

Abstract
Endonuclease G (EndoG) is a mitochondrial enzyme believed to be released during apoptosis to participate in the degradation of nuclear DNA. This paper describes a Drosophila protein, EndoGI, which inhibits EndoG specifically. EndoG and EndoGI associate with subpicomolar affinity, forming a 2:1 complex in which dimeric EndoG is bound by two tandemly repeated homologous domains of monomeric EndoGI. Binding appears to involve the active site of EndoG. EndoGI is present in the cell nucleus at micromolar concentrations. Upon induction of apoptosis, levels of the inhibitor appear to be reduced, and it is relocalized to the cytoplasm. EndoGI, encoded by the predicted open reading frame cg4930, is expressed throughout Drosophila development. Flies homozygous for a hypomorphic EndoGI mutation have a strongly reduced viability, which is modulated by genetic background and diet. We propose that EndoGI protects the cell against low levels of EndoG outside mitochondria.
Fragmentation of chromosomal DNA is a characteristic feature of apoptosis. The main enzyme responsible for apoptotic DNA degradation is CAD (or DFF40). CAD is controlled through its association with ICAD (or DFF45), which serves a dual function. First, ICAD plays a chaperone-like role for CAD; no functional CAD is produced without ICAD. Second, ICAD is an inhibitor of CAD. During apoptosis, ICAD is cleaved by caspases to liberate active CAD (1–3).
Although cells deficient for either CAD or ICAD have defects in apoptotic DNA fragmentation, they can still undergo apoptosis and degrade their DNA. This observation led to the purification of a second apoptotic nuclease, endonuclease G (EndoG)4 (4, 5). A role of EndoG in apoptosis was independently discovered in a genetic screen in Caenorhabditis elegans (6). EndoG is localized in the mitochondrial intermembrane space, sequestered from cellular nucleic acids. Upon induction of apoptosis, it is released from mitochondria, together with cytochrome C and other proapoptotic proteins, and transported to the cell nucleus (4, 7, 8). The enzyme is a metal ion-dependent homodimeric endonuclease of the ββα-Me finger superfamily (9). EndoG-like enzymes degrade single- and double-stranded DNA and RNA to oligonucleotides with 3′-OH and 5′-phosphate ends (10–12). At physiological salt concentrations, the preferred substrate is single-stranded nucleic acid, but the degradation of double-stranded DNA may be facilitated by cooperation with other proteins (11, 13–15).
A role of EndoG in apoptosis is somewhat controversial. EndoG knock-out mice have no defect in apoptotic DNA degradation (16, 17), but this might be due to redundancy with CAD and additional nucleases (1, 2, 18). RNA interference experiments and genetic data in Saccharomyces cerevisiae support a role of EndoG in apoptosis (8, 19, 20). EndoG is also thought to function in a non-apoptotic form of programmed cell death (21). Evidence for a positive function of EndoG in cell proliferation has been presented (8, 22, 23). A proposed role of mammalian EndoG in the genomic inversion of herpesvirus (22, 24) suggests the possibility that not all of EndoG is sequestered in mitochondria.
No inhibitor comparable with ICAD has been reported for EndoG, and no such control of the nuclease would seem essential, since it is sequestered in mitochondria. However, a prokaryotic EndoG family member, the secreted nuclease NucA of Anabaena, forms a 1:1 complex with an inhibitory protein (25, 26). Since other secreted nucleases like pancreatic RNase (27), barnase (28), or colicin E3 (29) also associate tightly with cytoplasmic inhibitors, export of such enzymes may not always be sufficient for the protection of cellular nucleic acids.
During our studies of enzymes involved in mRNA deadenylation, we rediscovered EndoG through its ability to degrade poly(A). More interestingly, we found an endogenous protein inhibitor of EndoG in Drosophila. This inhibitor, which we termed EndoGI, is located in the cell nucleus. We present evidence suggesting that it protects the cell from chromosomal damage inflicted by EndoG in nonapoptotic cells.
EXPERIMENTAL PROCEDURES
Plasmids—EndoGI, EndoG, and 14-3-3 ζ were amplified from cDNA clones 16I11, 21H12, and 66H2 (obtained from BACPAC; available on the World Wide Web), respectively. 14-3-3 ε was amplified from S2 cell cDNA. Appropriate restriction sites were introduced with the primers. 14-3-3 ε and ζ were ligated into the BamHI/KpnI sites, and EndoGI and its variants were ligated into the ClaI/EcoRI sites of the pRSET C vector (Invitrogen) in frame with an N-terminal His tag. For EndoGI deletion variants, artificial start or stop codons were introduced by PCR, and the products were cloned into pRSET C. For expression of nontagged EndoGI, the sequence encoding the His tag was removed from the pRSET C expression clone with NdeI and EcoRI. The EndoG coding sequence lacking the N-terminal 53 amino acids (EndoG ΔN53) was PCR-amplified with primers introducing an artificial start codon before codon 54 and appropriate restriction sites. The product was ligated into the NcoI/KpnI sites of pETM-11 (30) in frame with the N-terminal His tag. For the EndoG ΔN53 DAGA mutant, site-directed mutagenesis was performed on full-length cDNA, an artificial start codon replacing amino acids 1–53 was then introduced by PCR, and the product was ligated into the BamHI/EcoRI sites of pRSET C in frame with the N-terminal His tag. For the bicistronic co-expression construct, EndoGI was amplified and ligated into the NcoI/EcoRI sites of pET21d (Novagen). EndoG ΔN55 was amplified with a 5′ primer introducing an EcoRI site, a ribosome binding site, and a start codon (5′-GTG AAT TCA ATA ATT TTG TTT AAC TTT AAG AAG GAG ATA TAC ATATGT TGA TTC CCG CCC AAG-3′) and a suitable 3′ primer and ligated into the EcoRI/NotI sites of the pET21d-endoGI expression construct in frame with a C-terminal His tag.
For the full-length EndoG DAGA mutant, site-directed mutagenesis was performed on a full-length cDNA clone. The coding sequence was then amplified with a 3′ primer replacing the natural stop codon with a FLAG tag, followed by a stop codon and a suitable restriction site, and ligated into the KpnI and EcoRI sites of a pMT/V5-His C vector (Invitrogen). This vector allows expression in Drosophila S2 cells under the control of a Cu2+-regulated promoter. GFP-EndoGI fusions were made in the pAc5.1 vector (Invitrogen) containing the coding sequence for enhanced GFP (a gift from E. Izaurralde, Max-Planck-Institute, Tübingen, Germany). EndoGI coding sequences were amplified with primers introducing two EcoRI sites and removing the start codon. Products were ligated into the EcoRI-opened pAc5.1 enhanced GFP plasmid in frame with GFP. The fusion protein can be expressed in S2 cells under the control of a constitutive actin promoter. The intronless grim coding sequence was amplified from chromosomal DNA of Drosophila melanogaster w1118 with primers introducing restriction sites and a C-terminal FLAG tag and cloned into the KpnI and XhoI sites of pMT/V5-His C. All constructs were verified by sequencing.
Protein Purification—All column materials were from GE Healthcare except where noted. Preparation of S2 cell extract and DEAE chromatography have been described (31). Fractions containing EndoGI were pooled and applied to a MacroprepS column. EndoGI was present in the flow-through. Ammonium sulfate was added to 40% saturation, and, after centrifugation, the supernatant was applied to a phenyl-Superose column. The EndoGI-containing fractions were further purified by hydroxyapatite (Biogel-HTP; Bio-Rad) and MonoQ chromatography. The pattern of protein bands was not changed by subsequent gel filtration on Superdex 200. DEAE column fractions containing EndoG were also pooled and applied to a MacroprepS column. Enzyme eluted from the column was further purified by hydroxyapatite chromatography, ammonium sulfate precipitation (20% saturation), and Superdex 200 gel filtration. Further details of the procedures are available upon request.
