequences were determined using an automated DNA sequencer (CEQ2000, Beckman Coulter) according to the manufacturer's instructions. Dry cell weights were determined as described (24).
Quantitative PCR—Total RNA was extracted from strain A26 cells cultured in MMDN medium or in that containing 0.1-10 mm menadione, diamide, and hydrogen peroxide at 30 °C for 20 h using the RNeasy plant mini kit (Bio-Rad) according to the manufacturer's instructions. First-strand cDNA was synthesized by incubating total RNA (10 μg) in 10 μl of reaction buffer comprising oligo(dT) 20 (Toyobo Co., Ltd, Japan), 5× reverse transcriptase buffer, and reverse transcriptase Moloney murine leukemia virus (200 units) (Takara Bio, Inc.) at 42 °C for 90 min. First-strand cDNA (330 ng) synthesized in this reaction was amplified by quantitative PCR using iQ™ SYBR® Green Supermix (Bio-Rad) and MiniOpticon™ Version 3.1 (Bio-Rad) according to the manufacturer's instructions. The expression of glrA was normalized against that of actin-encoding actA. Data were calculated as relative expression. The primer sequences were 5′-GAGAAGGCGAAGAACCCAAC-3′ and 5′-CCAACACCGAGACCCAGAA-3′ for glrA and 5′-GAAGTCCTACGAACTGCCTGATG-3′ and 5′-AAGAACGCTGGGCTGGAA-3′ for actA.
Proteome Analysis—Samples for two-dimensional gel electrophoresis were prepared from A. nidulans A26 and DGR1 as described (35). Proteins (200 μg) were isoelectrically focused, loaded onto precast 11.0% homogeneous polyacrylamide (slab) gels (PAGE) (20 × 20 cm), and stained with SYPRO Ruby (Bio-Rad) as described (35). Spots were detected using a ChemiDoc XRS (Bio-Rad). Gel images were loaded into the Proteomeweaver software (Version 4.0, Bio-Rad), and processing, spot detection, quantitation, gel matching, and warping proceeded according to the manufacturer's instructions. The volumes of spots were normalized by dividing the volume of each spot by the sum of the total spot volume. All proteomic differential display experiments were independently repeated three times. Mean values of the normalized volumes from the three experiments determined the expression level of each protein and were used for statistical tests (Student's t test). We established the level of significance at p < 0.05.
Proteins were excised from the slab, digested with trypsin gold (Promega), and used for matrix-assisted laser desorption ionization time-of-flight mass spectrometry analysis as described (35). Proteins were identified by peptide mass finger analysis using the MASCOT (Matrix Science Ltd.) search engines of the entire NCBI protein data base, and a protein sequence library was constructed in-house with the most recent annotation of the A. nidulans genome (Version 3, Broad Institute, Cambridge, MA). The maximum deviation permitted for matching the peptide mass values was set at 100 ppm. Scores of >71 were considered significant (p < 0.005).
RESULTS
Glutathione Reductase Gene (glrA) from A. nidulans—Using the BLAST program, we discovered a hypothetical gene (gene ID, AN0932.3) in the gene set of the A. nidulans FGSC A4 genome (Broad Institute, Cambridge, MA) that encoded a similar protein to the GR from S. cerevisiae. The gene encoded a protein comprising 558 amino acid residues with a predicted sequence that was similar to those of GR from E. coli gorA (50% identical), S. cerevisiae GLR1 (YPL091W) (47%), S. pombe pgr1+ (48%), and humans (GSHR_HUMAN) (43%). In the amino-terminal region, we found a motif typical of the Rossmannfold superfamily (100GXGXXG, numbers started from translational initiation site, supplemental Fig. 1) followed by a highly conserved amino acid sequence among the GRs involving the catalytic Cys residue (132CVNVGC) (6). We also identified a variation of the Rossmann-fold (269GXGXXAXE) that is only found in the GR1 subfamily of proteins containing FAD that were classified by Dym and Eisenberg (6). Binding motifs for FAD (394(T/S)X6(F/Y)XXG(D/E)) and other highly conserved sequences among the GR family were also identified (supplemental Fig. 1). The entire amino acid sequence of the protein found by a search of the Pfam data base was in the thioredoxin reductase superfamily that involves conventional GR, indicating that the gene product is closely related to GR. We designated the gene glrA based on the biochemical and genetic properties described below.
When cultured in MMDN medium at 30 °C for 24 h, 80 ± 15 nmol min-1 mg protein-1 of GR activity was generated in the cell-free extract of A. nidulans A26. Subcellular fractions were prepared by differential centrifugation, and GR activity was analyzed. Most (99.5%) of the GR activity was distributed in the soluble fractions as well as the activity of glucose-6-phosphate dehydrogenase, a cytosolic marker enzyme. A little (0.5%), but significant (1.6 ± 0.3 nmol min-1 mg protein-1) GR activity was separated with particulate fractions in which mitochondrial cytochrome c oxidase was concentrated but not with mixed-membrane fractions, indicating that the GR was localized mostly in the cytosol (data not shown). Analysis of glrA by 5′-rapid amplification of cDNA ends resulted in major and minor amplified products. Nucleotide sequencing showed that the major product corresponded to the nucleotide that started from -22 (starting from the initiation codon) and which was followed by an initiation codon predicted as above. The minor product started from +172 that was followed by a second methionine codon (Met86) (supplemental Fig. 2). We assumed that the glrA gene contained two translation initiation sites and examined intracellular GlrA distribution using the strains producing GlrA-GFP fusions. Fig. 1A shows that fluorescence was emitted by GFP in mitochondria stained with MitoTracker as well as in the cytosol. By contrast, when the second Met86 was replaced with Ala, GFP fluorescence co-located with mitochondria and the cytosolic staining was lost (Fig. 1, C and D), which indicated that the GlrAM86A-GFP fusion protein localized in the mitochondria. These results indicated that the first methionine and Met86 are responsible for GlrA localization in the mitochondria and cytosol, respectively, and that the 85 amino acid residues before Met86 target GlrA into the mitochondria. Both cytosolic and mitochondrial localizations of A. nidulans GR are similar to the GR of S. cerevisiae and of S. pombe, which produce both mitochondrial and cytosolic GR from two translation initiation sites (10, 36). However, the activity of the mitochondrial GR differed in that A. nidulans produced considerably less of it than S. cerevisiae, which produces ∼35% more activity mg protein-1 in the mitochondria than in the cytosol (36).
FIGURE 1.

Localization of GlrA-GFP fusion in A. nidulans. Conidia of transformants harboring pGRgfp1 (A and B) or pGRgfp1-M86A (C and D) that, respectively, produce GlrA-GFP and GlrAM86A-GFP, were incubated in MMDN medium at 37 °C for 10 h. GlrA-GFP fusion proteins (A and C) and mitochondria stained with MitoTracker Red CMXRos (B and D) were observed by fluorescence microscopy. Scale bars, 10 μm.
