

Abstract
LP2086 is a family of outer membrane lipoproteins from Neisseria meningitidis, which elicits bactericidal antibodies and are currently undergoing human clinical trials in a bivalent formulation where each antigen represents one of the two known LP2086 subfamilies. Here we report the NMR structure of the recombinant LP2086 variant B01, a representative of the LP2086 subfamily B. The structure reveals a novel fold composed of two domains: a “taco-shaped” N-terminal β-sheet and a C-terminal β-barrel connected by a linker. The structure in micellar solution is consistent with a model of LP2086 anchored to the outer membrane bilayer through its lipidated N terminus. A long flexible chain connects the folded part of the protein to the lipid anchor and acts as spacer, making both domains accessible to the host immune system. Antibodies broadly reactive against members from both subfamilies have been mapped to the N terminus. A surface of subfamily-defining residues was identified on one face of the protein, offering an explanation for the induction of subfamily-specific bactericidal antibodies.
Neisseria meningitidis is a Gram-negative bacterial pathogen, which colonizes the upper respiratory tract, occasionally invading the bloodstream, causing sepsis, and crossing the blood-brain barrier, resulting in meningitis. Despite the availability of effective antibiotic treatment, the rapid progression of meningococcal disease still results in substantial morbidity and mortality (1). Five meningococcal serogroups, categorized according to the chemical structure of the bacterial capsular polysaccharides, A, B, C, Y, and W135, account for most of the disease (2). Although a vaccine against four of the five major serogroups of meningococci is currently available, a vaccine for the prevention of serogroup B disease is still an unmet clinical need (3). The development of vaccines against serogroup B meningococci has focused on subcapsular antigens, in order to avoid the risk of autoimmunity arising from structural similarities between the capsular polysaccharides and the sialic acid-modified surface of developing human brain (1, 4, 5).
Recently, a new family of lipidated outer membrane proteins, LP2086, was identified as a potential vaccine target (6). Members of the LP2086 family have been divided into two subfamilies, subfamily A and B, based on their genetic variation (6, 7). Since recombinant LP2086 (rLP2086)3 elicits a bactericidal response that is largely subfamily-specific, a bivalent vaccine containing one protein from each subfamily will offer protection against serogroup B meningococci (6, 8-11). LP2086 lipoproteins are lipidated at the N-terminal Cys with a tripalmitoyl lipid tail, which anchors the protein to the bacterial membrane (12). More recently, LP2086 was found to induce serum resistance via binding with human Factor H, a key regulator of the alternative complement pathway that prevents autologous complement attack (13).
Our work seeks to understand the structural elements of LP2086 responsible for inducing the subfamily specific immunological response. We determined the NMR structure of the full-length recombinant lipidated rLP2086-B01 in micellar solution, which revealed a novel protein fold comprising two domains. Alignment of 172 unique LP2086 sequences shows that invariant regions of the protein encompass the hydrophobic cores of two structural domains, suggesting conserved structural elements common for all LP2086 family members. Residues that define subfamily identity appear on one face of the protein that we propose would orient toward the extracellular space. Also, we identified a region of the protein that functions as an immunogenic determinant for two broadly reactive monoclonal antibodies. This work represents the first step in establishing the link between the structure and immunological properties of this protein and provides insights into the topology of the antigen relative to the bacterial surface.
