

Abstract
Angiogenesis is regulated by integrin-dependent cell adhesion and the activation of specific cell surface receptors on vascular endothelial cells by angiogenic factors. LPA and S1P are bioactive lysophospholipids which activate G-protein-coupled receptors that stimulate PI3-kinase, Ras, and Rho effector pathways involved in vascular cell survival, proliferation, adhesion, and migration. Previous studies have shown that anastellin, a fragment of the first type III module of fibronectin, functions as an anti-angiogenic peptide suppressing tumor growth and metastasis. We have previously shown that anastellin blocks serum-dependent proliferation of microvessel endothelial cells (MVEC) by affecting ERK-dependent G1/S transition (1). However, the mechanism by which anastellin regulates endothelial cell function remains unclear. In the present study, we mapped several lysophospholipid-mediated signaling pathways in MVEC and examined the effects of anastellin on LPA- and S1P-induced MVEC proliferation, migration and cytoskeletal organization. Both LPA and S1P activated PI3-kinase, Ras/ERK and Rho/Rho kinase pathways leading to migration, G1/S cell cycle progression and stress fiber formation, respectively. Stimulation of proliferation by LPA/S1P occurred through a Gi-dependent Ras/ERK pathway which was independent of growth factor receptors, PI3-kinase and Rho/Rho kinase signaling. Although LPA and S1P activated both PI3-kinase/Akt and Ras/ERK signaling through Gi, anastellin inhibited only the Ras/ERK pathway. Stress fiber formation in response to LPA was dependent on Rho/Rho kinase but independent of Gi and unaffected by anastellin. These results suggest that lysophospholipid mediators of Gi activation leads to PI3-kinase/Akt and Ras/ERK signaling bifurcate downstream of Gi and that anastellin selectively inhibits the Ras/ERK arm of the pathway.
INTRODUCTION
Angiogenesis is controlled by a complex series of coordinated signaling events that are regulated by integrin-dependent cell adhesion and the activation of specific cell surface receptors on vascular endothelial cells by angiogenic factors. The angiogenic response has both normal and pathological roles including tissue repair and regeneration during wound healing and growth of primary and metastatic tumors. Integrin receptor ligation to an extracellular fibronectin matrix has long been recognized to play a critical role in the regulation of endothelial cell adhesion, migration, proliferation, and survival [reviewed in (2)]. Lysophosphatidic acid (LPA) and sphingosine-1 phosphate (S1P) are membrane-derived bioactive lysophospholipids generated from phospholipid precursors of activated platelets, epithelial cells, macrophages, and some cancer cells with reported serum concentrations of 1 –10 µM and 0.2–0.5µM, respectively (3). LPA and S1P activate a variety of widely expressed G-protein-coupled receptors of the endothelial differentiation gene (Edg) family that regulate a broad range of cellular functions including survival, proliferation, adhesion, migration and chemotaxis suggesting potential roles in inflammation, wound healing and tumor progression (4). LPA and S1P receptors couple to at least three distinct G-protein subfamilies including G12/13, Gq/11, and Gi. Effects of LPA and S1P on cell survival and proliferation have been linked to Gi-dependent activation of PI3-kinase and Ras effector pathways, while activation of the Rho/Rho kinase (ROCK) pathway, implicated in the regulation of cell morphology, adhesion, and migration, has been linked to activation of G12/13-coupled Edg receptors (5–9).
LPA is produced in vivo through the action of autotaxin (ATX), an exoenzyme which functions in serum to convert lysophosphatidylcholine into bioactive LPA {2420}. Studies using ATX-deficient mice indicate that ATX is a major regulator of plasma LPA levels. Autotaxin-deficient mice exhibit impaired vessel formation suggesting that LPA production is essential for normal vascular development {2396, 2419}. LPA regulates the barrier function of the endothelium and also stimulates endothelial cell migration and proliferation [reviewed in (13)]. S1P is a proangiogenic factor which regulates endothelial cell proliferation and migration, tubulogenesis and the homing of bone marrow-derived endothelial cell precursors to sites of neovascularization [reviewed in {2390}]. Mice in which S1P receptors have been genetically disrupted exhibit vascular abnormalities indicating a role for S1P in maturation of the vascular system {2393}. In addition, antagonists of S1P and S1P receptors inhibit angiogenesis and tumor progression in mice, confirming a role for S1P in angiogenesis and suggesting that S1P is an important therapeutic target for the treatment of cancer {2394, 2391}.
Previous studies have shown that anastellin, a C-terminal fragment of the first type III homology repeat of fibronectin (III1C), functions as an anti-angiogenic peptide to suppress tumor growth and metastasis in mouse models of human cancer (18, 19). More recently, we have shown that anastellin blocks serum-dependent proliferation of microvessel endothelial cells by modulating extracellular signal-regulated mitogen-activated protein kinase (ERK)-dependent expression of cell cycle regulatory proteins and transition into S-phase (1). However, the mechanism by which anastellin regulates endothelial cell function remains unclear. Although the biological effects of LPA/S1P have been actively investigated over the past several years, surprisingly little is known about the signaling pathways regulated by these molecules in endothelial microvessel cells. In the present study, we mapped several lysophospholipid-mediated signal transduction pathways in human dermal microvessel endothelial cells and examined the effects of anastellin on LPA- and S1P-activated signal transduction pathways. Both LPA and S1P activated PI3-kinase/Akt, Ras/ERK and Rho/ROCK signaling pathways leading to increases in cell proliferation, migration and stress fiber formation. Endothelial cell transition into S-phase by LPA/S1P occurred through a Gi-dependent activation of ERK which was independent of growth factor receptor, PI3-kinase/Akt and Rho/ROCK signaling pathways. Although LPA and S1P activated both PI3 kinase/Akt and Ras/ERK pathways through Gi, anastellin specifically inhibited only the Ras/ERK pathway. These results suggest that the Gi effector pathways leading to activation of PI3k/Akt and ERK bifurcate downstream of Gi and that anastellin selectively inhibits the Ras/MEK/ERK arm of the pathway.
RESULTS
LPA and S1P Stimulate ERK-Dependent [3H]-Thymidine Uptake by Microvessel Cells
To examine the ability of LPA and S1P to stimulate proliferation of microvessel endothelial cells, serum-starved MVECs were treated with increasing concentrations of either LPA or S1P and assayed for incorporation of [3H]-thymidine. As shown in Fig. 1, both LPA and S1P treatment resulted in a 3–4 fold stimulation of [3H]-thymidine incorporation. The induction of cell proliferation by LPA and S1P was accompanied by an increase in the activation of ERK (Supplementary Fig. S1). ERK activation was observed in as little as 2.5 min and reached maximal stimulation within 5–10 min of lysophospholipid addition (Supplementary Fig. S1A,B). ERK phosphorylation decreased within 30 min and was nearly undetectable 60 min following addition. LPA activation of ERK was dose-dependent between 2.5 and 40 µM (Supplementary Fig. S1C). Maximum activation of ERK with S1P was seen at 0.5 µM (Supplementary Fig. S1D). The increases in ERK phosphorylation correlated well with the changes in thymidine incorporation observed in Fig. 1, suggesting that ERK activation plays a role in lysophospholipid-mediated endothelial transition into S-phase.
FIGURE 1. LPA and S1P stimulate ERK-dependent proliferation of microvessel endothelial cells.