For expression of EndoGI, Escherichia coli BL21 Codon+ was transformed with the expression plasmid, grown at 37 °C in LB medium, and induced in midlog phase (A600 = 0.6) by the addition of 1 mm isopropyl 1-thio-β-d-galactopyranoside, and the temperature was reduced to 25 °C. After 3 h, cells were harvested; washed with PBS; resuspended in ice-cold EndoGI buffer (50 mm Tris-HCl, pH 7.5, 10% (v/v) glycerol, 0.02% (v/v) Nonidet P-40) containing 300 mm KCl, 1 mm phenylmethylsulfonyl fluoride, 2 μg/ml pepstatin, 2 μg/ml leupeptin; and broken on ice by sonication. After centrifugation at 30,000 × g and 4 °C for 30 min, the supernatant was frozen in liquid nitrogen and stored at –80 °C. Fractionation was carried out at 4–8 °C. Extracts were applied to a Ni2+-nitrilotriacetic acid-agarose column (Qiagen). After washing with EndoGI buffer containing 300 mm KCl and then with EndoGI buffer containing 10 mm imidazole, protein was eluted with EndoGI buffer containing 100, 200, and 500 mm imidazole. Eluted proteins were diluted with EndoGI buffer, applied to a MonoQ column, and eluted with a 20-column volume gradient from 50 to 500 mm KCl in EndoGI buffer. Fractions containing the overexpressed proteins were pooled and concentrated by centrifugation through Amicon ultracentrifugal filter (Millipore). 2 ml of this concentrated protein was applied to a 120-ml Superdex 200 preparation grade column equilibrated in 50 mm potassium phosphate (pH 7.4), 100 mm KCl, 10% (v/v) glycerol. EndoGI variants and the 14-3-3 proteins were expressed according to the same procedure and purified by the same sequence of columns.
For expression of EndoG, E. coli BL21 Codon+ was transformed simultaneously with the EndoG plasmid and the EndoGI plasmid lacking a his tag, and double selection with ampicillin and kanamycin was carried out. Cell growth (in the continuous presence of both antibiotics) and induction, lysis, and Ni2+-nitrilotriacetic acid chromatography were as described above. EndoG-containing fractions were pooled and dialyzed against 50 mm Tris (pH 7.5), 20% (v/v) glycerol, 20 mm KCl, 0.5 mm DTT, 0.02% (v/v) Nonidet P-40, and an ammonium sulfate precipitation at 10% saturation was performed on ice for 1 h. After centrifugation, the supernatant was applied to a 1-ml phenyl-Superose column. The column was eluted by a descending ammonium sulfate gradient, and the peak fractions were pooled; dialyzed against 20 mm imidazole (pH 7.4), 50 mm KCl, 10% (v/v) glycerol, 0.5 mm DTT, 1 mm MgCl2; and applied to a 1-ml hydroxyapatite column. EndoG was eluted with a 10-ml gradient to 0.2 m potassium phosphate (pH 7.4) in the same buffer lacking MgCl2. Titrations of purified EndoG with EndoGI under stoichiometric conditions suggested that about 30% of the protein was active. Partial activity of the enzyme was not due to residual EndoGI; none was detectable by Western blotting.
For purification of EndoG ΔN53 DAGA, expression, cell lysis and centrifugation were done according to the same protocol. The pellet containing the majority of the desired protein was dissolved in urea buffer (8 m urea, 10 mm Tris-HCl, pH 8.0, 100 mm NaH2PO4) and incubated at room temperature for 2 h. After centrifugation at 20,000 × g for 15 min, the supernatant was applied to a Ni2+-nitrilotriacetic acid-agarose column (Qiagen). The column was washed with urea buffer, and protein was eluted with urea buffer brought to pH 4.0 with HCl. Eluted proteins were diluted with urea buffer to below 0.5 mg/ml and renatured by dialysis in a cold room against 20 mm Tris-HCl, pH 8.0, 50 mm KCl, 10% (v/v) glycerol, 0.02% (v/v) Nonidet P-40, 1 mm DTT, 0.7 m l-arginine overnight and then against the same buffer containing 0.4 m l-arginine for 12 h and finally against the same buffer lacking arginine overnight. The solution was clarified by centrifugation, and the supernatant was concentrated and applied to a 120-ml Superdex 200 column.
Co-expression of EndoG ΔN55 N187K and EndoGI from the bicistronic construct was done in BL21 (DE3) CodonPlus RIL (Stratagene). Cells were grown in LB medium to midlog phase (A600 = 0.6) at 37 °C and cooled to 20 °C, and expression was induced with 0.5 mm isopropyl 1-thio-β-d-galactopyranoside overnight. Cells were harvested, suspended in 50 mm Tris-HCl, pH 8.0, 250 mm NaCl, 2 mm MgSO4, 10 mm β-mercaptoethanol, and lysed by sonication. The lysate was cleared by centrifugation and loaded onto a 1-ml HisTrap HP column equilibrated with the same buffer. Proteins were eluted with a 10-column volume gradient to 250 mm imidazole in the same buffer; diluted into 50 mm Tris-HCl, pH 8.0, 2 mm MgSO4,2 mm dithioerythritol; and loaded onto a MonoQ column. Whereas EndoGI bound to the column, EndoG was found exclusively in the flow-through and subsequently loaded onto a 1-ml HiTrap heparin column equilibrated with 50 mm Tris-HCl, pH 7.3, 2 mm dithioerythritol. Bound EndoG was eluted with a 20-ml gradient to 1 m NaCl in the same buffer and finally purified on Superdex 75 equilibrated with 50 mm HEPES, pH 7.5, 100 mm NaCl, 2 mm MgSO4, 2 mm dithioerythritol.
Protein concentrations were determined photometrically, with extinction coefficients calculated from the amino acid composition. Because EndoG ΔN53 was available at low concentration and not homogeneous, its concentration was estimated by densitometry of a Coomassie-stained SDS-polyacrylamide gel and comparison with a bovine serum albumin standard. The concentration of active EndoG was determined by a stoichiometric titration with EndoGI and was 30% of the chemical concentration. This may reflect a fraction of inactive protein, inaccuracy of the concentration measurement, or both. In the figure legends, both the chemical concentration and the concentration of active dimer are indicated.
Activity Assays—Homogeneously labeled poly(A) was made by extension of an oligo(A) primer by poly(A) polymerase with [α-32P]ATP (32). L3pre-based RNAs were synthesized with [α-32P]UTP as described (33). Size-fractionated poly(A) was 5′-labeled with [γ-32P]ATP and T4 polynucleotide kinase under standard conditions. For 3′ labeling, 9 pmol (3′-ends) of size-fractionated poly(A) was incubated with 50 μCi of [α-32P]ATP (3000 Ci/mmol) and 2 pmol of bovine poly(A) polymerase as described (31).
Poly(A)-degrading activity was assayed by the release of trichloroacetic acid-soluble products from homogeneously labeled poly(A); 1 μl of protein sample was mixed at room temperature with 20 μl of reaction buffer (20 mm Hepes, pH 7.5, 1 mm magnesium acetate, 1 mm DTT, 10% (v/v) glycerol, 0.2 mg/ml bovine serum albumin, 0.02% (v/v) Nonidet P-40, 150 mm KCl) containing 20,000–50,000 cpm of homogeneously labeled poly(A) and 54 μm (mononucleotide concentration) of unlabeled poly(A) of heterogeneous size. The reaction was started immediately by incubation at 30 °C for the appropriate time. When required, EndoG and EndoGI were preincubated in the reaction mix for 90 min, and reactions were started by the addition of poly(A). Protein concentrations used are indicated in the figure legends. Reactions were stopped by the addition of 5 μl of 0.5 m EDTA (pH 8.0). 75 μl of cold 16% (w/v) trichloroacetic acid were added immediately before centrifugation at 20,000 × g for 10 min at 4 °C, and the amount of acid-soluble nucleotides was determined by scintillation counting. Soluble radioactivity in a sample incubated without enzyme was subtracted as background.
The in-gel activity assay used for the identification of EndoG was carried out as described (34) except that the reaction buffer was the one described above. The gel solution included 26 nm (as mononucleotides) of homogeneously labeled poly(A).