Biochemical Properties of GlrA—We prepared recombinant protein for glrA (rGlrA). The molecular mass (Mr) of rGlrA calculated from SDS-PAGE and from gel filtration chromatography was 52,000 and 100,000 (supplemental Fig. 3), indicating that rGlrA is dimeric. Yellow pigments released from rGlrA by boiling co-migrated with FAD on thin-layer chromatography (data not shown), indicating that rGlrA contains non-covalently bound FAD like other GR (6). The FAD content of the purified rGlrA was 0.60 ± 0.04. The absorption spectra of rGlrA exhibited peaks at 383 and 465 nm, which is consistent with those of typical flavoproteins. Titration of rGlrA with sodium dithionite under anaerobic conditions blue-shifted the absorption from 465 to 450 nm and decreased the size of the absorption peak (supplemental Fig. 3). An isosbestic point at 513 nm was observed during the titration. Sodium dithionite increased absorption around 540 nm, indicating the formation of the thiolate FAD charge transfer complex as observed with other GR and TR (21, 37, 38). Adding 0.1 mm NADPH to rGlrA (38 μm) caused a similar spectral change (supplemental Fig. 3), indicating that NADPH reduced the FAD moiety of rGlrA. The spectrum did not change after adding up to 1 mm NADH (data not shown). When excess GSSG (1 mm) was added, the spectral species of the NADPH-reduced rGlrA rapidly disappeared and generated a spectral species that was indistinguishable from oxidized rGlrA (supplemental Fig. 3). These findings indicated that rGlrA could transfer electrons to GSSG.
The initial velocity of NADPH oxidation by rGlrA in the presence of 5 mm GSSG and 0.1 mm NADPH was 5.5 × 103 mmol min-1 mmol FAD-1. A similar experiment replacing GSSG with A. nidulans thioredoxin (trxA product) at a physiological concentration (10 μm) resulted in <10 mmol min-1 mmol FAD-1 activity (data not shown). The amount of NADPH-O2 reductase (diaphorase) activity was <10 mmol min-1 mmol FAD-1. The apparent Michaelis-Menten constant (Km) for NADPH was 11 ± 1 μm (Table 1), and that for GSSG was 170 ± 25 μm in the presence of 0.1 mm NADPH, values that are comparable with those determined for S. cerevisiae GR (39) (Table 1). These results showed that A. nidulans glrA encoded glutathione reductase.
TABLE 1.
Kinetic parameters of rGlrA
Recombinant GlrA (rGlrA) was prepared as described under “Experimental Procedures.” Enzyme activity was determined by measuring increases in absorbance at 340 nm. Data are the means of three experiments.
| Enzyme | Substratea | Km | kcat | kcat/Km |
|---|---|---|---|---|
| μm | s−1 | mm−1 s−1 | ||
| A. nidulans rGR | GSSG | 170 ± 30 | 120 ± 20 | 0.71 |
| NADPH | 11 ± 1 | 130 ± 10 | 11 | |
| S. cerevisiae GRb | GSSG | 55 | 250 | 4.5 |
| NADPH | 3.8 | 250 | 66 |
Km and kcat values for GSSG were determined at 0.04-5 mm GSSG and at 0.1 mm NADPH, and those for NADPH were determined at 5 mm GSSG and 5-200 μm NADPH.
Data are from Massey and Williams (39).
GlrA Is Required for Normal Growth—We constructed a plasmid designed for double crossing over with the fungal chromosome at the 5′ and 3′ regions of glrA and introduced it into A. nidulans (Fig. 2A). Southern blotting of total DNA from the wild type strain and a transformant designated DGR1 revealed a specific 3.2-kilobase PstI DNA fragment for glrA in the wild type (WT) strain but not in DGR1. This transformant generated a 1.8-kilobase band, indicating a deletion of the glrA gene (Fig. 2B). The morphology of the mycelia as well as asexual (conidia) and sexual (cleistothecia and asci) reproduction organs were microscopically indistinguishable between WT and DGR1 after culture under standard conditions (data not shown), and the frequency of conidia and asci formation was normal. The DGR1 strain formed smaller colonies than WT due to a lower extension rate of the mycelia (Fig. 2C). The extent of the slow growth phenotype was dependent upon culture temperature and more prominent when cultured at higher temperatures (37 and 42 °C). The germination rates of conidia did not differ between WT and DGR1 when cultured at temperatures below 30 °C, whereas the numbers of germinated conidia decreased at 37 and 42 °C (Fig. 2D). These results indicated that glrA is required for normal growth and conidia germination at high temperatures.
FIGURE 2.

Construction of glrA mutant of A. nidulans. A, strategy for homologous recombination into glrA locus to construct glrA deletion mutants. kb, kilobases. B, Southern blot analysis of A. nidulans WT (FGSC A26) (lane 1) and DGR1 (lane 2). Total DNA from strains was digested with PstI before blotting and hybridization. C, strains were grown on MMDN agar plates with or without 10 mm GSH at indicated temperatures for 72 h. Radii of colonies are indicated in mm with S.E. D, conidia of WT (circles) and DGR1 (squares) strains of A. nidulans were spread onto MMDN agar plates with (open) or without (closed) 10 mm GSH and incubated at 30 °C for 120 h. Relative germination rates were determined by setting number of colonies on MMDN agar plates (without oxidant) as 100%. Data are the means of three experiments. Error bars represent S.E. E, mycelia of WT and DGR1 strains of A. nidulans were incubated for 6 h at 30 °C, and intracellular glutathione was determined. Closed and open bars represent reduced and oxidized glutathione, respectively. S.E. are <20%.
We quantified the cellular glutathione content in A. nidulans strains cultured at 30 °C. The results showed that total amounts of GSH/GSSG were increased 4-fold in DGR1 compared with WT (Fig. 2E). This indicated that GSH elimination induces de novo GSH synthesis in DGR1 (see below) as in yeasts (7, 8, 10). The WT strain contained 1.6 ± 0.1 nmol mg cell-1 of reduced GSH which occupied most (94%) of the intracellular GSH/GSSG pool (Fig. 2E). By contrast, the DGR1 cells contained less GSH than the WT. More than 95% of the GSH/GSSG pool in DGR1 existed as the oxidized GSSG form, indicating that GR is important for the reduction of GSSG to GSH. The temperature-sensitive growth and germination of DGR1 were partly restored by adding 10 mm GSH (Fig. 2, C and D) to the medium but not the same concentration of GSSG (data not shown). These results indicated that GSH is important for growth.
GlrA Is Required to Maintain Cellular Oxidation Levels—We
measured the steady-state concentration of ROS in the DGR1 and WT strains.