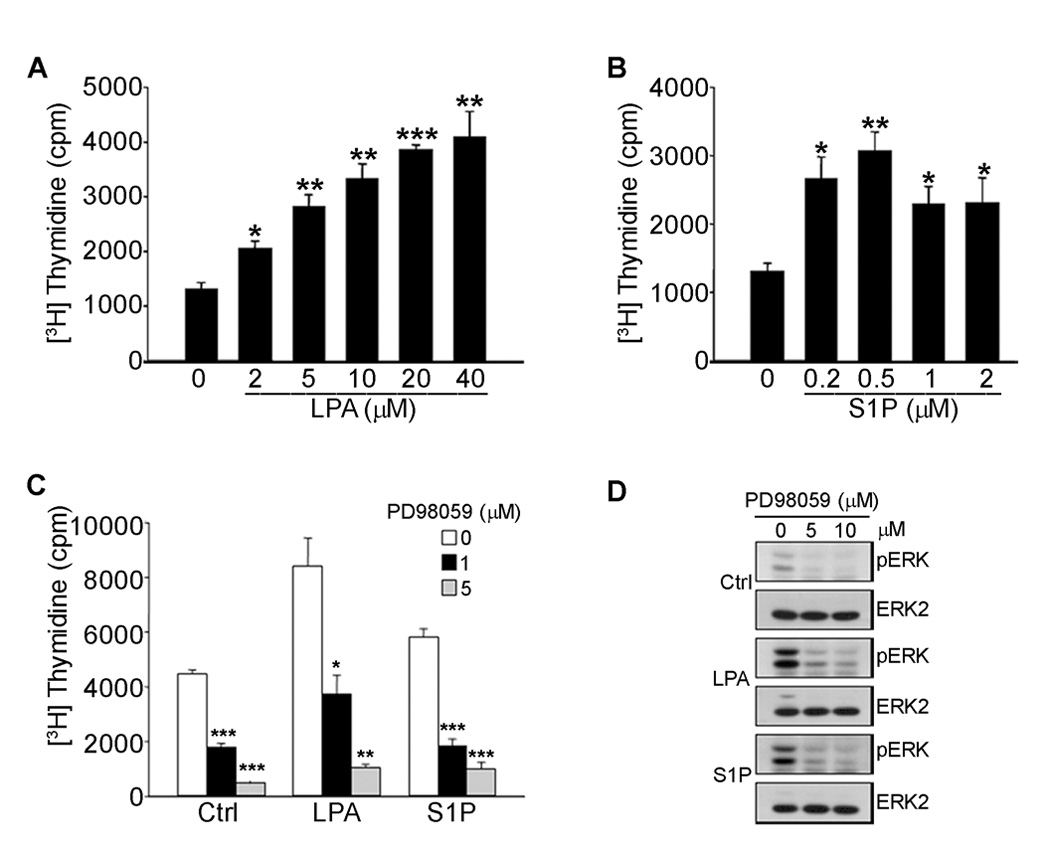
Serum starved human dermal microvessel endothelial cells were treated with LPA (A) or S1P (B) as indicated for 16 h and assayed for incorporation of [3H]-thymidine as described under Materials and Methods. In panel C, serum starved MVECs were treated with the MEK inhibitor PD98059 as indicated for 30 min prior to the addition of either 20 µM LPA or 1 µM S1P and assayed for [3H]-Thymidine incorporation. Alternatively, serum starved MVECs treated with PD98059 as indicted were stimulated with either 20 µM LPA or 1 µM S1P for 6 min (D). Cell lysates were subjected to SDS-PAGE and immunoblotted for changes in phospho-ERK. Stripped membranes were reprobed with an anti-ERK2 antibody for loading control. Statistical differences were measured by comparison with unstimulated cells. *p<0.05; **p< 0.01; ***p <0.001.
To test whether ERK activation is required for the effects of lysophospholipids on cell growth, MVECs were treated with MEK inhibitors PD98059 or U0126 prior to LPA or S1P stimulation and examined for effects on [3H]-thymidine incorporation. As shown in Fig. 1C and Supplementary Fig. S2A, both MEK inhibitors blocked lysophospholipid-mediated [3H]-thymidine incorporation at concentrations similar to those required to block ERK activation (Fig. 1D and Supplementary Fig. S2B). These data indicate that MEK-dependent ERK activation is required for the S1P/LPA-mediated stimulation of [3H]-thymidine uptake by MVECs.
Lysophospholipid-Mediated G1→S Transition Requires Gi-Dependent ERK Activation
LPA and S1P exert their effects by binding to a diverse group of cell surface G-protein-coupled receptors thereby leading to activation of a variety of downstream signaling events. To evaluate the role of Gi in LPA- and S1P-stimulated ERK activation in MVECs, cells were preincubated with increasing concentrations of pertussis toxin (PTx), a Gi-protein inhibitor. As shown in Supplementary Fig. S3A, as little as 0.1 ng/ml PTx blocked both LPA- and S1P-mediated ERK activation. Control experiments showed that PTx had no effect on ERK activation by either VEGF or EGF (data not shown). Similarly, PTx treatment completely blocked the ability of LPA to stimulate [3H]-thymidine uptake in MVECs indicating that LPA-mediated transition to S-phase occurs through the Gi-dependent activation of the ERK signaling pathway (Supplementary Fig. S3B).
Lysophospholipid-Mediated ERK Activation in MVECs Does Not Require EGF or VEGF Receptor Transactivation
Previous studies have shown that activation of the ERK signaling pathway by LPA can occur through a mechanism which involves transactivation of EGF or PDGF growth factor receptors (20, 21). Similarly, another report has demonstrated that S1P can activate ERK through transactivation of the VEGF receptor (22). To determine if growth factor receptor transactivation is required for LPA or S1P activation of ERK, MVECs were treated with inhibitors of EGFR and VEGFR kinase activity. As observed in Supplementary Fig. S4A, addition of LPA, S1P, and EGF each stimulated ERK activation. However, only EGF-mediated ERK activation was inhibited by the EGFR inhibitor, AG1478, suggesting that LPA and S1P activation of ERK does not involve EGFR kinase activity. This concept was examined further by evaluating the effect of LPA on EGFR phosphorylation. In Supplementary Fig. S4B, MVECs were treated with AG1478 prior to stimulation with either LPA or EGF. While EGF led to a significant increase in AG1478-sensitive EGFR phosphorylation, LPA treatment resulted in no detectable increase in EGFR phosphorylation. Similarly, we examined the role of VEGF receptor transactivation in LPA and S1P mediated ERK activation. MVECs were treated with the VEGF receptor inhibitor SU5416 followed by addition of LPA, S1P, and VEGF. As shown in Supplementary Fig. S4C, addition of each led to a robust increase in ERK phosphorylation. Only VEGF activation of ERK was inhibited by treatment with SU5416. Additionally, MVECs treated with LPA resulted in no detectable increase in VEGF receptor phosphorylation while VEGF was able to stimulate SU5416-inhibitable VEGF receptor Tyr phosphorylation (Supplementary Fig. S4D). Similar results were seen using an inhibitor (AG1295) of PDGF receptor kinases which had no effect on activation of ERK by either LPA or S1P (data not shown). Taken together, these data indicate that activation of ERK by LPA or S1P does not require transactivation of these growth factor receptor tyrosine kinases.
Lysophospholipid-Mediated Akt but not ERK Activation depends on PI3-Kinase
Earlier studies have demonstrated that lysophospholipids can activate phosphoinositide-3 kinase (PI3K)/Akt survival pathways in endothelial cells (23, 24). To determine if lysophospholipids activate Akt in MVECs, cells treated with LPA and S1P were examined for phospho-Akt by western blot analysis. As shown in Supplementary Fig. S5 (panels A, B), both LPA and S1P enhanced phosphorylation of Akt at Ser473 within 2.5–5 min of treatment. The kinetics of Akt phosphorylation paralleled those of ERK phosphorylation, peaking between 5 and 10 min and returning to basal levels within 60 min. To determine whether Akt phosphorylation depended on PI3K activity, MVECs were treated with the PI3K inhibitor LY294002 for 30 min prior to LPA and S1P stimulation. PI3K inhibition completely blocked the ability of both LPA and S1P to stimulate Akt phosphorylation at Ser473 but had no effect on ERK activation (Supplementary Fig. S5C). These results suggest that lysophospholipid-mediated Akt activation in microvessel cells occurs through a PI3K-dependent signaling mechanism while ERK activation is independent of PI3K activity. To determine which G-protein family member is required for PI3K/Akt activation, cells were treated with PTx (Supplementary Fig. S5D). These data show that PTx blocked both Akt and ERK activation, indicating that S1P/LPA activation of both Akt and ERK occur through a Gi-dependent signaling pathway.