For inhibition assays, EndoG and EndoGI were preincubated in the reaction mix for 90 min when necessary, and reactions were started by the addition of poly(A). Complete complex formation during the time of preincubation was experimentally verified. Protein concentrations used are indicated in the figure legends.
Affinity of the EndoG-EndoGI interaction was determined by a titration of a fixed amount of EndoG with EndoGI or EndoGI variants. Results were fitted to Equation 1,
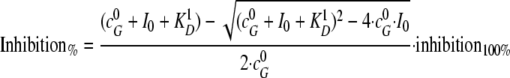 |
(Eq. 1) |
where 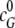 represents the initial concentrations
of EndoG dimers, and I0 represents the initial
concentrations of EndoGI monomers, respectively.
represents the initial concentrations
of EndoG dimers, and I0 represents the initial
concentrations of EndoGI monomers, respectively.
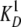 is the dissociation constant of the
complex.
is the dissociation constant of the
complex.
Since the concentration of active EndoG was not known precisely, a free fit of this parameter was allowed. However, the KD values obtained were quite insensitive to the concentration of EndoG. KD values are reported ± S.D. and are based on at least four independent titrations.
The affinity of EndoG ΔN55 N187K (EndoG N187K for short) for EndoGI
was determined by a competition (deinhibition) experiment in which a fixed
amount of EndoG was mixed with increasing amounts of EndoG N187K before EndoGI
was added, and the resulting nuclease activity was determined after complex
formation. Curve fitting was based on the assumption that no mixed dimers were
formed in the course of the experiment. The interaction of EndoG and EndoGI is
characterized by a 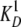 value of 0.2
pm, determined as above. The deinhibition approach revealed the
value of 0.2
pm, determined as above. The deinhibition approach revealed the
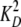 value of the inactive variant EndoG N187K and
EndoGI. Nuclease activity was measured at a substrate concentration of 53
μm (as mononucleotides; about Km). A
competitive inhibition according to DIXON results in
Equation 2,
value of the inactive variant EndoG N187K and
EndoGI. Nuclease activity was measured at a substrate concentration of 53
μm (as mononucleotides; about Km). A
competitive inhibition according to DIXON results in
Equation 2,
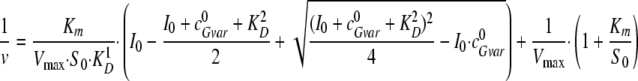 |
(Eq. 2) |
where v represents the substrate turnover rate at a specified initial
substrate concentration S0, Vmax is
the maximum substrate turnover rate at saturating substrate concentrations,
Km is the substrate concentration at half-maximum rate,
I0 the initial concentration of EndoGI, and
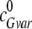 is the initial dimer concentration of the
inactive EndoG N187K competing with EndoG for EndoGI binding. All measurements
were performed at least in duplicate, and the data were averaged. Errors are
the S.E. values resulting from data fitting.
is the initial dimer concentration of the
inactive EndoG N187K competing with EndoG for EndoGI binding. All measurements
were performed at least in duplicate, and the data were averaged. Errors are
the S.E. values resulting from data fitting.
The rate constant of inhibitor release from the EndoG·EndoGI complex (koff) was determined by the gain in enzymatic activity after the addition of EndoG N187K to the preformed complex. EndoG N187K was used in excess to ensure the irreversibility of the reaction.
Equation 3 describes the relationship (proportionality) of the reaction rate and the concentration of the active enzyme (deinhibited enzyme), where v(c) is the constant rate after attaining the equilibrium, and v(t) is the rate at time t.
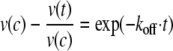 |
(Eq. 3) |
The product formation dependent on time yields the rate of the reaction. Integration of the differential equation following from Equation 3 yields the relationship of the time-dependent product formation and allows the determination of koff,
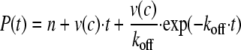 |
(Eq. 4) |
where P represents the product concentration at time t, and n is the offset. The progress curve of the gain in substrate turnover was investigated considering the maximum turnover rate determined for a simultaneous incubation of all components and yielded a value koff of (2.66 ± 0.12)·10–6 s–1. The error indicated is the S.E. value of the parameter resulting from data fitting.
Mass Spectrometry—For protein identification, excised bands were digested with trypsin (35). Peptides were separated by capillary liquid chromatography (Micromass), desalted online through a Symmetry300 C18 trap column (Waters), separated on a dC18 Atlantis NanoEase column (Waters), and analyzed on a Q-TOF 2 mass spectrometer (Micromass) equipped with a modified nanoelectrospray ionization source to hold a pico-tip (New Objective). MS/MS measurements were done on doubly and triply charged ions. Data were searched against the MSDB data base with MASCOT (available on the World Wide Web). For the analysis of mutant EndoG, the protein was desalted with a ZipTip C4 (Millipore), and protein mass was determined with a Q-TOF 2 mass spectrometer as described (35). A mass of 30,553 ± 3 Da was found, consistent with the predicted mass of the EndoG ΔN55 N187K mutant (30,550 Da compared with 30,536 Da for the wild-type protein). For peptide analysis, desalted mutant EndoG was digested with trypsin, peptides were reduced with DTT, and cysteines were alkylated with iodoacetamide. Peptide mass fingerprint spectra were recorded on an Ultraflex-II TOF/TOF mass spectrometer (Bruker) as described (35). A peptide with a mass of 2329.43 ± 0.4 ([M + H]+) was found, corresponding to cleavage behind the mutant Lys187 (amino acids 168–187 with a carbamidom-ethylated Cys169).
Data Base Searches—Homology searches were done with TBLASTN. Default parameters were used. Searches were carried out first with the complete sequence of D. melanogaster EndoGI and then with the consensus sequence obtained by an alignment of 24 homologous domains from the 12 EndoGI homologs identified in the first search.
Analytical Ultracentrifugation—Proteins were dialyzed into 50 mm potassium phosphate (pH 7.5), 100 mm KCl, 10% (v/v) glycerol. Sedimentation equilibrium experiments were performed with an Optima XL-A (Beckman Instruments) and an An50Ti rotor equipped with double sector cells. Sedimentation data were obtained at 10,000 rpm, 20 °C at a detection wavelength of 230, 280, or 290 nm. The apparent molecular masses of the protein samples were calculated using the software provided by Beckman Instruments; the density of the buffer was set to 1.026 ml/g due to glycerol content of the buffer. S.D. values are based on at least two different experiments, each analyzed at at least two different wavelengths.
S2 Cell Culture—All procedures were as described (36).
Antibodies and Immunological Procedures—Antibodies against EndoGI were generated by Eurogentec (Belgium). Other antibodies were as follows: monoclonal anti-tubulin clone DM 1A and anti-FLAG M2 (Sigma); rabbit anti-rox2 (37); rat serum directed against the last 16 amino acids of the splicing factor B52 (a generous gift from F. Juge). Western blots were performed as described (31). Secondary horseradish peroxidase-coupled anti-rabbit and anti-mouse antibodies (DAKO) and the Super Signal system (Pierce) were used for detection.
For microscopy, S2 cells were centrifuged onto coverslips coated with poly-l-lysine (1 mg/ml in PBS; Sigma) and washed once with PBS. After a 15-min fixation in 3.7% paraformaldehyde in PBS (Accustain; Sigma) at room temperature, cells were washed twice with PBS, permeabilized for 5 min with 0.3% (v/v) Triton X-100 in PBS, and washed twice with PBS. They were then incubated for 30 min in blocking solution (25 g/liter bovine serum albumin, 0.1% (v/v) Triton in PBS) for 1 h at 37 °C with the primary antibody diluted in blocking solution and again for 1 h at 37 °C with secondary antibodies (Alexa488-labeled anti-rabbit (Molecular Probes); Cy3-anti-mouse (DAKO)) at the recommended dilutions. Cells were mounted in Pro Long Gold antifade reagent with DAPI (Invitrogen) and analyzed with a Zeiss Axiovert 200M fluorescence microscope or a Zeiss Meta 510 LSM. For Mitotracker staining, S2 cells expressing FLAG-tagged EndoG DAGA were incubated with fresh medium containing 100–500 μm Mitotracker Orange CM-H2 TMRos (Invitrogen). After 15–30 min, the medium was removed, and cells were stained with an anti-FLAG antibody as above. Immunostaining of Drosophila embryos and ovaries was performed as described (31). Secondary antibodies were preadsorbed overnight to w1118 embryos or ovaries.