When cultured under normal conditions, DGR1 accumulated 25% more intracellular
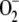 than the wild type strain
(Fig. 3A). This
implied that a depletion of GSH due to a lack of GlrA affects the
intracellular oxidation level. An excess of ROS causes oxidative stress, which
threatens the integrity of various biomolecules
(14). Indeed, we found that
the respiration activity of strain DGR1 was partially defective
(Fig. 3B). This is
likely due to the inactivation of Fe-S proteins in the mitochondrial
respiratory chain as
than the wild type strain
(Fig. 3A). This
implied that a depletion of GSH due to a lack of GlrA affects the
intracellular oxidation level. An excess of ROS causes oxidative stress, which
threatens the integrity of various biomolecules
(14). Indeed, we found that
the respiration activity of strain DGR1 was partially defective
(Fig. 3B). This is
likely due to the inactivation of Fe-S proteins in the mitochondrial
respiratory chain as 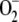 inactivates
Fe-S proteins by oxidizing their Fe-S cluster
(10,
14). The low respiration
activity could explain the slow growth due to GlrA depletion
(Fig. 2C). By
contrast, intracellular H2O2 levels in the glrA
mutant were decreased by 22% compared with those of wild type cells
(Fig. 3A), indicating
that A. nidulans expresses a compensation mechanism(s) in response to
GlrA depletion. The most likely candidates are thioredoxin peroxidases
(peroxiredoxins), cytochrome c peroxidase, and catalases, which we
found were increased in the DGR1 strain as discussed below.
inactivates
Fe-S proteins by oxidizing their Fe-S cluster
(10,
14). The low respiration
activity could explain the slow growth due to GlrA depletion
(Fig. 2C). By
contrast, intracellular H2O2 levels in the glrA
mutant were decreased by 22% compared with those of wild type cells
(Fig. 3A), indicating
that A. nidulans expresses a compensation mechanism(s) in response to
GlrA depletion. The most likely candidates are thioredoxin peroxidases
(peroxiredoxins), cytochrome c peroxidase, and catalases, which we
found were increased in the DGR1 strain as discussed below.
FIGURE 3.

Effects of glrA mutation on cellular processes. A, intracellular levels of superoxide and H2O2 in WT (closed bars) and DGR1 (open bars) strains of A. nidulans cultured at 30 °C for 20 h. Data are means of four experiments. Error bars represent S.E. p < 0.03 (superoxide) and p < 0.05 (H2O2). B, respiration activity of WT (closed bars) and DGR1 (open bars) strains of A. nidulans cultured at 30 °C for 6 h after pre-cultivating conidia at 30 °C for 20 h. Data are the means of four experiments. Error bars represent S.E. C, aconitase (left) and sulfite reductase (right) activity of WT (closed bars) and DGR1 (open bars) cultured at 30 °C for 6 h after pre-cultivating conidia at 30 °C for 20 h. Data are the means of four experiments. Error bars represent S.E.
Role of glrA in Oxidative Stress Tolerance—Expression of the glrA gene was quantified using PCR. Culture in the presence of reagents that cause oxidative stress resulted in the concentration-dependent generation of more glrA transcripts in A. nidulans (Fig. 4A). Menadione at 0.1 mm induced little glrA expression, but at 1 mm, 6.1-fold more glrA was expressed than in control cultures without menadione. Diamide at both 1 and 10 mm also induced up to 1.5-fold more glrA expression, which was less efficient than menadione. Hydrogen peroxide was the least effective (Fig. 4A) as 1 mm H2O2 did not affect glrA expression, and 10 mm was as effective as diamide. Increasing the culture temperature from 30 to 37 or 42 °C induced 1.4-fold more glrA transcription (Fig. 4A). This is consistent with a glrA requirement for normal cell growth at high temperatures (Fig. 2C).
FIGURE 4.

Effects of oxidative stress on A. nidulans DGR1. A, quantitative PCR analysis of glrA gene transcript. A. nidulans was incubated at 30 °C for 6 h, the indicated compounds were added or temperatures were shifted, and then incubation proceeded for further 3 h. Bars indicate values reported as relative expression rates obtained by RT-PCR and normalized to actA. B, strains were grown on MMDN agar plates with or without various oxidants at 30 °C for 120 h. Concentrations of oxidants: 30 μm (menadione (MD)), 2 mm (diamide), and 2.5 mm (hydrogen peroxide). Radii of colonies are indicated in mm with S.E. C-E, conidia of WT (circles) and DGR1 (squares) strains of A. nidulans were spread onto MMDN agar plates and incubated at 30 °C for 120 h. Relative germination rates were determined by setting number of colonies on MMDN agar plates (without oxidant) as 100%. Data are the means of three experiments. Error bars represent S.E.
We examined the growth of A. nidulans WT and DGR1 strains in the presence of various amounts of oxidants. The growth of both strains was almost completely suppressed by high levels of menadione, diamide, and H2O2 (data not shown). Fig. 3B shows examples of growth in the presence of moderate levels of these oxidants. The length of DGR1 hyphae after incubation on agar plates for 120 h at 30 °C was 74, 53, 63, and 72% that of WT in the presence of no oxidant, 30 μm menadione, 2 mm diamide, and 2.5 mm H2O2, respectively, indicating that menadione and diamide obviously suppressed DGR1 growth. Concentration ranges of H2O2 from 0.5 to 3 mm affected the length of DGR1 hyphae less than those of WT (data not shown), indicating that DGR1 is more sensitive to menadione and diamide than to H2O2. The results were essentially the same for DGR1germination rates; namely, the oxidants decreased the number of germinating conidia, and menadione and diamide inhibited DGR1 germination more effectively than H2O2 (Fig. 4, C-E). These results indicated that glrA plays critical roles in the presence of oxidative stress.