Effects of Rho/ROCK Signaling on Lysophospholipid-Mediated ERK Activation
Previous studies have shown that LPA is a strong inducer of Rho-dependent signaling pathways in many cell types and plays a central role in cell adhesion, migration, and cytoskeletal organization. Incubation of MVECs with either LPA or S1P resulted in an increase in myosin light chain 2 phosphorylation (pMLC2), a downstream effector of Rho/ROCK signaling (Fig. 2A). These data suggest that both lysophospholipids activate Rho signaling in microvessel cells, however, the effects were more pronounced in LPA-treated cells. To test whether LPA signaling to Rho occurred through a Gi-dependent mechanism, MVECs were treated with PTx overnight, stimulated with LPA or S1P, and assessed for changes in MLC2 phosphorylation levels (Fig. 2B). As demonstrated previously, PTx treatment completely blocked LPA and S1P-mediated Akt and ERK activation. However, PTx had no effect on the ability of LPA or S1P to stimulate MLC2 phosphorylation indicating that phospholipid-mediated Rho signaling proceeds through a Gi-independent mechanism (Fig. 2B). To determine the relationship of ERK activation to Rho signaling, MVECs were treated with either the ROCK inhibitor Y27632 or the MEK inhibitor U0126 prior to LPA or S1P stimulation. As shown in Fig. 2C, the ROCK inhibitor completely blocked the ability of LPA or S1P to phosphorylate MLC2, while the PI3K/Akt and MEK/ERK signaling pathways were unaffected. In addition, inhibition of MEK with U0126 had no effect on either the PI3K/Akt or Rho/ROCK signaling pathways. In support of these data, C3 exoenzyme, a Rho inhibitor, had no effect on the ability of LPA to activate ERK (data not shown). These data indicate that the activation of ERK by LPA does not depend on Rho signaling pathways. Furthermore, the data show that ERK activation is not required for lysophospholipid activation of either Akt or Rho/Rho kinase, suggesting that in microvessel cells, the PI3K/Akt, MEK/ERK and Rho/ROCK pathways are not inter-dependent.
FIGURE 2. Lysophospholipid-stimulated ERK activation is independent of Rho signaling.
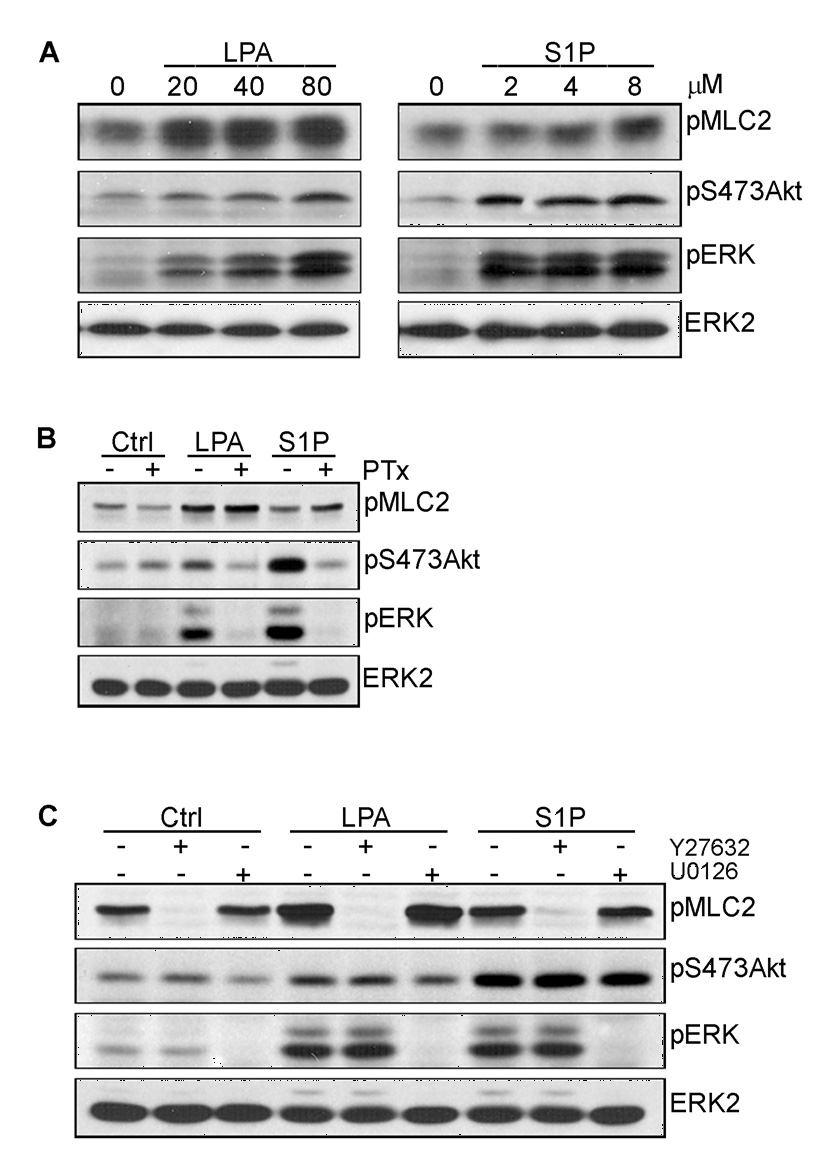
Extracts of MVECs treated with various concentrations of LPA and S1P as indicated for 6 min were subjected to SDS-PAGE followed by immunodetection with antibodies directed against phosphorylated Thr18/Ser19 of myosin light chain 2 (pMLC2), pS473Akt and pERK (A). Alternatively, cells were treated with 1 ng/ml PTx for 20 h (B), 2 µM Y27632, a specific inhibitor of Rho-associated kinase (ROCK) or 5 µM U0126 for 30 min (C) prior to stimulation with 40 µM LPA or 4 µM S1P for 6 min. Cell extracts were subjected to SDS-PAGE and immunoblots probed as described above. Membranes were stripped and reprobed with an anti-ERK2 antibody.
Anastellin (III1c) Works Upstream of Ras to Block Lysophospholipid-Mediated ERK Activation and Transition to S-Phase
To determine whether anastellin can alter lysophospholipid receptor signaling pathways, MVECs were treated with various concentrations of anastellin prior to LPA and S1P stimulation. As shown in Fig. 3A, anastellin reduced basal levels of phosphorylated ERK and blocked activation of ERK by LPA and S1P. The effects of anastellin on ERK activation were dose-dependent and seen with as little as 5 µM and reaching maximal inhibition at 20 µM anastellin. The control fibronectin module (III13) had no effect on LPA or S1P activation of ERK. The effect of anastellin on ERK activation was seen within 10 min with nearly a complete loss of active ERK at 60 min (Fig. 3B). As shown in Fig. 3C, anastellin blocked LPA or S1P mediated [3H]-thymidine uptake at concentrations similar to those required to block MAP kinase signaling.
FIGURE 3. Anastellin inhibits lysophospholipid-mediated ERK activation and proliferation in MVECs.

Serum starved cells were treated with increasing concentrations of anastellin for 60 min prior to stimulation with 20 µM LPA or 1 µM S1P for 6 min (A). In panel B, cells were treated with 20 µM anastellin for up to 60 min prior to the addition of 20 µM LPA or 1 µM S1P, lysed, and proteins immunoblotted for changes in phospho-ERK. Membranes were stripped and reprobed with an anti-ERK2 antibody. An unrelated type III module of FN (III13) was used as negative control (A). To evaluate the effect of anastellin on lysophospholipid-mediated endothelial cell proliferation, serum starved MVECs were treated with anastellin for 60 min prior to 20 µM LPA and 1 µM S1P stimulation and assessed for [3H]-thymidine incorporation (C) as described in Materials and Methods. Statistical differences were measured by comparison with cells in the absence of III1C. *p<0.05; **p<0.01; ***p<0.001.
To demonstrate a role for Ras in the activation of ERK by LPA/S1P, Ras activity was inhibited using an adenoviral construct of dominant-negative RasN17. Fig. 4A shows that ERK activation in response to either LPA or S1P was completely attenuated in cells expressing RasN17. In contrast, Akt activation by LPA or S1P was unaffected by RasN17 expression, suggesting that Akt activation was not dependent on Ras. EGF-stimulated ERK activation was also completely attenuated in RasN17 infected cells (data not shown). In contrast, there was no effect on either Akt or ERK activation in response to LPA or S1P stimulation in cells infected with control virus (adenovirus expressing GFP alone).
FIGURE 4. Anastellin inhibits lysophospholipid signaling to ERK upstream of Ras.

Adherent microvessel endothelial cells were infected with adenoviral constructs of GFP or dominant negative Ras (RasN17) under serum-free conditions for 24 h. Infected cells were then treated with 20 µM LPA or 1 µM S1P for 6 min. Cell lysates were prepared and immunoblotted with phospho-specific antibodies to Akt and ERK. Antibodies directing against the hemaglutinin epitope (HA) were used to detect adenoviral expression of HA-tagged RasN17. Membranes were stripped and reprobed with an anti-ERK2 antibody (A). In panel B, the effect of anastellin on Ras activation by LPA, S1P and EGF was examined by active Ras ELISA as described under Materials and Methods. Statistical differences were measured by comparison with the untreated control cells (a) or with stimulated cells in the absence of III1C. *p<0.05; **p<0.01; ***p<0.001.