Drosophila Stocks and Genetics—w1118 flies were used as a control. The EndoGI mutant cg4930e00915 was generated by Exelixis (38) and obtained from the Harvard Exelixis Stock Collection. The mutant allele was rebalanced over a CyO GFP.TM3 Ser GFP balancer (39) (a gift from G. Reuter) and a SM5.TM6 Hu Tb balancer. Flies were kept at room temperature on 1% agar, 3.5% raisins, 5% yeast, 2.5% molasses, 3% cornmeal, 0.1% Nipagin. Alternatively, a less rich medium was used: 0.65% agar, 5.8% glucose, 4.68% cornmeal, 1.9% yeast, 0.43 g/liter CaCl2, 5.38 g/liter K-Na-tartrate, 0.15% Nipagin. For the collection of embryos and larvae, apple juice-agar plates supplemented with yeast were used.
RESULTS
Identification of an Inhibitor for Endonuclease G—In the course of experiments concerning mRNA turnover, an inhibitor of a poly(A)-degrading activity was identified in column fractions derived from Drosophila Schneider cell extracts (31). Two observations suggested that this inhibitor was specific. First, the amount of inhibitor required was insensitive to the amount of substrate RNA but varied in proportion to the amount of nuclease, suggesting that the inhibitor acted on the enzyme and not on the RNA substrate. Second, the inhibitor was specific for poly(A)-degrading activity detected in neighboring column fractions; a mammalian poly(A)-degrading enzyme, PARN (32), was not inhibited (data not shown). The inhibitor was purified by chromatography, inhibition of a partially purified nuclease fraction serving as an assay. Gel electrophoresis of the final column fractions showed a small group of bands co-eluting with the inhibitory activity (Fig. 1A). Mass spectrometry identified three Drosophila proteins: the two 14-3-3 proteins, ε and ζ, and an unknown protein predicted from the genome sequence, CG4930. All three proteins were expressed separately in E. coli, purified, and tested for inhibition of the crude nuclease fraction. The two 14-3-3 proteins proved inactive, whereas the CG4930 protein inhibited poly(A) degradation to the same extent as the inhibitor purified from Schneider cells (see below).
FIGURE 1.

A, analysis of the purified inhibitor. Coomassie-stained 10% SDS-polyacrylamide gel of the final gel filtration column. The gel was used to identify the individual polypeptides. The proteins with the highest score in the mass spectrometry analysis are indicated. Sizes of marker proteins are indicated on the left. B, identification of the poly(A)-degrading nuclease by an in-gel activity assay. Aliquots from a column fraction (Superdex 200) from the purification of the nuclease were loaded on two lanes of a 10% SDS-polyacrylamide gel polymerized with homogenously 32P-labeled poly(A). After the run, the gel was cut in two pieces. One was silver-stained (right), and the other was renatured and incubated in reaction buffer. After the reaction, the gel was rinsed in water, dried, and analyzed with a PhosphorImager (left). Protein with nuclease activity is marked with an arrow on the left. Markers seen in the silver stain are indicated on the right.
Purification of the corresponding poly(A)-degrading activity was monitored by inhibitor-sensitive release of acid-soluble radioactivity from homogeneously 32P-labeled poly(A). The inhibitor-sensitive nuclease was separated from the CCR4-NOT complex, a known mRNA deadenylase (31), but homogeneity was not achieved due to limiting material. Therefore, an in-gel activity assay was applied to the most highly purified material. This assay identified a single band of 29 kDa, which was also visible in the stained gel (Fig. 1B) and contained the protein encoded by the predicted gene CG8862, the closest homolog of endonuclease G in D. melanogaster. The data presented below confirm that EndoG is indeed the nuclease inhibited by the CG4930 protein. Therefore, the latter protein is referred to as EndoGI.
Characterization of Drosophila EndoG—The EndoG gene encodes a protein of 310 amino acids with a predicted molecular mass of 34.6 kDa. Comparison with the bovine sequence (40) suggests that the first ∼50 amino acids serve as a mitochondrial targeting signal; the apparent size of the purified mature protein is consistent with removal of this presequence. Drosophila EndoG is 42% identical with its human orthologue and contains the catalytically important DRGH motif characteristic for this class of enzymes (12) (supplemental Fig. 1A). Cloning of EndoG was successful only when the inhibitor was co-expressed. The mature part of EndoG (amino acids 54–310) was expressed, and a purification protocol was developed that allowed a separation from the co-expressed inhibitor. This EndoG ΔN53 variant was used for all functional assays in this paper and is called EndoG for simplicity. “Wild-type EndoG” refers to the same protein as opposed to variants carrying point mutations.
Under standard assay conditions with homogeneously 32P-labeled poly(A) as a substrate, 1 mg of the enzyme (∼5 nmol of active dimers) released ∼100 μmol (as mononucleotides) of acid-soluble product/min. The purified enzyme degraded 5′- and 3′-end-labeled poly(A) through similar continuous patterns of intermediates; thus, the enzyme is an endonuclease. The end products of digestion were oligonucleotides of approximately six nucleotides (supplemental Fig. 2A), although synthetic A6 could be digested even further upon prolonged incubation (data not shown). Digestion of RNAs with an internal or 3′-terminal poly(A) tract of 45 nucleotides showed some preferred cleavage within the poly(A) tract, but the RNA was further digested with a continuous distribution of intermediates and end products of about six nucleotides (supplemental Fig. 2B) (data not shown). Thus, the enzyme has but a weak preference for either poly(A) or unstructured RNA and probably little sequence specificity overall. Single- or double-stranded 5′-labeled DNA was degraded as efficiently as RNA substrates (supplemental Fig. 2C). All of these properties are in agreement with those determined for other EndoG enzymes (11, 12, 40).
The EndoG Inhibitor and Its Interaction with EndoG—EndoGI is composed of 359 amino acids with a predicted molecular mass of 40.6 kDa (supplemental Fig. 1B). Orthologues of EndoGI are present in all 12 sequenced genomes of the genus Drosophila with sequence identities between 60 and over 96%, but proteins with obvious similarities were not found in any other genome, not even in other insect genomes. The protein was expressed in soluble form in E. coli with an N-terminal His tag and purified to homogeneity by metal affinity chromatography followed by anion exchange chromatography and gel filtration. In nuclease assays carried out at relatively high protein concentrations, recombinant EndoGI blocked the activity of EndoG immediately and completely, suggesting that simple association is sufficient for inhibition (Fig. 2). Titrations of EndoGI into EndoG activity assays at very low EndoG concentrations showed a very tight interaction with a KD of 0.19 ± 0.14·10–12 m (Fig. 3A).
FIGURE 2.

Rapid and complete inhibition of EndoG by EndoGI. 26 pm EndoG (calculated monomer concentration; ∼4 pm estimated concentration of active dimer) was incubated in reaction buffer at 30 °C under standard conditions. At the indicated time points, aliquots were taken into the stop solution. After 60 min, 130 nm EndoGI was added, and additional time points were taken. Regression lines drawn through the two sets of data points intersect at the point of EndoGI addition.
FIGURE 3.

Affinity of EndoGI and its variants for EndoG. A, EndoG at a monomer concentration of 13 pm (estimated concentration of active dimer ∼2 pm) was titrated with 0.1–5 pm EndoGI. The samples were preincubated at 30 °C for 90 min before substrate was added. The reactions were stopped 4 h later. B, EndoG at a monomer concentration of 65 pm (estimated concentration of active dimer ∼10 pm) was titrated with 0.05–2.07 μm EndoGI 1–191. The proteins were mixed in reaction buffer, and reactions were started through the addition of poly(A) and stopped after 2 h. In this experiment, a preincubation was unnecessary because of the high protein concentrations. C, EndoG at a monomer concentration of 33 pm (estimated concentration of active dimer ∼5 pm) was titrated with 0.05–20 nm EndoGI 192–359. The samples were preincubated at 30 °C for 90 min before substrate was added. Reactions were stopped after 3 h.