Global Protein Production Altered by glrA Deletion—We resolved the intracellular proteins of A. nidulans WT and DGR1 by two-dimensional gel electrophoresis and compared global protein production between them. We identified more than 600 proteins in each gel (supplemental Figs. 4 and 5), and an analysis of gel images revealed 13 and 7 proteins that were >2-fold increased and decreased in the DGR1strain, respectively. Their peptide mass fingerprints showed that the increased proteins included thioredoxin reductase (TrrA, AN3581.3), cytochrome c peroxidase (AN1630.3), and catalase B (CatB, AN9339.3) (Table 2). Levels of their enzyme activities in cell-free extracts were higher in DGR1 than in the wild type strain (Table 3), which confirmed that a glrA deletion up-regulated these proteins. Up-regulation of the H2O2-degrading enzymes (CatB and cytochrome c peroxidase) was consistent with the lower H2O2 concentration in DGR1 (Fig. 4A). A peroxiredoxin homologous to yeast Ahp1p (AN8692.3, 40% identical) as well as elongation factor 1B (AN9304.3), of which the Aspergillus fumigatus ortholog (elfA) reportedly has peroxiredoxin activity (40), were more than 2-fold up-regulated in the DGR1 strain. We previously constructed A. nidulans proteome maps comprising 300 intracellular proteins (35). They include catalase A (CatA, AN8553.3) (41), catalase peroxidase (catalase D) (CpeA, AN7388.3) (42), manganese-dependent superoxide dismutase (SodM, AN0785.3) (43), and proteins homologous to yeast Cu,Zn-dependent superoxide dismutase (Sod1p) (AN0241.3, 73% identical) and to yeast peroxiredoxins Prx1p (AN10223.3, 59%) and Dot5p (AN4301.3, 30%) (Table 2). The Prx1p homolog was 1.8-fold more abundant in DGR1 than in the wild type strain, whereas CatA and CpeA was less induced in DGR1 (Table 2). The amounts of SodM and the Sod1p homolog did not significantly differ between the strains. No Dot5p homolog was detected in this study, probably due to low expression levels. Because peroxiredoxins are physiologically of significance for the thioredoxin-dependent reduction of H2O2 in DGR1, this finding together with TrrA up-regulation (Tables 2 and 3) and a low H2O2 level (Fig. 4A) indicated that GR depletion up-regulated the thioredoxin system and that the glutathione- and thioredoxin-dependent anti-oxidant systems are functionally relevant.
FIGURE 5.

Characterization of novel GST from A. nidulans. A, SDS-PAGE of purified enzyme. Purified enzymes (1 μg) were resolved by SDS-PAGE on 10% polyacrylamide gels and stained with Coomassie Brilliant Blue. Lane 1, markers (Novex Sharp Protein Standard kit, Invitrogen); lane 2, rGstB. B, Quantitative PCR analysis of gstB gene transcript. A. nidulans A26 was grown at 30 °C for 20 h, the indicated compounds were added or temperatures were shifted, and then incubation proceeded for further 3 h. Bars indicate values reported as relative expression rates obtained by RT-PCR and normalized to 18 S rRNA. C, phylogenetic relationships among GstB-like GSTs from various organisms. Multiple alignment and neighbor joining were performed using Molecular Evolutionary Genetics Analysis (MEGA) Software Version 4.0. Accession numbers are from Broad Institute. GstA involved in cluster 2 GST from A. nidulans served as outgroup.
TABLE 2.
Putative proteins involved in oxidative responses in A. nidulans identified in two-dimensional PAGE
| No.a | Annotation name | Gene IDb | Genec | -Foldd | tpIe | tMWf |
|---|---|---|---|---|---|---|
| Oxidative stress response | ||||||
| 1 | Thioredoxin reductase | AN3581.3 | trrA | 6.2 | 5.2 | 36.1 |
| 2 | Cytochrome c peroxidase | AN1630.3 | CCP1 | 2.0 | 8.9 | 39.9 |
| 3 | Catalase B | AN9339.3 | catB | 4.8 | 4.9 | 79.2 |
| 4 | Catalase-peroxidase (catalase D) | AN7388.3 | cpeA | 1.2 | 5.9 | 82.1 |
| 5 | Catalase A | AN8553.3 | catA | 1.3 | 7.7 | 60.2 |
| 6 | Manganese superoxide dismutase | AN0785.3 | sodM | 1.1 | 6.2 | 23.4 |
| 7 | Cu,Zn superoxide dismutase | AN0241.3 | SOD1 | 0.9 | 5.8 | 15.9 |
| 8 | Peroxiredoxin/Prx5-like /allergen Asp f3 | AN8692.3 | AHP1 | 4.2 | 5.6 | 18.5 |
| 9 | Peroxiredoxin | AN4301.3 | DOT5 | - | 9.5 | 21.8 |
| 10 | Peroxiredoxin | AN10223.3 | PRX1 | 1.8 | 5.5 | 23.3 |
| 11 | Elongation factor 1B | AN9304.3 | elfA | 2.5 | 5.4 | 24.0 |
| 12 | Hypothetical protein (glutathione S-transferase) | AN6024.3 | gstB | 2.6 | 5.9 | 24.9 |
| 13 | Glutathione reductase | AN0932.3 | glrA | g | 6.2 | 51.6 |
| Other | ||||||
| Up-regulated proteins | ||||||
| 14 | Sulfite reductase | AN1752.3 | - | 1.9 | 5.0 | 114.8 |
| 15 | Glucose-methanol-Choline oxidoreductase | AN7267.3 | - | 3.7 | 5.7 | 59.2 |
| 16 | M20A metallohydrolase family | AN3459.3 | DUG1 | 2.0 | 5.4 | 52.6 |
| 17 | Tropomyosin | AN5686.3 | TPM2 | 2.1 | 5.0 | 18.8 |
| 18 | Proteasome subunit α 2 | AN6726.3 | PRE8 | 2.2 | 5.6 | 30.1 |
| 19 | Zinc binding dehydrogenase | AN7914.3 | - | 2.3 | 5.0 | 36.0 |
| 20 | Reduced viability upon starvation protein 161 | AN8831.3 | RVS161 | 2.8 | 5.6 | 31.2 |
| 21 | Serine hydroxymethyl transferase | AN3058.3 | SHM2 | 1.7 | 7.6 | 50.3 |
| 22 | Ubiquinol cytochrome c reductase core protein 2 | AN8273.3 | QCR2 | 2.3 | 9.0 | 47.6 |
| 23 | Woronin body major protein | AN4695.3 | hexA | 1.9 | 6.7 | 25.1 |
| 24 | Aconitase | AN5525.3 | acoA | 1.7 | 6.0 | 83.1 |
| Down-regulated proteins | ||||||
| 25 | Hypothetical protein | AN3674.3 | - | 0.58 | 4.9 | 58.2 |
| 26 | O-Methyl transferase | AN9233.3 | - | 0.50 | 6.1 | 42.9 |
| 27 | Short chain dehydrogenase | AN0179.3 | TMA29 | 0.35 | 6.2 | 29.3 |
| 28 | UDP-N-acetylglucosamine pyrophosphorylase | AN9094.3 | QRI1 | 0.48 | 6.0 | 55.8 |
| 29 | Pyruvate dehydrogenase complex E2 component | AN6708.3 | LAT1 | 0.46 | 6.1 | 51.8 |
| 30 | S-Adenosylmethionine synthetase | AN1222.3 | SAM2 | 0.61 | 5.3 | 42.2 |
| 31 | Bifunctional purine biosynthesis protein | AN4464.3 | purH | 0.55 | 6.3 | 65.6 |
| 32 | 6-phosphogluconate dehydrogenase | AN3954.3 | GND2 | 0.52 | 5.7 | 54.0 |
| 33 | Phosphoglycerate kinase | AN1246.3 | pgkA | 0.45 | 6.4 | 44.9 |
| 34 | Flavohemoglobin | AN7169.3 | YHB1 | 0.60 | 6.1 | 44.9 |
| 35 | Alcohol dehydrogenase | AN8979.3 | alcA | 0.49 | 7.6 | 37.1 |
| 36 | Peptidyl-tRNA hydrolase | AN10249.3 | PTH2 | 0.60 | 5.5 | 57.5 |
| 37 | ATP synthase α -chain | AN1523.3 | ATP1 | 0.39 | 9.1 | 61.9 |
| 38 | Porin | AN4402.3 | POR1 | 0.41 | 9.0 | 29.9 |
| 39 | Constitutive protein, actin | AN6542.3 | actA | 1.1 | 5.5 | 41.7 |
Numbers in this correspond to the protein spots in supplemental Fig. 4 and 5.