To determine whether anastellin alters lysophospholipid-mediated Ras activation, anastellin-treated MVECs were stimulated with LPA and S1P and examined for changes in active Ras using a Ras-GTPase ELISA assay. EGF served as a positive control. LPA induced a significant increase in Ras activation, however S1P did not increase Ras activity when compared to control cells (Fig. 4B). As shown in Fig. 4B, anastellin blocked the ability of LPA to induce Ras activation. Conversely, anastellin had no significant effect on EGF-stimulated Ras activation which was consistent with control experiments showing that anastellin did not inhibit the activation of ERK by EGF (data not shown). These data indicate that the activation of ERK by LPA/S1P is dependent on Ras activity and that the inhibitory effect of anastellin on ERK lies upstream of Ras.
Effects of Anastellin on Rho/ROCK Signaling and Cytoskeletal Organization
To determine the effects of anastellin on lysophospholipid signaling through Rho, MVECs were treated with 20 µM anastellin or the control module III13 prior to stimulation with LPA and S1P. Cell lysates were immunoblotted for anastellin-mediated effects on LPA-induced phosphorylation of ERK, Akt, and myosin light chain-2 (MLC2). Although anastellin treatment effectively blocked ERK activation, anastellin had no effect on S1P or LPA-mediated phosphorylation of either Akt or MLC2 (Fig. 5A). These results suggest that anastellin does not interfere with the ability of S1P or LPA to initiate downstream signaling to both Rho/ROCK and PI3K/Akt signaling pathways. Rho pull-down assays confirmed that anastellin had no effect on the ability of LPA to activate Rho (data not shown).
FIGURE 5. Effect of anastellin on lysophospholipid-mediated Rho activation.

Serum starved MVECs were treated with 20 µM anastellin or 20 µM III13 as control for 60 min prior to stimulation with either 40 µM LPA or 1 µM S1P for 6 min. Cells were lysed and extracts subjected to SDS-PAGE followed by immunodetection with antibodies directed against pMLC2, pS473Akt, and pERK (A). Membranes were stripped and reprobed with an anti-ERK2 antibody. In panel B, serum starved endothelial cells were treated with 2 µM Y27632 (30 min) or 20 µM anastellin (60 min) prior to 1 h stimulation with 20 µM LPA. Cells were fixed and immuno-stained with phospho-specific antibodies to MLC2 (green). F-actin was visualized with Alexa Fluor 594-conjugated phalloidin (red). LPA-induced formation of cortical stress fibers (arrows) and regions of pMLC2 colocalization (arrow heads) are indicated.
We also examined the effects of LPA and S1P on the formation of stress fibers in confluent monolayers of endothelial cells (Fig. 5B). LPA stimulation dramatically induced the polymerization of actin into cortical stress fibers when compared to control cells (arrows, Fig 5B) and enhanced immunodetection of pMLC2, consistent with the biochemical data presented in Fig. 2 and Fig. 5A. The increase in pMLC2 was predominantly associated with its co-localization to the newly organized cortical stress fibers following LPA stimulation (arrow heads and merge, Fig 5B). S1P had little effect on stress fiber formation (data not shown) consistent with earlier results showing that S1P only weakly activated Rho/ROCK signaling (Fig. 2). Inhibition of Rho kinase with Y27632 dramatically reduced immunodetection of pMLC2 and blocked the formation of cortical stress fibers following LPA stimulation. Moreover, anastellin had no effect on the ability of LPA to induce stress fiber formation or MLC2 phosphorylation (Fig. 5B). These results are consistent with the data presented in Fig. 2C and Fig. 5A and indicate that LPA activation of Rho/ROCK signaling leading to increased stress fiber formation is not affected by anastellin.
PI3-kinase Signaling Plays a Role in Lysophospholipid-mediated Endothelial Cell Migration
To determine whether lysophospholipid signaling through Ras/ERK, PI3K/Akt, or Rho/ROCK play any role in cell migration, endothelial cells were seeded onto collagen-coated transwell tissue culture inserts and stimulated with LPA or S1P. As shown in Fig 6A, both lipids increased endothelial cell migration, although S1P was a more potent stimulator than LPA. Migration was pertussis toxin-sensitive suggesting that Gi activation is required (Fig. 6B). Cells pretreated with the ROCK inhibitor Y27632 or the MEK inhibitor U0126, prior to stimulation with LPA or S1P, exhibited no significant changes in migration (Fig. 6C, D, respectively), indicating that neither the Rho pathway nor the ERK pathway was required for LPA/S1P induced migration. Conversely, LY294002 significantly reduced migration suggesting a role for PI3-kinase signaling in endothelial cell migration (Fig. 6E). Similar results were obtained with the PI3K inhibitor wortmanin (data not shown). To determine whether PI3K-mediated Akt activation was specifically required for migration, cells were treated with an Akt-selective inhibitor prior to stimulation with LPA and S1P. Although lysophospholipid-dependent phosphorylation of Akt was completely blocked (data not shown), migration was unaffected (Fig 6E), suggesting that Gi-dependent activation of PI3-kinase is required for lysophospholipid-mediated endothelial cell migration while subsequent activation of Akt by PI3-kinase is not. Anastellin had no significant effect on migration in response to LPA/S1P (Fig. 6F).
FIGURE 6. Lysophospholipid-mediated endothelial cell migration is PI3-kinase-dependent.

Microvessel endothelial cells were seeded onto collagen-coated tissue culture inserts and stimulated with LPA or S1P as indicated (A). Alternatively, cells were treated with either 25 ng/ml PTx (B), 2 µM Y27632 (C), 5 µM U0126 (D), 10 μM LY294002 or 10 µM AktI-1/2 (E), or 20 µM anastellin (F) for 30–60 min in suspension prior to seeding and stimulation with 20 µM LPA or 1 µM S1P. After 4 h, migratory cells were fixed and stained with Hoechst dye, visualized with a digital fluorescent microscope, and counted. The number of migratory cells/10x field obtained from 2–5 independent experiments carried out in triplicate were normalized to untreated control cells. Statistical differences were measured by comparison with cells in the absence of inhibitor. *p<0.05; **p<0.01; ***p<0.001.
As shown in the schematic (Fig. 7), our results indicate that LPA/S1P activates both PI3K/Akt and ERK signaling through Gi-dependent pathways. Activation of Akt but not ERK required PI3K, suggesting that the ERK and Akt signaling pathways are independent. This was confirmed by studies showing that inhibitors of MEK and Ras blocked ERK phosphorylation but did not block Akt phosphorylation. Activation of PI3-kinase led to an increase in cell migration, while ERK activation stimulated cell cycle progression. Anastellin had no effect on cell migration, but completely inhibited cell proliferation. LPA and S1P (but to a lesser extent) activated the Rho signaling pathway resulting in the phosphorylation of MLC2 and formation of stress fibers. This pathway was not dependent on Gi and did not impact on either the activation of ERK or Akt. Activation of Rho by LPA resulted in an increase in stress fibers which was unaffected by anastellin. These results suggest that the LPA/S1P signaling pathway initiated through Gi bifurcates downstream of Gi and that anastellin specifically inhibits the Ras/ERK arm of the pathway. The data further indicate that the site of anastellin action lies upstream of Ras but downstream of Gi.
FIGURE 7. Lysophospholipid-mediated signaling pathways in MVECs.

LPA and S1P activate specific G-protein-coupled receptors (LPA1–4 and S1P1–5, respectively) and initiate Ras/ERK, PI3K/Akt, and Rho/ROCK signaling pathways in microvessel endothelial cells. Results presented here have demonstrated that both Ras/ERK and PI3K/Akt signaling is mediated by a Gi-dependent pathway while Rho signaling occurs through an alternate G-protein, possibly G12/13 or Gq as has been shown in previous studies (5–9). In addition, Gi-dependent activation of Ras/ERK and PI3K/Akt occur through independent pathways and are unaffected by inhibition of Rho signaling. These results are further supported by the finding that anastellin suppresses Ras activation by LPA and prevents ERK activation without affecting PI3K/Akt or Rho/ROCK signaling. The dashed line linking S1P receptors to G12/13 indicates that a weak induction by S1P was observed in MVECs.