MATERIALS AND METHODS
Expression and Purification—The rP2086 gene, cloned in the pET9a vector, was expressed in Escherichia coli BLR(DE3)-pLysS, as previously reported (6). For the uniformly 2H,15N,13C-labeled rP2086, cells were grown at 37 °C in 99% D2O M9 minimum media, enriched with 2 g/liter 15N-labeled ammonium sulfate, 2 g/liter [U-2H,13C]glucose (Cambridge Isotope Laboratories), and 50 μg of kanamycin. The protein was induced with 1 mm isopropyl β-d-thiogalactopyranoside and expressed for 14 h. The rLP2086 gene was cloned in a pBAD18Cm vector in E. coli BLR, and expression was induced with 1% l-arabinose. Protein selectively 1H,15N,13C-labeled at Leu on a deuterated background was produced as reported (14-16). The cell pellet was suspended in 10 mm HEPES buffer at pH 7.4 and lysed with 90 μm lysozyme. Cells were grown in M9 media supplemented with 2 g/liter 15N-labeled ammonium sulfate, 2 g/liter d7-glucose, 50 mg/liter 1H,15N,13C-leucine (Cambridge Isotope Laboratories) in 99% D2O. Expression was induced with 1 mm isopropyl β-d-thiogalactopyranoside and continued for 5 h. Nonlipidated rP2086 was purified following precipitation with 75% saturated ammonium sulfate by sequential chromatographic steps on cation (Poros HS50) and size exclusion (Superdex 75) columns. Highly concentrated protein was prepared by adsorption on a strong cation exchange column (Mono-S) and elution with a gradient of NaCl in 30 mm phosphate buffer at pH 4.8.
Sample Conditions—Final sample conditions for the nonlipidated protein were 0.5-1.0 mm LP2086-B01 in 30 mm sodium phosphate, 90 mm NaCl, pH 4.7, 0.03% NaN3, 5% D2O. For the lipidated protein sample, conditions were as follows: 0.5-1.0 mm, 30 mm sodium phosphate, 90 mm NaCl, 1% Zwittergent 3-12 (Calbiochem), pH 7.4, 0.03% NaN3.
NMR Spectroscopy—All NMR experiments were recorded at 30 °C on a Bruker Avance 700 MHz spectrometer equipped with a cryoprobe. Assignment of backbone resonances was performed through a standard combination of TROSY-based HNCA, HN(CO)CA, HNCO, and HN(CA)CO experiments (17, 18). The entire backbone was assigned with the exception of His32 (the assigned two-dimensional TROSY spectrum is reported in supplemental Fig. 2). α and β protons and carbons were identified using HNHA (19) and CBCA(CO)NH experiments (secondary structure and chemical shift index is reported in supplemental Fig. 1A). Approximately 90% of the side chain protons and carbons were assigned through a HCCH-TOCSY experiment. Distance constraints were obtained from three-dimensional 15N nuclear Overhauser effect spectroscopy (NOESY) HMQC, and 13C-edited NOESY, both recorded with mixing times of 100 ms. The overlap between the methyls in the hydrophobic cores was resolved through 15N NOESY on a sample where Leu residues were selectively labeled with 1H,15N, whereas the backbone was 15N-labeled on a deuterated background (14, 15). In this experiment, the Leu spin system displays both intra- and interresidue contacts with other Leu, whereas nonleucine residues only display interresidue NOEs contact with leucines. Backbone dynamics was studied, recording T1, T2 relaxation measurements and heteronuclear 1H-15N steady-state NOE experiments. R1 and R2 relaxation rates were obtained through exponential fitting of duplicate experiments recorded with delays ranging from 10 to 1300 ms for T1 and from 4 to 150 ms for the T2 experiment. Data were processed with NMRPipe/NMRDraw (21) and analyzed with Sparky (22). Molecular visualization and graphics where prepared with PyMol (23).
Quenching with Paramagnetic Reagents—Solvent-protected contacts between the two domains were confirmed through paramagnetic quenching with MnCl2 (0.3, 0.75, 2.9, and 7.25 mm) and 3-carboxy-proxyl (Aldrich) (2, 5, 10, and 15 mm), which induce selective line broadening of solvent-accessible residues. Since at identical pH, the two reagents carry opposite electrostatic charges, they are affected by opposite local electrostatic properties of the protein surface, allowing discrimination between the effects of steric protection from electrostatic repulsion (24). The side-chain HNε of Arg159 survives quenching with 3-carboxy-proxyl up to 15 mm and MnCl2 up to 7.5 mm and also retains its intensity in 60% D2O, suggesting that Arg159 is protected and probably hydrogen-bonded to the neighboring side chain of Thr157.