Gene IDs are according to A. nidulans genome database.
Gene names for orthologs in S. cerevisiae are shown when relevant Aspergillus genes were uncharacterized. -, no orthologous gene was identified in S. cerevisiae.
Levels of expression in A. nidulans DGR1 (glrA disruptant) were compared with FGSC A26 (wild type). Standard deviations of data were <31%. p < 0.05.
Theoretical pI.
Theoretical molecular mass.
No GlrA was detected in strain DGR1.
TABLE 3.
Enzyme activity of anti-oxidant enzymes A. nidulans strains
A. nidulans strains were incubated at 30 °C for 6 h after precultivating conidia at 30 °C for 20 h, and enzyme activity in cell-free extracts was determined as described under “Experimental Procedures.” -Fold induction in DGR1 is presented in parentheses. Data are the means of three experiments ± S.D.
|
Specific activity
|
|||
|---|---|---|---|
| Strain | Thioredoxin reductase | Catalase | Cytochrome c peroxidase |
| μmol min−1 mg protein−1 | |||
| WT | 7.6 ± 1.5 × 10−3 (1) | 2.3 ± 0.3 × 104 (1) | 1.3 ± 0.2 × 101 (1) |
| DGR1 | 2.7 ± 0.3 × 10−2 (3.6) | 1.1 ± 0.1 × 105 (4.8) | 4.0 ± 0.2 × 101 (3.1) |
Proteomic analyses identified other proteins that were up- and down-regulated by GlrA depletion (Table 3). Some house-keeping proteins were up-regulated such as sulfite reductase (AN1752.3) (1.9×) for sulfate assimilation and aconitase (AN5525.3) (1.7×), constituting the tricarboxylic acid cycle. The enzyme activity of these two proteins was increased 9.3- and 1.9-fold in cell-free extracts of the DGR1 strain, confirming their up-regulation (Fig. 3C). Other housekeeping proteins, for example, a subunit of pyruvate dehydrogenase (AN6708.3), 6-phosphogluconate dehydrogenase (AN3954.3), phosphoglycerate kinase (AN1246.3), alcohol dehydrogenase (alcA, AN8979.3), and the ATP synthase catalytic subunit (AN1523.3), were down-regulated, indicating that carbon and energy metabolic mechanisms were altered due to GlrA depletion. Higher levels of proteins related to glutathione metabolism were also detected in DGR1. Proteins orthologous to human tropomyosin that is post-translationally modified with glutathionylation (44) and to S. cerevisiae Dug1p that constitutes an alternative GSH degradation mechanism (45) are such examples. Together with increased de novo GSH synthesis (Fig. 2), these result indicated that glutathione turnover was accelerated in the absence of glrA.
Identification of Novel Fungus-specific GST—We discovered a hypothetical protein that was expressed at 2.6-fold higher levels in the DGR1 than in the wild type strains (Table 1 and spot 12 in supplemental Fig. 4). Peptide mass fingerprints of the protein after trypsin digestion revealed that it was the gene product of AN6024.3 with sequence coverage of 60% and a Mascot score of 140. The theoretical Mr and pI values or the AN6024.3 product were 25,000 and 5.4, which were almost identical to our empirical results (23,000 and 5.2). A search of the Pfam data base revealed a potential protein motif for the carboxyl terminal domain of GST buried in the amino acid sequence. The Tyr residue located near the amino terminus is characteristic of some GSTs. Fraser et al. cloned and analyzed the gstA gene of A. nidulans (AN4905.2) and showed that it encoded a glutathione S-transferase-like protein that is involved in resistance against xenobiotics and heavy metals (46). The gene product of AN6024.3 was obviously different from GstA, and they were only 17.2% homologous with each other. A purified recombinant protein for the AN6024.3 product generated in the E. coli expression system as described under “Experimental Procedures” that resolved as a single band on SDS-PAGE at Mr 25,000 (Fig. 5A) was comparable with the Mr of the native protein detected by two-dimensional gel electrophoresis (supplemental Fig. 4). The Mr estimated from gel filtration chromatography was 51,000, indicating that the protein was dimeric (data not shown). The protein exhibited significant ability (7.6 ± 1.0 nmol min-1 mg-1) to transfer GSH to 1-chloro-2,4-dinitrobenzene (Table 4); the Km for GSH was 32 ± 3 μm. These results indicated that the gene AN6024.3 encoded a novel fungal GST, and we designated it gstB. Both 1,2-dichloro-4-nitrobenzene and ethacrynic acid are conventional substrates for GST assays, but they were not substrates for GstB. We measured glutathione peroxidase activity by measuring the amount of GSSG produced by the cumene hydroperoxide-dependent oxidation of GSH. The specific activity of recombinant GstB (rGstB) for the respective reactions was 90 ± 18 nmol min-1 mg-1 (Table 4).
TABLE 4.
Glutathione peroxidase activities and GST of purified rGstB protein
We prepared rGstB and determined enzyme activity as described under “Experimental Procedures.” Assays included 1 mm GSH and 15 μg of rGstB. Data are the means of three experiments ± S.D.
| Substrate | Specific activity |
|---|---|
| nmol min−1 mg protein−1 | |
| 1-Chloro-2,4-dinitrobenzene | 7.6 ± 1.0 |
| 1,2-Dichloro-4-nitrobenzene | <0.5 |
| Ethacrynic acid | <0.5 |
| Cumene hydroperoxide | 90 ± 18 |
We investigated the regulation of gstB transcription using quantitative PCR. More gstB transcripts accumulated in DGR1 than in the wild type strain after cultivation for 3 h (×12 ± 1) and 6 h (×15 ± 3) at 30 °C (data not shown), which was consistent with the amounts of GstB protein identified by two-dimensional gel electrophoresis (Table 2) and confirmed GstB up-regulation in the absence of GlrA. The expression of gstB was induced by exposing the fungus to various oxidants for 3 h (Fig. 5B). The extent of induction varied among the oxidants with menadione being the most effective. This is similar to the expression profile of glrA (Fig. 3). Menadione, diamide, and H2O2 caused more effective expression of gstB than of glrA. These oxidants also induce the expression of gstA of A. nidulans (46), indicating that the glutathione system plays a role(s) in the fungal response to oxidative stress.