Biophysical assays of complex formation were hampered by lack of sufficient quantities of EndoG, which was poorly soluble and had to be separated from co-expressed EndoGI. An EndoG variant with the DRGH motif (amino acids 152–155) mutated to DAGA could be expressed in the absence of EndoGI, although most of it formed inclusion bodies. After purification and renaturation, dimeric material was obtained by gel filtration (data not shown). As expected, no catalytic activity was detectable in EndoG DAGA (specific activity less than 10–7-fold that of wild-type). The protein was structured, since its CD spectrum was very similar to that of EndoGI N187K, which was shown to have a native structure (see below; supplemental Fig. 3), and both proteins showed a cooperative unfolding transition at the same temperature of 45 °C (data not shown). If EndoG DAGA were able to bind EndoGI, it should compete with wild-type EndoG for EndoGI binding and thus protect the enzyme from inhibition. However, inhibition of wild-type EndoG by EndoGI was not affected in the presence of a 10-fold excess of EndoG DAGA. As a control, the addition of a similar excess of wild-type EndoG shifted the EndoGI titration curve to higher concentrations (Fig. 4). Even when the mutant was present at more than 5000-fold excess over the wild-type enzyme, no competition for EndoGI was observed (data not shown). This suggests that binding of EndoGI involves the active site of EndoG.
FIGURE 4.

EndoG DAGA is unable to bind EndoGI. EndoG (32 pm monomer concentration, ∼5 pm active dimers) was titrated with EndoGI either alone (triangles) or in the presence of 320 pm EndoG DAGA (filled circles). As a control, the titration was also carried out with 352 pm wild-type EndoG (∼55 pm active dimers; filled squares). Inhibition of EndoG was measured by a trichloroacetic acid precipitation assay under standard conditions. Reactions were stopped after 3 h (with 32 pm EndoG) or 25 min (with 352 pm EndoG). Note that the proteins were not preincubated before the addition of substrate. The apparent binding curves therefore do not reflect equilibrium conditions.
Surprisingly, bicistronic co-expression of EndoGI and a slightly modified version of EndoG (amino acids 56–310 with a C-terminal His tag; EndoG ΔN55) yielded large amounts of soluble EndoG in excess of EndoGI. This EndoG preparation turned out to be inactive (specific activity ∼50,000-fold lower than wild type). Sequencing of the plasmid from the expression culture revealed a spontaneous point mutation, Asn187 → Lys. The mutation was confirmed by mass spectrometry of purified EndoG (see “Experimental Procedures”). Asn187 corresponds to Asn174 of bovine EndoG, which is involved in binding the catalytic Mg2+ ion (12). In contrast to the DAGA mutant, the N187K mutant was able to compete with active wild-type enzyme for binding of the inhibitor; increasing amounts of EndoG N187K relieved the inhibition (Fig. 5A). The competition with wild-type EndoG could be modeled with a relatively weak affinity of 37.7 ± 4.8 pm, ∼200-fold weaker than the wild-type enzyme (Fig. 5B), supporting the idea that EndoGI binds in the active site of EndoG.
FIGURE 5.

EndoG N187K binds weakly to EndoGI. A, EndoG at a calculated monomer concentration of 13 pm (estimated concentration of active dimer ∼0.5 pm) and 2 pm EndoGI were titrated with 10 pm to 10 nm EndoG N187K. EndoG and EndoG N187K were mixed before EndoGI was added, and all three proteins were preincubated together for 90 min in reaction buffer before substrate was added. The reactions were stopped after 3 h. B, the competition experiment was evaluated as described under “Experimental Procedures.” The line represents the calculated fit.
When excess EndoG N187K was added to a preformed EndoG·EndoGI complex, time-dependent recovery of EndoG activity showed that the inactive enzyme acted as a trap for EndoGI dissociating from wild-type EndoG. This demonstrated the reversibility of the inhibition and allowed the dissociation rate to be measured. The complex proved to be extremely stable, with an estimated half-life of about 3 days (dissociation rate constant 2.66 ± 0.12·10–6 s–1) (Fig. 6). Together with the equilibrium constant (see above), this leads to an estimate of the association rate constant of ∼107 m–1 s–1, which is close to the diffusion limit.
FIGURE 6.

Kinetics of EndoG·EndoGI complex dissociation. A, all reactions contained 26 pm wild-type EndoG (calculated monomer concentration). Proteins were preincubated for 90 min in reaction buffer in order to allow complex formation. Reactions were then started by the addition of substrate (t = 0), and aliquots were taken at the times indicated. Analysis of wild-type EndoG by itself (circles) represents the rate of uninhibited nuclease activity. Incubation with 10 pm EndoGI (inverted triangles) shows nearly complete inhibition. The addition of 26 nm EndoG N187K (squares) to wild-type EndoG shows that the mutant enzyme had no detectable activity at these concentrations. The addition of both 10 pm EndoGI and 26 nm EndoG N187K (triangles) represents the extent of protection of active EndoG by the excess of inactive mutant enzyme. In a fifth tube, EndoG was preincubated with 10 pm EndoGI, and 26 nm EndoG N187K was added together with the substrate (diamonds). The increasing rate of nuclease activity reflects time-dependent dissociation of the EndoG·EndoGI complex. Left, the entire time course is shown for all five reactions. Right, the lower section of the diagram shown on the left is enlarged to show the dissociation of the EndoG·EndoGI complex. B, enlargement of the lower part of A. C, the experiment shown in A was evaluated as described under “Experimental Procedures.” The line represents the calculated fit.
EndoG N187K was used for analytical ultracentrifugation assays of the EndoG-EndoGI association. Under equilibrium conditions, EndoGI behaved as a monomer (native molecular mass of 45 ± 2 kDa) at concentrations of 1–6 μm. EndoG N187K, at a monomer concentration of 2 μm, was a dimer (native molecular mass of 58 ± 6 kDa), as expected (12). When 5 μm EndoGI was titrated with EndoG N187K, complex formation was observed. The highest mass of the complex (101 ± 3.8 kDa) was reached at a 2-fold molar excess of EndoG monomers over EndoGI (Fig. 7). Both the stoichiometry and the molecular weight suggest a 2:1 complex of EndoG and EndoGI. This stoichiometry was confirmed in isothermal titration calorimetry (data not shown). Formation of these well defined complexes showed that EndoG N187K had a native structure and justified its use as a point of reference for the DAGA variant.
FIGURE 7.

EndoG and EndoGI form a 2:1 complex. EndoGI at a concentration of 5 μm was titrated with 0–20 μm (monomeric protein) EndoG N187K. The samples were centrifuged at 10,000 rpm, 20 °C until equilibrium was reached. The apparent molecular masses of the respective protein mixtures were calculated from data collected at 230 and 280 nm. Data from two independent experiments are plotted.
EndoGI consists of two tandemly repeated domains, which have 50% sequence identity over a stretch of 140 amino acids (29–168 and 213–352) (supplemental Fig. 1B). This structure is conserved in all orthologues of EndoGI. The two domains, amino acids 1–191 and 192–359, were expressed separately in E. coli and purified. Each domain inhibited EndoG on its own, but the affinity was much lower than that of full-length EndoGI; the KD was 0.04 ± 0.006·10–9 m for the C-terminal domain and 85 ± 33·10–9 m for the N-terminal domain (Fig. 3, B and C). Thus, the 2:1 stoichiometry revealed by analytical ultracentrifugation corresponds to a 1:1 stoichiometry of EndoGI domains and EndoG monomers, suggesting that each domain of EndoGI inhibits one subunit in the EndoG dimer (see “Discussion”).
Because 14-3-3 proteins were present in the original EndoGI preparation, a possible interaction was examined. Analytical ultracentrifugation revealed a weak association with 14-3-3 ε (KD in the micromolar range) (supplemental Fig. 4), which was supported by co-immunoprecipitation from S2 cell extract (data not shown). 14-3-3 did not affect the inhibition of EndoG by EndoGI at concentrations up to 15 μm. The localization of EndoGI was not different in embryos homozygous for a 14-3-3 ε null mutation (41), and no change in the EndoGI-14-3-3 association was apparent during apoptosis.