DISCUSSION
Anastellin, a peptide derived from the first Type III domain of fibronectin has been shown to exhibit anti-angiogenic properties in vivo and to block serum dependent growth of endothelial cells in vitro (1, 19, 25). Several studies have documented multiple cellular effects of anastellin, including changes in cell signaling pathways, reorganization of actin filaments and conformational changes in extracellular fibronectin fibrils (26–28). In the present study, we address the mechanism by which anastellin might regulate the angiogenic phenotype. We show that anastellin inhibits signaling pathways initiated by both LPA and S1P, two bioactive phospholipids which have been implicated in the regulation of vascular development and angiogenesis (29, 30). Our studies indicate that in human microvessel endothelial cells, LPA and S1P regulate similar parallel pathways leading to the activation of ERK, Akt and Rho kinase and that anastellin is a selective inhibitor of the pathway leading to ERK activation.
Both S1P and LPA bind to members of the Edg family of G-protein coupled receptors to affect several aspects of endothelial cell biology including proliferation, migration, survival and barrier function [reviewed in (13)]. Our data indicate that LPA and S1P can stimulate ERK-dependent [3H]-thymidine incorporation in endothelial microvessel cells. Activation of ERK by lysophospholipids is Ras-dependent and required for the movement of cells from G1 into S-phase. In several cell types, activation of ERK by LPA or S1P has been linked to transactivation of growth factor receptors or to Rho signaling (22, 31–33). We found no evidence to support a role for either growth factor receptors or Rho in the activation of ERK by LPA/S1P in human microvessel cells.
In agreement with this, anastellin had no effect on LPA induced stress fiber formation which was dependent on Rho-kinase activity. LPA and S1P both stimulated cell migration which was dependent on PI3-kinase. Although both LPA and S1P stimulated phosphorylation on Akt by PI3-kinase, Akt was not required for cell migration. This suggests that PI3-kinase pathway bifurcates downstream into separate pathways regulating migration and other Akt-dependent pathways such as cell survival. Anastellin had no effect on cell migration or Akt activation suggesting that the antiangiogenic activity of anastellin does not result from effect of anastellin on migration or survival.
Activation of both ERK and Akt by LPA/S1P was inhibited by pertussis toxin, indicating a role for Gi in the LPA/S1P signaling pathways. Although both ERK and Akt lie down stream of Gi, anastellin inhibits only the activation of ERK, suggesting that anastellin acts on one arm of a signaling pathway which bifurcates downstream of Gi. These data suggest that anastellin selectively inhibits only those biological effects which lie downstream of ERK, such as cell proliferation, without affecting other lysophospholipid regulated activities such as migration, survival, endothelial NO synthase (eNOS) activity, or endothelial barrier function which lie downstream of PI3-kinase, Akt or Rho kinase (34–37).
Several studies have shown fragments of extracellular matrix molecules to be fairly potent regulators of angiogenesis. These endogenous angiogenic regulators may be presented to the cell as either matrix derived fragments released through proteolysis or as unfolded cryptic domains which become exposed following changes in the organization of the extracellular matrix (38, 39). Steered molecular dynamics and NMR have shown that the fibronectin III-1 domain unfolds in response to cellular tension to a mechanically stable, intermediate which is similar to the structure of anastellin (40). Therefore, anastellin may regulate endothelial cell behavior by reproducing the biological effects of conformationally-regulated sequences within matrix fibronectin. Alternatively, anastellin may mimic the effects of soluble peptides containing the III-1 domain of fibronectin. Truncated forms of fibronectin which contain the III-1 domain are synthesized by both tumors and tumor-associated stromal cells (41).
The molecular basis for the anti-angiogenic action of anastellin is not well understood. One possibility is that anastellin competes for cellular receptors which interact with the III-1 domain present in matrix fibronectin. Earlier studies have shown that β1 integrin and proteoglycans mediate adhesion of smooth muscle cells to substrate-attached anastellin which results in the activation of ERK (27). Additionally, our previous studies have shown that anastellin binds to matrix fibronectin causing conformational changes in the EDA domain (28) suggesting that anastellin may affect the association of α4β1 integrins with the EDA-containing fibronectin present in matrix (42, 43). Anastellin induced changes in fibronectin matrix organization may also regulate VEGF activity as recent studies have shown that VEGF binds to conformationally labile sites present in the fibronectin matrix (44, 45). In addition to its effects on angiogenesis, anastellin might exert anti-tumor effects by regulating lysophospholipid signaling in the tumor cell. LPA autocrine loops have been described in several tumor types (46–48). LPA present in ascites fluid contributes to the progression of both ovarian and pancreatic cancers (49, 50). It is also possible that anastellin may have direct effects on other cell types present in the tumor microenvironment, as earlier studies have shown that anastellin regulates p38 MAP kinase and Cdc42 activity in dermal fibroblasts (26, 28)
Anastellin triggers polymerization of soluble fibronectin into higher order polymers termed "superfibronectin" (51). Superfibronectin exhibits distinct effects on cell behavior and appears to be important in mediating the anti-angiogenic effects of anastellin (52). Numerous studies have indicated neosynthesis of fibronectin isoforms during tumor progression and these isoforms are currently being evaluated as targets for tumor therapy [(reviewed in (53)]. Angiogenesis associated with solid tumor progression is marked by the presence of alternatively spliced forms of fibronectin which contain extra Type III modules, EDA and EDB (54–56) and in vivo studies have shown the fibronectin matrix to be a critical component of angiogenic programs (57–60). The specific functions of these isoforms are not well understood, but recent studies have shown that deletion of the EDA/EDB modules from fibronectin results in embryonic lethality due to multiple cardiovascular defects which include defects in vascular remodeling and angiogenesis (61). These findings provide compelling evidence that the EDA/EDB isoforms of fibronectin are necessary for appropriate vascular remodeling and suggest that the polymerizing fibronectin matrix may be a useful target for therapeutic modalities directed at controlling tumor progression.
MATERIALS and METHODS
Reagents
Unless indicated otherwise, reagents were obtained from Sigma-Aldrich Co. (St. Louis, MO). Fetal bovine serum (FBS) was from Hyclone (Logan, UT). LPA and tyrphostin AG1478 were obtained from Biomol Research Laboratories. Rabbit polyclonal antibodies to ERK2 and Flk-1 (VEGFR-2) were obtained from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Monoclonal antibody to phospho-ERK (pERK), phospho-Ser19 myosin light chain 2 (pMLC2), and the MEK inhibitors U0126 and PD98059 were obtained from Cell Signaling Technology (Beverly, MA). Monoclonal antibody to phosphotyrosine (clone 4G10) was obtained from Upstate Biotechnology. Alexa Fluor 594 phalloidin and monoclonal antibody to phospho-Ser473Akt were obtained from Invitrogen. The Akt-selective inhibitor VIII (AktI-1/2), pertussis toxin (PTx), SU5416, LY294002, and Y27632 were obtained from Calbiochem. Vitrogen-100 (type I collagen) was from Cohesion Technologies (Palo Alto, CA). Recombinant human VEGF and EGF were obtained from R&D Systems. Recombinant fragments of human fibronectin, III1C (anastellin) and III13, were generated and purified as previously described (28). Adenovirus expressing dominant-negative RasN17 was a generous gift of Kevin Pumiglia (Albany Medical College). The GFP adenovirus used as control was obtained from Q-Biogene (Carlsbad, CA).
Cell Culture
Human dermal microvessel endothelial cells (MVECs) were obtained from VEC Technologies Inc. (Rensselaer, NY). Endothelial cells were cultured in complete medium [MCDB-131, 20% defined FBS, 2 mM GlutaMAX (Gibco) and EGM-2MV (Cambrex Corp, East Rutherford, NJ)] supplemented with 10 µg/ml heparin and cultured in a humidified incubator at 37°C/5% CO2 on collagen-coated (20 µg/ml) tissue culture dishes. For most experiments, endothelial cells were plated onto collagen-coated dishes in complete medium at a density of 8 × 104 cells/well (12-well tissue culture dish), cultured overnight, and serum-starved in MCDB-131, 0.5% BSA for 24 h prior to treatment.