A search of the NCBI data base and the fungal genome data base at the Broad Institute using the BLAST algorithm uncovered putative proteins with high identity to A. nidulans GstB that were distributed in Aspergillus terreus (ATEG_06528.1, 69% identical), Aspergillus niger (e_gw1_7.1165, 64%), Aspergillus oryzae (AO090102000005, 65%; AO090003001469, 57%), Fusarium graminearum (anamorph of Gibberella zeae) (FGSG_03802, 58%; FGSG_11062, 53%), Fusarium verticillioides (FVEG_12937, 61%; FVEG_12433, 56%), and Fusarium oxysporum (FOXG_13104, 57%). Phylogenetic analysis showed that these proteins and A. nidulans GstB were included in a single branch (Fig. 5C), whereas proteins that were more distantly related to GstB were clustered in another branch. The latter included hypothetical proteins from Rhizopus oryzae (RO3G_01266.1, 33% identical to GstB; RO3G_08558.1, 32%) as well as Cunninghamella elegans GST1-1 (33%) and GST2-2 (32%) (16). Given that both of the C. elegans GSTs are of the γ class (16) and these four GSTs had <33% identity to the GstB-like proteins located in the same branch as A. nidulans GstB (Fig. 5C), these results indicated that the GstB-like proteins comprise a novel class of GSTs that are distinct from the γ class. Glutathione S-transferases are commonly distributed from most aerobic bacteria to higher eukaryotes, and they constitute a superfamily based on their primary sequences as well as other criteria. Mammalian and plant GSTs have been studied in detail and grouped into at least eight structurally distinct families (47). A comparison of the amino acid sequences of these GSTs and GstB resulted in only 20-25% sequence identity (data not shown). Although others have proposed that GSTs with more than 40% identity should be categorized into the same class (48), GstB does not belong to any known GST family according to this criterion, which we confirmed by phylogenetic analysis (supplemental Fig. 6).
DISCUSSION
We investigated the glutathione system of A. nidulans by
constructing a fungal GR-encoding glrA gene knock-out strain. The
strain (DGR1) almost completely lacked intracellular GSH as well as GR
activity and accumulated more GSSG than the wild type strain, showing that GR
plays a critical role for GSSG reduction to GSH in this fungus. We further
prepared rGlrA and showed that its dimeric and kinetic properties as well as
nucleotide specificity resembled those of GR from S. cerevisiae and
S. pombe, implying that the GR-glutathione system is conserved among
these fungi. The present results showed that GR depletion reduced the growth
rate of A. nidulans under standard (normoxic) conditions. In
comparison, a deletion of the GR gene did not affect growth in S.
cerevisiae (8), whereas it
was lethal in S. pombe
(7), indicating that
GR-glutathione system functions differently among fungal species. Meanwhile,
extensive studies of S. cerevisiae have indicated that the yeast
accumulates the same level of GSH in the absence of GR
(8), whereas S. pombe
and A. nidulans strains defective in GR accumulate only 12%
(7) and 21%
(Fig. 2E) of GSH
relative to the wild type strain. These observations indicate that the extent
of fungal growth defects correlates with a decrease in intracellular GSH. The
growth defect in A. nidulans was complemented by adding GSH to the
media (Fig. 2, C and
D), suggesting that defective ROS removal due to a
decrease of GSH and/or a cellular redox state imbalanced because of a lowered
GSH/GSSG ratio caused the growth defect. The partial O2 respiration
deficiency and higher intracellular 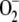 concentration in DGR1 (Fig. 4, A
and B) is consistent with excessive ROS accumulation in
the GR-deficient cells, damaging the fungal respiration mechanism.
concentration in DGR1 (Fig. 4, A
and B) is consistent with excessive ROS accumulation in
the GR-deficient cells, damaging the fungal respiration mechanism.
A small amount of GSH accumulated in the DGR1 cells, although the cells exhibited no significant intracellular GR activity. Intracellular thioredoxin is the most likely candidate protein to produce GSH as a combination of NADPH, TR, and thioredoxin of A. nidulans reduces GSSG in vitro (21). This is also true of S. cerevisiae when GSH under the genetic background of Δglr1 is supplied by TR (3, 8). These imply that both fungi share a TR-thioredoxin-dependent mechanism that compensates for the GR (or GSH) deficiency. Indeed, the concentrations of GSH in the GR-deficient cells of A. nidulans, S. cerevisiae, and S. pombe considerably differ, suggesting that the TR-thioredoxin system contributes to maintain cellular GSH concentration to different extents among these fungi. Because the properties of both fungal thioredoxins are similar, this difference seems to be a consequence of variable production of TR and/or thioredoxin. To date little is known about TR production in response to GR depletion in S. cerevisiae and S. pombe. However, the present study demonstrated that A. nidulans upregulated TR in response to GR depletion (Tables 2 and 3), indicating that TR production is regulated in these yeasts. Alternatively, the steady-state turnover level of enzymes that consume GSH could differ among fungi to support normal growth, which could determine (or regulate) the intracellular GSH concentration and, thus, the growth of A. nidulans. The GstB found in this study (see following discussion) and other GSH-dependent antioxidant proteins such as glutathione peroxidases and glutaredoxins identified in the gene set of A. nidulans (data not shown) should be candidates for such proteins. Our findings that gstB expression is inducible by exogenous oxidants like other fungal GSTs and that GstB exhibited activity for transferring not only GSH but also glutathione-dependent peroxidases like C. elegans GST1-1 and GST2-2 (16), and A. fumigatus GstA, GstB, and GstC (18) suggests that they play a role in the fungal response to oxidative stress.
We showed that exogenous oxidants induced glrA expression and
inhibited cell growth and conidial germination more effectively in the DGR1
than in the wild type strain, indicating that GR (or GSH) functions as an
antioxidant in A. nidulans as it does in yeasts and other organisms
(7-10).
Menadione was the most powerful inducer of glrA expression among the
tested oxidants. The results also indicated that GR depletion caused
hypersensitive growth and germination of A. nidulans in response to
menadione (Fig. 3). This
phenomenon is explained as follows. Fungal cells accumulated more
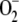 in the absence of GR, probably due
to a dysfunction of the respiratory chain
(Fig. 4), whereas levels of
in the absence of GR, probably due
to a dysfunction of the respiratory chain
(Fig. 4), whereas levels of
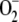 -scavenging superoxide dismutase
remained stable (Table 2).
Because menadione is generally thought to generate
-scavenging superoxide dismutase
remained stable (Table 2).