EndoGI Is a Ubiquitous Nuclear Protein—Western blotting of Drosophila extracts with a rabbit antiserum raised against EndoGI detected expression at all developmental stages even before the onset of zygotic transcription (0–2 h). Expression was relatively high throughout embryogenesis and larval development and in adult females and slightly lower in adult males (Fig. 8A). The absence of a detectable signal in adult flies homozygous for the endoGIe00915 mutation (see below) confirmed the specificity of the antibody (Fig. 8B). In immunofluorescence experiments with the same antibody, EndoGI was detected in the nuclei of embryos and Schneider cells (Figs. 8D and 9). In ovaries, strong nuclear staining was detected in nurse cells and follicle cells, whereas staining in the oocyte nucleus was absent (Fig. 8, C and E). Staining was reduced to almost zero in ovaries from homozygous endoGIe00915 females; thus, the signal in wild-type cells is specific (Fig. 8C). From estimates of nuclear volume and of EndoGI abundance by Western blotting and comparison with a recombinant protein standard, we calculate that the nuclear concentration of EndoGI is ∼4 μm.
FIGURE 8.

Expression and subcellular distribution of EndoGI in Drosophila. A, Western blot probed with anti-EndoGI showing the presence of EndoGI in different developmental stages. w1118 protein extracts loaded were from one ovary, 20 embryos, 20 first instar and 10 second instar larvae, one third instar larva, and one adult. The blots were probed with anti-α-tubulin as a loading control. B, Western blot probed with anti-EndoGI showing the absence of EndoGI in the endoGIe00915 mutant. 4–5 flies were homogenized in 10 μl of SDS sample buffer/fly. The equivalent of one fly was loaded per lane. +/+, w1118; +/–, endoGIe00915/CyO; –/–, endoGIe00915/endoGIe00915. C, immunodetection of EndoGI in wild-type ovaries and its absence in endoGIe00915 ovaries. Ovaries from either heterozygous endoGIe00915/CyO GFP.TM3 Ser GFP females or homozygous endoGIe00915 females were stained with anti-EndoGI antibody and DAPI. Both samples were imaged with the same microscope settings. D, nuclear localization of EndoGI in embryos. A confocal image of a stage 4 embryo is shown. EndoGI is labeled green, and tubulin as a cytoplasmic marker is in red. E, nuclear localization of EndoGI in ovaries. A confocal image of a stage 10 egg chamber is shown. Green, EndoGI; blue, DAPI.
FIGURE 9.

Behavior of EndoGI during apoptosis. A, endogenous EndoGI. Confocal images of S2 cells stably transfected with the inducible Grim-FLAG plasmid before (0 h) or 2 h after Grim induction. Cells were labeled with antibodies against EndoGI (green) and B52 (red) and with DAPI. B, EndoGI-GFP. Shown are confocal images of S2 cells stably transfected with two plasmids expressing EndoGI-GFP from a constitutive promoter and Grim-FLAG from an inducible promoter. Cells were harvested before (0 h) or 2 h after Grim induction. Rox2 was detected using an antibody (red), and EndoGI-GFP was detected via the GFP fluorescence. DAPI staining was used as a nuclear marker.
When full-length EndoG carrying the DAGA mutation and a C-terminal FLAG tag was transiently expressed in S2 cells, immunofluorescence microscopy showed its localization in mitochondria, identified by Mitotracker staining (supplemental Fig. 5).
EndoGI Is Lost from the Nucleus during Apoptosis—If EndoG is to degrade nuclear DNA during apoptosis, EndoGI has to be removed or inactivated under these conditions. A stable cell line was generated expressing the proapoptotic protein Grim (42) under the control of a regulated metallothionein promoter. Induction of Grim expression resulted in a large fraction of cells undergoing apoptosis and dying within 3 h. Immunofluorescence experiments showed that, under these conditions, EndoGI left the nucleus at a stage when apoptosis was clearly detectable by blebbing of the plasma membrane. At the same time, the nucleus still contained condensed chromatin, as judged by DAPI staining, and a control protein, the splicing factor B52 (43) (Fig. 9A). Thus, EndoGI is selectively lost from the nucleus prior to complete destruction of the organelle. When Grim-expressing cells were transiently transfected with a plasmid encoding a GFP-EndoGI fusion protein under the control of a constitutive promoter, GFP fluorescence was visible mostly in the nucleus. Upon induction of apoptosis, loss of nuclear fluorescence was observed. Again, two control proteins, B52 and the nuclear poly(A)-binding protein (37), were still visible in the cell nucleus (Fig. 9B) (data not shown). In these experiments, all GFP fluorescence was due to the fusion protein; no GFP lacking EndoGI was detectable in Western blots with an anti-GFP antibody (data not shown).
A comparison of apoptotic and nonapoptotic cells in the same sample showed a weaker EndoGI signal in the apoptotic cells, suggesting the possibility that EndoGI is degraded (Fig. 9A). However, we have been unable to confirm this impression by Western blots in which the abundance of EndoGI was compared with that of control proteins in the entire cell population. Possibly, EndoGI is degraded during a short time window before the degradation of control proteins, so these cells represent only a small fraction of the entire population.
Mutations in EndoGI Reduce Viability—The gene for EndoGI is localized at 35F1 on the left arm of chromosome 2. A publicly available stock, endoGIe00915, contains a PiggyBac transposable element insertion in the 5′-untranslated region of the gene. As shown above, EndoGI protein was not detectable in flies homozygous for this mutation (Fig. 8), so the allele is strong although probably not a null. We outcrossed the insertion allele to generate a number of balanced stocks carrying the endoGIe00915 allele. When balanced with a compound second and third chromosome balancer, SM5.TM6, several independent stocks were homozygous lethal or semilethal (one homozygote in many hundreds of progeny). These chromosomes were also lethal in combination with two deficiencies that uncover endoGI, Df(2L)RA5 and Df(2L)r10 (no transheterozygotes in more than 200 progeny), suggesting that the lethality was caused by a mutation in the chromosomal region containing endoGI. Unfortunately we were unable to excise the endoGIe00915 insertion with a genomic source of PiggyBac transposase to confirm that this lethality was caused by the insertion, presumably because there had been a change in the sequence of the element or neighboring sequences during transposition.
When the insertion chromosome was combined with other balancers (e.g. CyO GFP.TM3 Ser GFP), more homozygous progeny were obtained but at lower than expected frequency (11% homozygotes in 3622 progeny instead of the predicted 33%). Combinations of the insertion allele with chromosomal deficiencies for the region also showed lower viability than expected. Interestingly, culturing these stocks on less rich medium led to the complete absence of homozygotes, suggesting that these mutant flies were particularly sensitive to diet (results summarized in Table 1). Homozygous mutant adult males produced on rich medium proved infertile when crossed with wild-type females. In contrast, homozygous mutant females were fertile. When offspring from an endoGIe00915 stock balanced with the compound, GFP-tagged CyO.TM3 Ser balancer were examined during development, homozygous mutant L1 larvae, identified by the absence of green fluorescence, were observed at the expected frequency (75 in 250 progeny). However, when homozygous mutant larvae were collected at the L1 and L2 stage, only 40–50% proceeded to the pupal stage, and only 20–30% hatched in comparison with 70–80% of heterozygous L1 and L2 larvae hatching from the same cross. In summary, the data show that homozygous endoGI mutant individuals show lower viability than normal flies, dying during late development but at no specific stage, and that their viability is strongly influenced by genetic background and diet.
TABLE 1.