Immunoblot and Expression Analysis
Cell layers were washed twice with ice-cold PBS containing 1 mM Na3VO4 before solubilization in RIPA buffer [20 mM Tris-Cl, pH 7.4, 1% Triton X-100, 0.5% Nonidet P-40, 0.1 M NaCl, 40 mM NaF, 30 mM Na4P2O7, 2 mM EGTA, 1 mM Na3VO4, and 0.5 mM PMSF containing Complete Mini protease inhibitors (Roche Biochemical, Indianapolis, IN)]. Cell lysates were cleared by centrifugation (14,000 rpm for 10 min at 4°C) and stored at −80°C until use. Protein concentrations were determined with a BCA protein assay reagent kit (Pierce, Rockford, IL) using BSA as standard. In some experiments, whole cell lysates were prepared. Briefly, cell layers were first washed three times with ice-cold PBS containing 1 mM Na3VO4 followed by solubilization in Laemmli buffer. Samples were separated on SDS polyacrylamide gels, transferred onto nitrocellulose membranes (Schleicher and Schuell Bioscience, Keene, NH), blocked overnight in TBST (Tris-Cl, pH 7.4, 150 mM NaCl, 0.1% Tween-20) containing 5% (w/v) BSA, and hybridized with primary antibodies. Bound antibodies were hybridized with horseradish peroxidase-conjugated secondary antibodies and detected using an enhanced chemiluminescence reagent (Amersham Biosciences; Piscataway, NJ). Blots were reprobed after stripping in 62.5 mM Tris-Cl, pH 6.7 and 2% SDS containing 10 mM β-mercaptoethanol for 30 min at 60°C.
Measurement of DNA Synthesis
MVECs were plated onto collagen-coated 24-well dishes at 1.25 × 104 cells per well in the presence of complete medium and allowed to adhere overnight. Cells were serum-starved for 30 h, treated with various pharmacological agents as indicated prior to stimulation with either 20 µM LPA or 2 µM S1P for an additional 16 h. S-phase nuclei were labeled by incubating cells with 1 µCi of [3H]-thymidine for 6 h. Cells were treated with 10% TCA and recovered in 1N NaOH. Samples were neutralized with 1N HCl and transferred to Ecoscint A (National Diagnostics, Atlanta, GA) scintillation fluid. Incorporation of [3H]-thymidine was determined by liquid scintillation.
Ras Activation Assays
MVECs were plated onto collagen-coated 10 cm dishes (Ras pull down) or 60 mm dishes (Ras activity) at a density of 1 × 104 cells/cm2 in complete media and allowed to adhere overnight. Cells were then serum-starved for 20 h prior to addition of 40 mM LPA, 4 mM S1P, or 10 ng/ml EGF for 3 min. Precipitation of activated Ras (Ras-GTP) was carried out using EZ-Detect Ras Activation Kit (Pierce) according to manufacturers protocol. Alternatively, active Ras was measured using a Ras GTPase Chemiluminescent ELISA Kit (Active Motif, Carlsbad, CA).
Fluorescence Microscopy
Microvessel endothelial cells were cultured overnight on collagen-coated (20 µg/mL) glass cover slips, serum-starved for 4h, (2% FBS, MCDB-131) and treated 30 min with 2 µM Y27632 or 60 min with 20 µM anastellin prior to 1h stimulation with 20 µM LPA or 1 µM S1P. Cells were fixed for 20 min in 3% paraformaldehyde, permeabilized in 0.5% Triton X-100 for 10 min, blocked in 1% BSA, and immunostained with polyclonal antibodies to pMLC2. F-actin was visualized with Alexa Fluor 594-conjugated phalloidin. Cell layers were examined using an Olympus BMX-60 microscope equipped with a cooled CCD sensi-camera (Cooke, Auburn Hills, MI) and images acquired using Slidebook software (Intelligent Imaging Innovation, Denver, CO).
Cell Migration
Transwell tissue culture inserts (Costar, Corning, NY; 6.5mm diameter, 8.0 µm pore size) were coated with 20 µg/ml collagen for 1 h at 37°C, rinsed once, and blocked with 1% BSA. Microvessel cells were trypsinized, washed once and suspended in MCDB-131 containing 0.2% fatty acid-free BSA, treated with inhibitors for 30–60 min at 37°C and seeded at 5 × 105 cells per insert. Tissue culture inserts were then placed into wells containing 20 µM LPA or 1 µM S1P with inhibitor concentrations matching that of the inner well. After 4 h, cells were fixed in 3.7% paraformaldehyde, PBS for 15 min and stained with 0.5% crystal violet. Non-migratory cells attached to the top surface were removed with a cotton tip applicator. Migratory cells adhered to the bottom surface were permeabilized with 0.1% Triton X-100, PBS for 15 min and stained with Hoechst 33258 (1 µg/ml for 20 min). Cell nuclei were visualized with a fluorescence microscope and three random fields (10× magnification) were collected. The average cell number per field was determined by automated counting using ImageJ imaging software.
Data Analysis
Unless indicated otherwise, results are presented as the mean ± S.E. of at least three independent experiments performed in duplicate or triplicate. Statistical significance was determined by one-way analysis of variance; p values <0.05 were considered significant.
Supplementary Material
Acknowledgments
These studies were supported by CA-69612 from the National Institutes of Health. The authors wish to thank Dr. Kevin Pumiglia (Albany Medical College, Albany, NY) for providing the RasN17 adenoviral construct used in this study and Lin Yu for technical assistance with purification of recombinant fibronectin fragments.
Footnotes
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
REFERENCES
- 1.Ambesi A, Klein RM, Pumiglia KM, McKeown-Longo PJ. Anastellin, a fragment of the first type III repeat of fibronectin, inhibits extracellular signal-regulated kinase and causes G1 arrest in human microvessel endothelial cells. Cancer Res. 2005;65:148–156. [PubMed] [Google Scholar]
- 2.Hynes RO. Cell matrix adhesion in vascular development. J Thromb Haemost. 2007;5 Suppl 1:32–40. doi: 10.1111/j.1538-7836.2007.02569.x. [DOI] [PubMed] [Google Scholar]
- 3.Goetzl EJ, An S. Diversity of cellular receptors and functions for the lysophospholipid growth factors lysophosphatidic acid and sphingosine 1-phosphate. FASEB J. 1998;12:1589–1598. [PubMed] [Google Scholar]
- 4.Xu Y, Shen Z, Wiper DW, Wu M, Morton RE, Elson P, Kennedy AW, Belinson J, Markman M, Casey G. Lysophosphatidic acid as a potential biomarker for ovarian and other gynecologic cancers. J Am Med Assoc. 1998;280:719–723. doi: 10.1001/jama.280.8.719. [DOI] [PubMed] [Google Scholar]
- 5.Hart JJ, Jiang X, Kozasa T, Roscoe W, Singer WD, Gilman AG, Sternweis PC, Bollag G. Direct stimulation of the guanine nucleotide exchange activity of p115 RhoGEF by Gα13. Science. 1998;280:2112–2114. doi: 10.1126/science.280.5372.2112. [DOI] [PubMed] [Google Scholar]
- 6.Fukuhara S, Murga C, Zohar M, Igishi T, Gutkind JS. A novel PDZ domain containing guanine nucleotide exchange factor links heterotrimeric G proteins to Rho. J Biol Chem. 1999;274:5868–5879. doi: 10.1074/jbc.274.9.5868. [DOI] [PubMed] [Google Scholar]
- 7.Booden MA, Siderovski DP, Der CJ. Leukemia-associated Rho guanine nucleotide exchange factor promotes Gαq-coupled activation of RhoA. Mol Cell Biol. 2002;22:733–738. doi: 10.1128/MCB.22.12.4053-4061.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Suzuki N, Nakamura S, Mano H, Kozasa T. Gα12 activates Rho FTPase through tyrosine-phosphorylated leukemia-associated RhoGEF. Proc Natl Acad Sci USA. 2003;100:733–738. doi: 10.1073/pnas.0234057100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dutt P, Nguyen N, Toksoz D. Role of Lbc RhoGEF in Gα12/13-induced signals to Rho GTPase. Cell Signal. 2004;16:201–209. doi: 10.1016/s0898-6568(03)00132-3. [DOI] [PubMed] [Google Scholar]