Because menadione is generally thought to generate
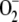 under normoxic conditions, the cells
were faced with more
under normoxic conditions, the cells
were faced with more 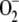 in the presence
of menadione under the genetic background of ΔglrA, and thus,
DGR1 consequently grows more slowly than the wild type. Meanwhile,
H2O2 induces glrA transcription less
effectively than menadione and minimally inhibits DGR1 growth, which is in
sharp contrast to menadione. This phenomenon is similar in a S. pombe
mutant devoid of GR (7),
although mechanistic insight into the difference remains elusive. We showed
that GR depletion in A. nidulans induced catalases A and B,
catalase-peroxidase (catalase D), cytochrome c peroxidases (Tables
2 and
3), and peroxiredoxins
(Table 2), all of which remove
intracellular H2O2. The intracellular concentration of
H2O2 was lower in the GR mutant than in the wild type
strain, which implies that a glrA deletion activates the
anti-H2O2 mechanism, which confers more tolerance upon
the cells to H2O2.
in the presence
of menadione under the genetic background of ΔglrA, and thus,
DGR1 consequently grows more slowly than the wild type. Meanwhile,
H2O2 induces glrA transcription less
effectively than menadione and minimally inhibits DGR1 growth, which is in
sharp contrast to menadione. This phenomenon is similar in a S. pombe
mutant devoid of GR (7),
although mechanistic insight into the difference remains elusive. We showed
that GR depletion in A. nidulans induced catalases A and B,
catalase-peroxidase (catalase D), cytochrome c peroxidases (Tables
2 and
3), and peroxiredoxins
(Table 2), all of which remove
intracellular H2O2. The intracellular concentration of
H2O2 was lower in the GR mutant than in the wild type
strain, which implies that a glrA deletion activates the
anti-H2O2 mechanism, which confers more tolerance upon
the cells to H2O2.
To date, information about fungal GST is limited. The only GSTs characterized in detail are those in cluster 2 and the ω class from S. cerevisiae (Gtt2p, Gto1p, Gto2p, Gto3p) (49), the θ class from Issatchenkia orientalis (GST Y-1 and Y-2) (17), the γ class from C. elegans (GST1-1, GST2-2) (16), cluster 2 from A. nidulans (GstA) (46), and cluster 2 (GstA and GstB) and EF1Bγ from A. fumigatus (ElfA) (18). However, this suggests that fungi produce divergent classes of GST dependent on the species. In addition, the present proteomic analysis indicated that the GST (GstB) discovered herein belongs to a novel class. This implies that an evolutionarily distant class of GST must be uncovered in fungal genomes by re-evaluating phylogenetic relationships among fungal GST orthologs. Indeed, we found genes for hypothetical proteins similar to α, κ, ω, cluster 2, EF1Bγ, Ure2p-like, MAK16, and MAPEG classes of GST in the A. nidulans genome (supplemental Table 1) in addition to GstB (note that A. nidulans also produces a maleylacetoacetate isomerase (AN1895.3) which is a classical ζ GST (50)) (supplemental Fig. 4). Among them, orthologs for clusters 2, ω class, EF1Bγ class, and MAK16 class GST are widely distributed in fungi in Ascomycetes, Basidiomycetes, and Zygomycetes, whereas Ure2p class GST were found only in Ascomycetes (supplemental Table 1). Meanwhile, fungi harboring the GstB orthologs belong exclusively to Ascomycetes and especially to the Aspergillus and Fusarium genera, whereas we found no protein orthologous to GstB with >40% sequence identity in other Ascomycetes fungi such as Candida lusitaniae, Candida tropicalis, Chaetomium globosum, Coccidioides immitis, Histoplasma capsulatum, Magnaporthe grisea, Neurospora crassa, Sclerotinia sclerotiorum, S. cerevisiae, and in Basidiomycetes (Coprinus cinerea, Cryptococcus neoformans, Puccinia graminus, Ustilago maydis) and Zygomycetes fungi (Botryotinia fuckeliana, R. oryzae) among known genomic DNA sequences. This indicated that GstB-like GSTs are distributed in restricted fungal species. γ class GSTs are another example of GSTs found only in the Zygomycetes C. elegans and R. oryzae. That these specific GST are distinct from the five clusters proposed by McGoldrick et al. (15) is notable.
Here we presented an initial proteomic differential display from GR-depleted and wild type fungal strains and found that a deletion of the GR gene (glrA) resulted in global changes in cellular protein production in A. nidulans. The change in the proteome also demonstrated interplay between glutathione- and thioredoxin-dependent antioxidant systems and led to the discovery of a novel class of fungal GSTs, which shed light upon hidden GSTs distributed among fungal species. The present findings suggest that a combination of targeted gene disruption and proteomics is a powerful approach to molecular analyses of A. nidulans in the post-genome era.
Supplementary Material
Acknowledgments
We thank Norma Foster for critical reading the manuscript.
This study was supported in part by the Bio-oriented Technology Research Advancement Institution and a grant-in-aid for Scientific Research from the Ministry of Education, Science, Culture, and Sports, Japan. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. 1-6 and Table 1.
Footnotes
The abbreviations used are: ROS, reactive oxygen species; GR, glutathione reductase; GST, glutathione S-transferase; TR, thioredoxin reductase; WT, wild type; GFP, green fluorescence protein; r-, recombinant.