Summary of phenotypes of endoGI mutant flies
| Genotype | Viability on rich food | Viability on restricted diet | Fertility of homozygotes |
|---|---|---|---|
| endoGIe00915/endoGIe00915 from SM5.TM6 stock | Lethal | Lethal | NAa |
| endoGIe00915/Df(2L)RA5 from SM5.TM6 stock | Lethal | Lethal | NA |
| endoGIe00915/endoGIe00915 from CyO.TM3 Ser GFP stock | Reduced viability | Lethal | Male sterile |
| endoGIe00915/Df(2L)RA5 from CyO.TM3 Ser GFP stock | Reduced viability | Lethal | Male sterile |
NA, not applicable.
DISCUSSION
We have described a novel protein inhibitor of endonuclease G. In contrast to the apoptotic nuclease CAD, which is kept inactive by stoichiometric binding of the inhibitor ICAD, EndoG is controlled by sequestration in the mitochondrial intermembrane space; constitutive binding of an inhibitor is thus unnecessary. This agrees with the fact that EndoGI is localized mostly in the cell nucleus, well separated from the bulk of EndoG. Therefore, we propose that EndoGI acts as a life insurance for the cell against stray EndoG produced by failure of mitochondrial import or leakage from the organelle. The low viability phenotype of the homozygous endoGI mutant, which also depends on genetic background and diet, is consistent with the gene having a protective role in specific circumstances, allowing some mutant flies to survive to adulthood. In its protective role, EndoGI is comparable with cytoplasmic inhibitors of secreted nucleases (see Introduction). EndoGI may be required, since EndoG is probably genotoxic even at extremely low levels, as suggested by a comparison with the related homodimeric Serratia nuclease; the Serratia enzyme cleaves both strands of double-stranded DNA in close proximity, introducing in effect double strand breaks. Moreover, the enzyme appears to have a degree of processivity, cleaving one DNA molecule multiple times before dissociating (44). This type of DNA damage would be hard to repair. The simultaneous presence of uncleaved substrate and oligonucleotide end products in the Drosophila EndoG activity assays (supplemental Fig. 2) is consistent with a processive mechanism.
EndoGI inhibits EndoG by simple complex formation, as indicated by the rapid onset of inhibition at sufficiently high concentrations, its independence of any cofactors, and the reversibility of inhibition. The high concentration of EndoGI in the nucleus, a very high thermodynamic and kinetic stability of the EndoG·EndoGI complex, and rapid, diffusion-limited association guarantee efficient scavenging of any EndoG that has escaped its proper location. Interestingly, the Drosophila genome contains additional genes for EndoG-like proteins. In one of them (CG32483; 43% sequence identity with EndoG), the catalytic DRGH motif is conserved. Related enzymes with a SRGH active site motif have recently been shown to be also located in mitochondria and have 5′-exonuclease activity (45). These proteins might be additional targets for EndoGI. Although EndoGI is conserved in all sequenced Drosophila genomes, we have not found any orthologues in other genomes. These organisms might have EndoG inhibitors with very different primary structures; for example, NuiA, the inhibitor of the Anabaena EndoG ortholog NucA, has no detectable sequence similarity with EndoGI. Alternatively, other organisms survive without EndoG inhibitors (e.g. due to more efficient repair of DNA strand breaks or more reliable retention of EndoG in mitochondria).
EndoG is a homodimer, and EndoGI consists of two tandemly repeated highly similar domains. We propose that, in the complex, each of the duplicated domains of EndoGI binds one subunit of the EndoG homodimer. This arrangement is supported not only by the corresponding subunit structures of the two proteins but also by the experimentally determined 2:1 stoichiometry of the complex. EndoG is similar to the secreted nuclease from Serratia (12), which is known to have two well separated and functionally independent active sites in the homodimer (44, 46). Each separate domain of EndoGI can inhibit the activity of EndoG completely (Fig. 3) and, therefore, block both active sites. We infer that, in the EndoG·EndoGI complex, the two inhibitor domains make similar contacts with the two subunits of the nuclease. These contacts are likely to be in the active site, as shown by the inability of the EndoG DAGA mutant to compete with wild-type enzyme for EndoGI binding and by the reduced affinity of the EndoG N187K mutant for EndoGI. Anabaena NuiA also engages the active site of NucA, being directly coordinated with the catalytic Mg2+ ion (26).
The nuclear localization of EndoGI suggests that nuclear DNA is a target for EndoG, in agreement with its proposed role during apoptosis. The presence of EndoGI in the nucleus also argues that, in Drosophila, EndoG can only function in this organelle when EndoGI is removed or inactivated, but not under normal conditions. Thus, proposed functions of EndoG in the nucleus of normal, nonapoptotic cells (22, 24) may be specific for mammalian cells. Mammalian cells can be killed by extramitochondrial expression of active EndoG (12). Since the intracellular concentration of EndoG required for killing was not determined, the result does not permit an unambiguous conclusion as to whether mammalian cells have a nuclear inhibitor of EndoG. In S. cerevisiae, partial deletion of the mitochondrial targeting sequence of Nuc1 leads to a large fraction of the nuclease being localized outside the mitochondria. Nevertheless, these cells are viable unless apoptosis is induced, so the nuclease may not be active in the nucleus under nonapoptotic conditions (8).
EndoGI leaves the nucleus during apoptosis. However, since the affinity of the EndoG-EndoGI interaction is extremely high, the nuclease, on its way from the mitochondrion to the cell nucleus, would be expected to form a stable complex with the inhibitor; therefore, the relocalization of EndoGI from the nucleus to the cytoplasm is unlikely to be sufficient to allow EndoG activity. Possibilities remaining to be explored for the regulation of EndoGI activity during apoptosis include effects of 14-3-3 or proteolysis at a late stage of cell death. Although DNA degradation is initiated by nucleases within the apoptotic cell, the process is continued by lysosomal enzymes after the dying cell has been engulfed by phagocytosis and also by secreted nucleases if DNA has been released into the extracellular space (1, 2, 47). It is possible that inactivation of EndoGI to permit EndoG activity is not required until a late stage.
Supplementary Material
Acknowledgments
We are grateful to Stefan Hüttelmaier for advice on immunofluorescence experiments and for reading the manuscript. We thank Francois Juge, Elisa Izaurralde, and Efthimios Skoulakis for fly strains, plasmids, and reagents and Gregor Meiss and Alfred Pingoud for information on EndoG-like enzymes. The Wahle group acknowledges the generous help of Gunter Reuter and Thomas Rudolph with fly work. C. T. is grateful to Martine Simonelig for teaching her how to stain ovaries and embryos. A. M. thanks Maike Gebhardt for technical assistance.
This work was supported by grants from the Deutsche Forschungsgemeinschaft and the Fonds der Chemischen Industrie (to E. W.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement”in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. 1–5.
Footnotes
The abbreviations used are: EndoG, endonuclease G; EndoGI, EndoG inhibitor; GFP, green fluorescent protein; PBS, phosphate-buffered saline; DTT, dithiothreitol; TOF, time of flight; DAPI, 4′,6-diamidino-2-phenylindole; CAD, caspase-activated DNase; ICAD, inhibitor of caspase-activated DNase.