- 10.Tokumura A, Majima E, Kariya Y, Tominaga K, Kogure K, Yasuda K, Fukuzawa K. Identification of human plasma lysophospholipase D, a lysophosphatidic acid-producing enzyme, as autotaxin, a multifunctional phosphodiesterase. J Biol Chem. 2002;277:39436–39442. doi: 10.1074/jbc.M205623200. [DOI] [PubMed] [Google Scholar]
- 11.van Meeteren LA, Ruurs P, Stortelers C, Bouman P, van Rooijen MA, Pradere JP, Pettit TR, Wakelam MJO, Saulnier-Blache JS, Mummery CL, Moolenaar WH, Jonkers J. Autotaxin, a secreted lysophospholipase D, is essential for blood vessel formation during development. Mol Cell Biol. 2006;26:5015–5022. doi: 10.1128/MCB.02419-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tanaka M, Okudaira S, Kishi Y, Ohkawa R, Iseki S, Ota M, Noji S, Yatomi Y, Aoki J, Arai H. Autotaxin stabilizes blood vessels and is required for embryonic vasculature by producing lysophosphatidic acid. J Biol Chem. 2006;281:25822–25830. doi: 10.1074/jbc.M605142200. [DOI] [PubMed] [Google Scholar]
- 13.Panetti JS. Differential effects of sphingosine 1-phosphate and lysophosphatidic acid on endothelial cells. Biochim et Biophys Acta. 2002;1582:190–196. doi: 10.1016/s1388-1981(02)00155-5. [DOI] [PubMed] [Google Scholar]
- 14.Sabbadini RA. Targeting sphingosine-1-phosphate for cancer therapy. Brit J Cancer. 2006;95:1131–1135. doi: 10.1038/sj.bjc.6603400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kono M, Mi Y, Liu Y, Sasaki T, Allende ML, Wu YP, Yamashita T, Proia RL. The sphingosine-1-phosphate receptors S1P1, S1P2, and S1P3 function coordinately during embryonic angiogenesis. J Biol Chem. 2004;279:29367–29373. doi: 10.1074/jbc.M403937200. [DOI] [PubMed] [Google Scholar]
- 16.Visentin B, Vekich JA, Sibbald BJ, Cavalli AL, Moreno KM, Matteo RG, Garland WA, Lu Y, Yu S, Hall HS, Kundra V, Mills GB, Sabbadini RA. Validation of an anti-sphingosine-1-phosphate antibody as a potential therapeutic in reducing growth, invasion, and angiogenesis in multiple tumor lineages. Cancer Cell. 2006;9:225–238. doi: 10.1016/j.ccr.2006.02.023. [DOI] [PubMed] [Google Scholar]
- 17.LaMontagne K, Littlewood-Evans A, Schnell C, O'Reilly T, Wyder L, Sanchez T, Probst B, Butler J, Wood A, Liau G, Billy E, Theuer A, Lla T, Wood J. Antagonism ofsphingosine-1-phosphate receptors by FTY720 inhibits angiogenesis and tumor vascularization. Cancer Res. 2006;66:221–231. doi: 10.1158/0008-5472.CAN-05-2001. [DOI] [PubMed] [Google Scholar]
- 18.Pasqualini R, Bourdoulous S, Looivunen E, Woods VL, Jr, Ruoslahti E. A polymeric form of fibronectin has antimetastatic effects against multiple tumor types. Nat Med. 1996;2:1197–1203. doi: 10.1038/nm1196-1197. [DOI] [PubMed] [Google Scholar]
- 19.Yi M, Ruoslahti E. A fibronectin fragment inhibits tumor growth, angiogenesis, and metastasis. Proc Natl Acad Sci. 2001;98:620–624. doi: 10.1073/pnas.98.2.620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Daub H, Wallasch CLA, Herrlich A, Ullrich A. Signal characteristics of G protein-transactivated EGF receptor. EMBO J. 1997;16:7032–7044. doi: 10.1093/emboj/16.23.7032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang L, Cummings R, Zhao Y, Kazlauskas A, Sham JK, Morris A, Georas S, Brindley DN, Natarajan V. Involvement of phospholipase D2 in lysophosphatidate-induced transactivation of platelet-derived growth factor receptor-β in human bronchial epithelial cells. J Biol Chem. 2003;278:39931–39940. doi: 10.1074/jbc.M302896200. [DOI] [PubMed] [Google Scholar]
- 22.Tanimoto T, Jin Z-G, Berk BC. Transactivation of vascular endothelial growth factor(VEGF) receptor Flk-1/KDR is involved in sphingosine 1-phosphate-stimulated phosphorylation of Akt and endothelial nitric-oxide synthase (eNOS) J Biol Chem. 2002;277:42997–43001. doi: 10.1074/jbc.M204764200. [DOI] [PubMed] [Google Scholar]
- 23.Radeff-Huang J, Seasholtz TM, Matteo RG, Brown JH. G protein mediated signaling pathways in lysophospholipid induced cell proliferation and survival. J Cell Biochem. 2004;92:949–966. doi: 10.1002/jcb.20094. [DOI] [PubMed] [Google Scholar]
- 24.Spiegel S, Milstien S. Exogenous and intracellularly generated sphingosine 1-phosphate can regulate cellular processes by divergent pathways. Biochem Soc Trans. 2003;31:1216–1219. doi: 10.1042/bst0311216. [DOI] [PubMed] [Google Scholar]
- 25.Yi M, Sakai T, Fassler R, Ruoslahti E. Antiangiogenic proteins require plasma fibronectin or vitronectin for in vivo activity. Proc Natl Acad Sci USA. 2003;100:11435–11438. doi: 10.1073/pnas.1635112100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bourdoulous S, Orend G, MacKenna DA, Pasqualini R, Ruoslahti E. Fibronectin matrix regulates activation of RHO and CDC42 GTPases and cell cycle progression. J Cell Biol. 1998;143:267–276. doi: 10.1083/jcb.143.1.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mercurius K, Morla A. Cell adhesion and signaling on the fibronectin 1st type III repeat; requisite roles for cell surface proteoglycans and integrins. BMC Cell Biol. 2001;2:18–30. doi: 10.1186/1471-2121-2-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Klein RM, Zheng M, Ambesi A, van de Water L, McKeown-Longo PJ. Stimulation of extracellular matrix remodeling by the first type III repeat in fibronectin. J Cell Sci. 2003;116:4663–4674. doi: 10.1242/jcs.00778. [DOI] [PubMed] [Google Scholar]
- 29.van Meeteren LA, Moolenaar WH. Regulation and biological activities of the autotaxin-LPA axis. Prog Lipid Res. 2007;46:145–160. doi: 10.1016/j.plipres.2007.02.001. [DOI] [PubMed] [Google Scholar]
- 30.Argraves KM, Argraves WS. HDL serves as a S1P signaling platform mediating a multitude of cardiovascular effects. J Lipid Res. 2007;48:2325–2333. doi: 10.1194/jlr.R700011-JLR200. [DOI] [PubMed] [Google Scholar]
- 31.Sukocheva O, Wadham C, Holmes A, Albanese N, Verrier E, Gend F, Bernal A, Derian CK, Ullrich A, Vadas MA, Xia P. Estrogen transactivates EGFR via the sphingosine 1-phosphate receptor Edg-3: the role of sphingosine kinase-1. J Cell Biol. 2006;173:301–310. doi: 10.1083/jcb.200506033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gschwin A, Prenzel N, Ullrich A. Lysophosphatidic acid-induced squamous cell carcinoma cell proliferation and motility involves epidermal growth factor receptor signal transactivation. Cancer Res. 2002;62:6329–6336. [PubMed] [Google Scholar]
- 33.Budnik LT, Brunswig-Spickenheier B, Mukhopadhyay AK. Lysophosphatidic acid signals through mitogen-activated protein kinase-extracellular signal regulated kinase in ovarian theca cells expressing the LPA1/edg2-receptor: Involvement of a nonclassical pathway. Mol Endocrinol. 2003;17:1593–1606. doi: 10.1210/me.2002-0371. [DOI] [PubMed] [Google Scholar]
- 34.Yokomori H, Yoshimura K, Funakoshi S, Nagai T, Fujimaki K, Nomura M, Ishii H, Oda M. Rho modulates hepatic sinusoidal endothelial fenestrae via regulation of the actin cytoskeleton in rat endothelial cells. Lab Invest. 2004;84:857–864. doi: 10.1038/labinvest.3700114. [DOI] [PubMed] [Google Scholar]