References
- 1.Meister, A., and Anderson, M. E. (1983) Annu. Rev. Biochem. 52 711-760 [DOI] [PubMed] [Google Scholar]
- 2.Cannel-Harel, O., and Storz, G. (2000) Annu. Rev. Microbiol. 54 439-461 [DOI] [PubMed] [Google Scholar]
- 3.Trotter, E. W., and Grant, C. M. (2003) EMBO Rep. 4 185-188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Holmgren, A. (1985) Annu. Rev. Biochem. 54 237-271 [DOI] [PubMed] [Google Scholar]
- 5.Mustacich, D., and Powis, G. (2000) Biochem. J. 346 1-8 [PMC free article] [PubMed] [Google Scholar]
- 6.Dym, O., and Eisenberg, D. (2001) Protein Sci. 10 1712-1728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee, J., Dawes, I. W., and Roe, J. (1997) J. Biol. Chem. 272 23042-23049 [DOI] [PubMed] [Google Scholar]
- 8.Muller, E. G. (1996) Mol. Biol. Cell 7 1805-1813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Teggle, C. K., and Fuchs, J. A. (1985) J. Bacteriol. 162 448-450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Song, J., Cha, J., and Roe, J. (2006) Eukaryot. Cell 11 1857-1865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hayes, J. D., Flanagan, J. U., and Jowsey, I. R. (2005) Annu. Rev. Pharmacol. Toxicol. 45 51-88 [DOI] [PubMed] [Google Scholar]
- 12.Klein, M., Mamnun, Y. M., Eggmann, T., Schüller, C., Wolfger, H., Martinoia, E. E., and Kuchler, K. (2002) FEBS Lett. 520 63-67 [DOI] [PubMed] [Google Scholar]
- 13.Beer, S. M., Taylor, E. R., Brown, S. E., Dahm, C. C., Costa, N. J., Runswick, M. J., and Murphy, M. P. (2004) J. Biol. Chem. 279 47939-47951 [DOI] [PubMed] [Google Scholar]
- 14.Herrero, E., Ros, J., Belli, G., and Cabiscol, E. (2008) Biochim. Biophys. Acta 1780 1217-1235 [DOI] [PubMed] [Google Scholar]
- 15.McGoldrick, S., O'Sullivan, S. M., and Sheehan, D. (2005) FEMS Microbiol. Lett. 242 1-12 [DOI] [PubMed] [Google Scholar]
- 16.Cha, C., Kim, S., Kim, Y., Stingley, R., and Cerniglia, C. E. (2002) Biochem. J. 368 589-595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tamaki, H., Yamamoto, K., and Kumagai, H. (1999) J. Bacteriol. 181 2958-2962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Burns, C., Geraghty, R., Neville, C., Murphy, A., Kavanagh, K., and Doyle, S. (2005) Fungal Genet. Biol. 43 319-327 [DOI] [PubMed] [Google Scholar]
- 19.Kim, H. G., Park, K. N., Cho, Y. W., Park, E. H., Fuchs, J. A., and Lim, C. J. (2001) Biochim. Biophys. Acta 1520 179-185 [DOI] [PubMed] [Google Scholar]
- 20.Cho, Y. W., Park, E. H., Fuchs, J. A., and Lim, C. J. (2002) Biochim. Biophys. Acta 1574 399-402 [DOI] [PubMed] [Google Scholar]
- 21.Thön, M., Al-Abdallah, Q., Hortschansky, P., and Brankhage, A. A. (2007) J. Biol. Chem. 282 27259-27269 [DOI] [PubMed] [Google Scholar]
- 22.Barratt, R. W., Johnson, G. B., and Ogata, W. N. (1965) Genetics 52 233-246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sambrook, J., Fritch, E. F., and Maniatis, T. (1989) in Molecular Cloning: A Laboratory Manual, Vol. 2, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY
- 24.Takasaki, K., Shoun, H., Yamaguchi, M., Takeo, K., Nakamura, A., Hoshino, T., and Takaya, N. (2004) J. Biol. Chem. 279 12414-12420 [DOI] [PubMed] [Google Scholar]
- 25.Beers, R. F., and Sizer, I. W. (1952) J. Biol. Chem. 195 133-140 [PubMed] [Google Scholar]
- 26.Goodhew, C. F., Wilson, I. B. H., Hunter, D. J. B., and Pettigrew, G. W. (1990) Biochem. J. 271 707-712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Habic, W. H., Pabst, M. J., and Jakoby, W. B. (1974) J. Biol. Chem. 249 7130-7139 [PubMed] [Google Scholar]
- 28.Kennedy, M. C., Emptageg, M. H., Dreyerll, J. L., and Beinert, H. (1983) J. Biol. Chem. 258 11098-11105 [PubMed] [Google Scholar]
- 29.Siegel, L. M., Davis, P. S., and Kamin, H. (1974) J. Biol. Chem. 249 1572-1586 [PubMed] [Google Scholar]
- 30.Hankinson, O., and Cove, D. J. (1974) J. Biol. Chem. 249 2344-2353 [PubMed] [Google Scholar]
- 31.Cooperstein, S. J., and Lazarow, A. (1951) J. Biol. Chem. 189 665-670 [PubMed] [Google Scholar]
- 32.Anderson, M. E. (1985) Methods Enzymol. 113 548-555 [DOI] [PubMed] [Google Scholar]
- 33.Faeder, E. J., and Siegel, L. M. (1973) Anal. Biochem. 53 332-336 [DOI] [PubMed] [Google Scholar]
- 34.Oracz, K., Bouteau, H. E., Farrant, J. M., Cooper, K., Belghazi, M., Job, C., Job, D., Corbineau, F., and Bally, C. (2007) Plant J. 50 452-465 [DOI] [PubMed] [Google Scholar]
- 35.Shimizu, M., Fujii, T., Masuo, S., Fujita, K., and Takaya, N. (2009) Proteomics 9 7-19 [DOI] [PubMed] [Google Scholar]
- 36.Outten, C. E., and Culotta, V. C. (2004) J. Biol. Chem. 279 7785-7791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sahlman, L., and Williams, C. H. (1989) J. Biol. Chem. 264 8033-8038 [PubMed] [Google Scholar]
- 38.Huang, H. H., Arscott, L. D., Ballou, D. P., and Williams, C. H. (2008) Biochemistry 47 1721-1731 [DOI] [PubMed] [Google Scholar]
- 39.Massey, V., and Williams, C. H. (1965) J. Biol. Chem. 240 4470-4480 [PubMed] [Google Scholar]
- 40.Carberry, S., Neville, C., Kavanagh, K., and Doyle, S. (2006) Biochem. Biophys. Res. Comm. 341 1096-1104 [DOI] [PubMed] [Google Scholar]
- 41.Navarro, R. E., Stringer, M. A., Hansberg, W., Timberlake, W. E., and Aguirre, J. (1996) Curr. Genet. 29 352-359 [PubMed] [Google Scholar]
- 42.Scherer, M., Wei, H., Liese, R., and Fischer, R. (2002) Eukaryot. Cell 1 725-735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Oberegger, H., Zadra, I., Schoeser, M., and Haas, H. (2000) FEBS Lett. 485 113-116 [DOI] [PubMed] [Google Scholar]
- 44.Fratelli, M., Demol, H., Puype, M., Casagrande, S., Eberini, I., Salmona, M., Bonetto, V., Mengozzi, M., Duffieux, F., Miclet, E., Bachi, A., Vandekerckhove, J., Gianazza, E., and Ghezzi, P. (2002) Proc. Natl. Acad. Sci. U. S. A. 99 3505-3510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ganguli D., Kumar, C., and Bachhawat, A. K. (2007) Genetics 175 1137-1151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fraser, J., Davis, M., and Hynes, M. (2002) Appl. Environ. Microbiol. 68 2802-2808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sheehan, D., Meade, G., Foley, V., and Dowd, C. (2001) Biochem. J. 360 1-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hayes, J. D., and Pulford, D. J. (1995) CRC Crit. Rev. Biochem. Mol. Biol. 30 445-600 [DOI] [PubMed] [Google Scholar]
- 49.Garcera, A., Barreto, L., Piedrafita, L., Tamarit, J., and Herrero, E. (2006) Biochem. J. 398 187-196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fernandez, M., and Penalva, A. (1998) J. Biol. Chem. 273 329-337 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.