References
- 1.Parrish, J. Z., and Xue, D. (2006) Chromosoma 115 89–97 [DOI] [PubMed] [Google Scholar]
- 2.Samejima, K., and Earnshaw, W. C. (2005) Nat. Rev. Mol. Cell. Biol. 6 677–686 [DOI] [PubMed] [Google Scholar]
- 3.Widlak, P., and Garrard, W. T. (2005) J. Cell. Biochem. 94 1078–1087 [DOI] [PubMed] [Google Scholar]
- 4.Li, L. Y., Luo, X., and Wang, X. (2001) Nature 412 95–99 [DOI] [PubMed] [Google Scholar]
- 5.van Loo, G., Schotte, P., van Gurp, M., Demol, H., Hoorelbeke, B., Gevaert, K., Rodriguez, I., Ruiz-Carrillo, A., Vandekerckhove, J., Declerq, W., Beyaert, R., and Vandenabeele, P. (2001) Cell Death Differ. 8 1136–1142 [DOI] [PubMed] [Google Scholar]
- 6.Parrish, J., Li, L., Klotz, K., Ledwich, D., Wang, X., and Xue, D. (2001) Nature 412 90–94 [DOI] [PubMed] [Google Scholar]
- 7.Zhao, T., Zhang, H., Guo, Y., and Fan, Z. (2007) J. Biol. Chem. 282 12104–12111 [DOI] [PubMed] [Google Scholar]
- 8.Büttner, S., Eisenberg, T., Carmona-Gutierrez, D., Ruli, D., Knauer, H., Ruckstuhl, C., Sigrist, C., Wissing, S., Kollroser, M., Fröhlich, K.-U., Sigrist, S., and Madeo, F. (2007) Mol. Cell 25 233–246 [DOI] [PubMed] [Google Scholar]
- 9.Kühlmann, U. C., Moore, G. R., James, R., Kleanthous, C., and Hemmings, A. M. (1999) FEBS Lett. 463 1–2 [DOI] [PubMed] [Google Scholar]
- 10.Gerschenson, M., Houmiel, K. L., and Low, R. L. (1995) Nucleic Acids Res. 23 88–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Widlak, P., Li, L. Y., Wang, X., and Garrard, W. T. (2001) J. Biol. Chem. 276 48404–48409 [DOI] [PubMed] [Google Scholar]
- 12.Schäfer, P., Scholz, S. R., Gimadutinow, O., Cymerman, I. A., Bujnicki, J. M., Ruiz-Carrillo, A., Pingoud, A., and Meiss, G. (2004) J. Mol. Biol. 338 217–228 [DOI] [PubMed] [Google Scholar]
- 13.Wang, X., Yang, C., Chai, J., Shi, Y., and Xue, D. (2002) Science 298 1587–1592 [DOI] [PubMed] [Google Scholar]
- 14.Kalinowska, M., Garncarz, W., Pietrowska, M., Garrard, W. T., and Widlak, P. (2005) Apoptosis 10 821–830 [DOI] [PubMed] [Google Scholar]
- 15.Parrish, J., Yang, C., Shen, B., and Xue, D. (2003) EMBO J. 22 3451–3460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Irvine, R. A., Adachi, N., Shibata, D. K., Cassell, G. D., Yu, K., Karanjawala, Z. E., Hsieh, C.-L., and Lieber, M. R. (2005) Mol. Cell. Biol. 25 294–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.David, K. K., Sasaki, M., Yu, S.-W., Dawson, T. M., and Dawson, V. L. (2006) Cell Death Differ. 13 1147–1155 [DOI] [PubMed] [Google Scholar]
- 18.Parrish, J., and Xue, D. (2003) Mol. Cell 11 987–996 [DOI] [PubMed] [Google Scholar]
- 19.Bahi, N., Zhang, J., Llovera, M., Ballester, M., Comella, J. X., and Sanchis, D. (2006) J. Biol. Chem. 281 22943–22952 [DOI] [PubMed] [Google Scholar]
- 20.Ishihara, Y., and Shimamoto, N. (2006) J. Biol. Chem. 281 6726–6733 [DOI] [PubMed] [Google Scholar]
- 21.Niikura, Y., Dixit, A., Scott, R., Perkins, G., and Kitagawa, K. (2007) J. Cell Biol. 178 283–296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huang, K.-J., Ku, C.-C., and Lehman, I. R. (2006) Proc. Natl. Acad. Sci. U. S. A. 103 8995–9000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Büttner, S., Carmona-Gutierrez, D., Vitale, I., Castedo, M., Ruli, D., Eisenberg, T., Kroemer, G., and Madeo, F. (2007) Cell Cycle 6 1072–1076 [DOI] [PubMed] [Google Scholar]
- 24.Huang, K.-J., Zemelman, B. V., and Lehman, I. R. (2002) J. Biol. Chem. 277 21071–21079 [DOI] [PubMed] [Google Scholar]
- 25.Muo-Pastor, A. M., Herrero, A., and Flores, E. (1997) J. Mol. Biol. 268 589–598 [DOI] [PubMed] [Google Scholar]
- 26.Ghosh, M., Meiss, G., Pingoud, A., London, R. E., and Pedersen, L. C. (2007) J. Biol. Chem. 282 5682–5690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dickson, K. A., Haigis, M. C., and Raines, R. T. (2005) Prog. Nucleic Acids Res. Mol. Biol. 80 349–374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hartley, R. W. (1989) Trends Biochem. Sci. 14 450–454 [DOI] [PubMed] [Google Scholar]
- 29.Carr, S., Walker, D., James, R., Kleanthous, C., and Hemmings, A. M. (2000) Struct. 8 949–960 [DOI] [PubMed] [Google Scholar]
- 30.Sauter, C., Basquin, J., and Suck, D. (2003) Nucleic Acids Res. 31 4091–4098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Temme, C., Zaessinger, S., Simonelig, M., and Wahle, E. (2004) EMBO J. 23 2862–2871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Körner, C. G., Wormington, M., Muckenthaler, M., Schneider, S., Dehlin, E., and Wahle, E. (1998) EMBO J. 17 5427–5437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kerwitz, Y., Kühn, U., Lilie, H., Knoth, A., Scheuermann, T., Friedrich, H., Schwarz, E., and Wahle, E. (2003) EMBO J. 22 3705–3714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.LaGrandeur, T. E., and Parker, R. (1998) EMBO J. 17 1487–1496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fronz, K., Otto, S., Kölbel, K., Kühn, U., Friedrich, H., Schierhorn, A., Beck-Sickinger, A. G., Ostareck-Lederer, A., and Wahle, E. (2008) J. Biol. Chem. 283 20408–20420 [DOI] [PubMed] [Google Scholar]
- 36.Bönisch, C., Temme, C., Moritz, B., and Wahle, E. (2007) J. Biol. Chem. 282 21818–21828 [DOI] [PubMed] [Google Scholar]
- 37.Benoit, B., Nemeth, A., Aulner, N., Kühn, U., Simonelig, M., Wahle, E., and Bourbon, H.-M. (1999) Nucleic Acids Res. 27 3771–3778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Parks, A. L., Cook, K. R., Belvin, M., Dompe, N. A., Fawcett, R., Huppert, K., Tan, L. R., Winter, C. G., Bogart, K. P., Deal, J. E., Deal-Herr, M. E., Grant, D., Marcinko, M., Miyazaki, W. Y., Robertson, S., Shaw, K. J., Tabios, M., Vysotskaia, V., Zhao, L., Andrade, R. S., Edgar, K. A., Howie, E., Killpack, K., Milash, B., Norton, A., Thao, D., Whittaker, K., Winner, M. A., Friedman, L., Margolis, J., Singer, M. A., Kopczynski, C., Curtis, D., Kaufman, T. C., Plowman, G. D., Duyk, G., and Francis-Lang, H. L. (2004) Nat. Genet. 36 288–292 [DOI] [PubMed] [Google Scholar]
- 39.Rudolph, T., Lu, B., Westphal, T., Szidonya, J., Eissenberg, J., and Reuter, G. (1999) Drosophila Information Service 82 99–100 [Google Scholar]
- 40.Côté, J., and Ruiz-Carrillo, A. (1993) Science 261 765–769 [DOI] [PubMed] [Google Scholar]
- 41.Acevedo, S. F., Tsigkari, K. K., Grammenoudi, S., and Skoulakis, E. M. C. (2007) Genetics 177 239–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen, P., Nordstrom, W., Gish, B., and Abrams, J. M. (1996) Genes Dev. 10 1773–1782 [DOI] [PubMed] [Google Scholar]
- 43.Fic, J. W., Juge, F., Soret, J., and Tazi, J. (2007) PLoS ONE 2 e253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Franke, I., Meiss, G., and Pingoud, A. (1999) J. Biol. Chem. 274 825–832 [DOI] [PubMed] [Google Scholar]
- 45.Cymerman, I. A., Chung, I., Beckmann, B. M., Bujnicki, J. M., and Meiss, G. (2008) Nucleic Acids Res. 36 1369–1379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Miller, M. D., and Krause, K. L. (1996) Protein Sci. 5 24–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mukae, N., Yokoyama, H., Yokokura, T., Sakoyama, Y., and Nagata, S. (2002) Genes Dev. 16 2662–2671 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.