- 35.Wojciak-Stothard B, Ridley AJ. Rho GTPases and the regulation of endothelial permeability. Vasc Pharmacol. 2002;39:187–199. doi: 10.1016/s1537-1891(03)00008-9. [DOI] [PubMed] [Google Scholar]
- 36.van Nieuw Amerongen GP, Vermeer MA, van Hinsbergh VW. Role of RhoA and Rho kinase in lysophosphatidic acid-induced endothelial barrier dysfunction. Arterioscler Thromb Vasc Biol. 2000;20:E127–E133. doi: 10.1161/01.atv.20.12.e127. [DOI] [PubMed] [Google Scholar]
- 37.Kou R, Igarashi J, Michel T. Lysophosphatidic acid and receptor-mediated activation of endothelial nitric-oxide synthase. Biochemistry. 2002;41:4982–4988. doi: 10.1021/bi016017r. [DOI] [PubMed] [Google Scholar]
- 38.Wickstrom SA, Alitalo K, Keski-Oja J. Endostatin associates with lipid rafts and induces reorganization of the actin cytoskeleton via down-regulation of RhoA activity. J Biol Chem. 2003;278:37895–37901. doi: 10.1074/jbc.M303569200. [DOI] [PubMed] [Google Scholar]
- 39.Nyberg P, Xie L, Kalluri R. Endogenous inhibitors of angiogenesis. Cancer Res. 2005;65:3967–3979. doi: 10.1158/0008-5472.CAN-04-2427. [DOI] [PubMed] [Google Scholar]
- 40.Gao M, Craig D, Lequin O, Campbell ID, Vogel V, Schulten K. Structure and functional significance of mechanically unfolded fibronectin type III1 intermediates. Proc Natl Acad Sci. 2003;100:14784–14789. doi: 10.1073/pnas.2334390100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schor SL, Ellis IR, Jones S, Baillie R, Seneviratne K, Clausen J, Motegi K, Vojtesek B, Kanova K, Furrie E, Sales MJ, Schor AM, Kay RA. Migration-stimulating factor: a genetically truncated onco-fetal fibronectin isoform expressed by carcinoma and tumor-associated stromal cells. Cancer Res. 2003;63:8827–8836. [PubMed] [Google Scholar]
- 42.Liao Y-F, Gotwals PJ, Koteliansky VE, Sheppard D, van de Water L. The EIIIA segment of fibronectin is a ligand for integrins α9β1 and α4b1 providing a novel mechanism for regulating cell adhesion by alternative splicing. J Biol Chem. 2002;277:14467–14474. doi: 10.1074/jbc.M201100200. [DOI] [PubMed] [Google Scholar]
- 43.Okamura Y, Watari M, Jerud ES, Young DW, Ishizaka ST, Rose J, Chow JC. Strauss JR3. The extra domain A of fibronectin activates Toll-like receptor 4. J Biol Chem. 2001;276:10229–10233. doi: 10.1074/jbc.M100099200. [DOI] [PubMed] [Google Scholar]
- 44.Mitsi M, Hong Z, Costello CE, Nugent MA. Heparin-mediated conformational changes in fibronectin expose vascular endothelial growth factor binding sites. Biochemistry. 2006;45:10319–10328. doi: 10.1021/bi060974p. [DOI] [PubMed] [Google Scholar]
- 45.Wijelath ES, Rahman S, Namekata M, Murray J, Nishimura T, Mostafavi-Pour Z, Patel Y, Suda Y, Humphries MJ, Sobel M. Heparin-II domain of fibronectin is a vascular endothelial growth factor-binding domain: enhancement of VEGF biological activity by a singular growth factor/matrix protein synergism. Circ Res. 2006;99 doi: 10.1161/01.RES.0000246849.17887.66. 863-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kishi Y, Okudaira S, Tanaka M, Hama K, Shida D, Kitayama J, Yamori T, Aoki J, Fujimaki T, Arai H. Autotaxin is overexpressed in glioblastoma multiforme and contributes to cell motility of glioblastoma by converting lysophosphatidylcholine to lysophosphatidic acid. J Biol Chem. 2006;281:17492–17500. doi: 10.1074/jbc.M601803200. [DOI] [PubMed] [Google Scholar]
- 47.Xie Y, Gibbs TC, Mukhin YV, Neier KE. Role for 18:1 lysophosphatidic acid as an autocrine mediator in prostate cancer cells. J Biol Chem. 2002;277:32516–32526. doi: 10.1074/jbc.M203864200. [DOI] [PubMed] [Google Scholar]
- 48.Chen M, O'Connor KL. Integrin α6β4 promotes expression of autotaxin/ENPP2 autocrine motility factor in breast carcinoma cells. Oncogene. 2005;24:5125–5130. doi: 10.1038/sj.onc.1208729. [DOI] [PubMed] [Google Scholar]
- 49.Ren J, Xiao YJ, Singh LS, Zhao X, Xhao Z, Feng L, Rose TM, Prestwich GD, Xu Y. Lysophosphatidic acid is constitutively produced by human peritoneal mesothelial cells and enhances adhesion, migration, and invasion of ovarian cancer cells. Cancer Res. 2006;66:3006–3014. doi: 10.1158/0008-5472.CAN-05-1292. [DOI] [PubMed] [Google Scholar]
- 50.Yamada T, Sato K, Komachi M, Malchinkhuu E, Tobo M, Kimura T, Kuwabara A, Yanagita Y, Ikeya T, Tanahashi Y, Ogawa T, Ohwada S, Morishita Y, Ohta H, Im D-S, Tamoto K, Tomura H, Okajima F. Lysophosphatidic acid (LPA) in malignant ascites stimulates motility of human pancreatic cancer cells through LPA1. J Biol Chem. 2004;279:6595–6605. doi: 10.1074/jbc.M308133200. [DOI] [PubMed] [Google Scholar]
- 51.Morla A, Zhang Z, Ruoslahti E. Superfibronectin is a functionally distinct form of fibronectin. Nature. 1994;367:193–196. doi: 10.1038/367193a0. [DOI] [PubMed] [Google Scholar]
- 52.Akerman ME, Pilch J, Peters D, Ruoslahti E. Angiostatic peptides use plasma fibronectin to home to angiogenic vasculature. Proc Natl Acad Sci USA. 2005;102:2040–2045. doi: 10.1073/pnas.0409844102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kaspar M, Zardi L, Neri D. Fibronectin as target for tumor therapy. Int J Cancer. 2006;118:1331–1339. doi: 10.1002/ijc.21677. [DOI] [PubMed] [Google Scholar]
- 54.Zardi L, Carnemolla B, Siri A, Petersen E, Paolella G, Sebastio G, Baralle FE. Transformed human cells produce a new fibronectin isoform by preferential alternative splicing of a previously unobserved exon. EMBO J. 1987;6:2337–2342. doi: 10.1002/j.1460-2075.1987.tb02509.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Borsi L, Carnemolla B, Castellani P, Rosellini C, Vecchio D, Allemanni G, Chang SE, Taylor-Papadimitriou J, Pande H, Zardi L. Monocloncal antibodies in the analysis of fibronectin isoforms generated by alternative splicing of mRNA precursors in normal and transformed human cells. J Cell Biol. 1987;104:595–600. doi: 10.1083/jcb.104.3.595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Carnemolla B, Balza E, Siri A, Zardi L, Nicotra MR, Bigotti A, Natali PG. A tumor-associated fibronectin isoform generated by alternative splicing of messenger RNA precursors. J Cell Biol. 1989;108:1139–1148. doi: 10.1083/jcb.108.3.1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Boudreau NJ, Varner JA. The homeobox transcription factor Hox D3 promotes integrin α5β1 expression and function during angiogenesis. J Biol Chem. 2004;279:4862–4868. doi: 10.1074/jbc.M305190200. [DOI] [PubMed] [Google Scholar]
- 58.George EL, Georges-Labouesse EN, Patel-King RS, Rayburn H, Hynes RO. Defects in mesoderm, neural tube and vascular development in mouse embryos lacking fibronectin. Development. 1993;119:1079–1091. doi: 10.1242/dev.119.4.1079. [DOI] [PubMed] [Google Scholar]
- 59.Kim S, Harris M, Varner JA. Regulation of integrin αvβ3-mediated endothelial cell migration and angiogenesis by integrin α5β1 and protein kinase A. J Biol Chem. 2000;275:33920–33928. doi: 10.1074/jbc.M003668200. [DOI] [PubMed] [Google Scholar]
- 60.Yang JT, Rayburn H, Hynes RO. Embryonic mesodermal defects in α5 integrin-deficient mice. Development. 1993;119:1093–1105. doi: 10.1242/dev.119.4.1093. [DOI] [PubMed] [Google Scholar]
- 61.Astrof S, Crowley D, Hynes RO. Multiple cardiovascular defects caused by the absence of alternatively spliced segments of fibronectin. Dev Biol. 2007;311:11–24. doi: 10.1016/j.ydbio.2007.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.