The topology of the protein with respect to the micelle was assessed using 5-doxyl-stearic acid (Aldrich) in concentrations of 0.5 and 1.0 mm. Deuterium oxide exchange experiments were performed through dialysis against 60% D2O, 30 mm sodium phosphate, 90 mm NaCl, pH 4.7. A total of 31 hydrogen bonds were obtained for the N-domain, and 57 were obtained for the C-domain.
Structural Determination in Micellar Solution—The structure of the nonlipidated protein was first determined at pH 4.7, without the detergent. Since the lipidated protein was unstable in Zwittergent 3-12 (Calbiochem) solution at this pH, it was studied at pH 7.4 in 30 mm Zwittergent 3-12. In order to extend the backbone assignment to the higher pH and to verify that the protein folding does not undergo pH-dependent structural changes, three-dimensional 15N-edited NOESY experiments were compared for the two conditions (nonlipidated LP2086-B01 at pH 4.7, in the absence of detergent, versus lipidated LP2086-B01 at pH 7.4 in detergent micelles). The pattern of NOE peaks for each residue was identical in the two conditions, for both number and intensity of the NOE cross-peaks, confirming that the protein folding is affected neither by the pH nor by the presence of the detergent. A few residues, mainly belonging to unstructured regions of the protein, were not observable at the higher pH due to the increased rate of solvent exchange.
Epitope Mapping by Differential Line Broadening—Differential line broadening was measured, recording HSQC spectra on a 200 μm sample of labeled rP2086-B01 in the presence of Fab MN86-440-18 at concentrations of 20 and 40 μm (antigen/Fab ratios of 10:1 and 5:1), in 30 mm sodium phosphate, 90 mm NaCl, pH 7.4, 0.03% NaN3, 5% D2O. The magnitude of line broadening Δ was measured as the decrease in peak intensities relative to the average intensity, as reported by Wagner and co-workers (25, 26),
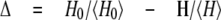 |
(Eq.1) |
where H and H0 are the peak intensities, and 〈H〉 and 〈H0〉 are the average peak intensities, in the presence and absence of monoclonal Fab fragment, respectively.
To ascertain the reproducibility of our results, three identical samples were prepared at each concentration, and three experiments were recorded on each sample. For each concentration, the values of Δ derived from a total of nine HSQCs were summed in order to reduce experimental noise. Attenuation of the random errors arising from the sample preparation and NMR measurements and the high reproducibility are evidenced by the low S.D. values observed for the residues experiencing above average line broadening. Experiments performed at the two concentrations produced identical results.
Structure Calculations—Structure calculations where performed employing 2113 interresidue and 1210 intraresidue NOEs from 15N-edited NOESY and 13C-edited NOESY spectra. NOE distances were calibrated, taking as a reference the intensities of characteristic NOEs of antiparallel β-sheets. Distances of 3.3, 3.0, and 2.2 Å were assigned to interstrand HNi-HNj, HNi-HAj, and HNi-HAi-1 NOEs, respectively (27). NOEs were grouped as strong, medium, weak, and very weak with equilibrium distances of 2.5, 3.0, 4.0, and 6.0 Å, respectively. Additional distance adjustments for pseudoatoms were applied as reported by Wuthrich and co-workers (28). 208 dihedral angles were calculated from backbone chemical shifts using the program TALOS (29). 88 hydrogen bond restraints were assigned for peaks that did not exchange back with H2O from the protein expressed in 100% deuterated media and from exchange experiments conducted in 60% D2O. Two distances were assigned for each hydrogen bond with equilibrium values of 1.9 and 2.9 Å. Notably, identical protein folds were achieved when the hydrogen bond constraints were omitted from the calculation, supporting the quality of the NOE assignment.
Standard input files of simulated annealing and refinement from the XPLOR-NIH package were used to generate an ensemble of 100 structures starting from an extended fold (30). 20 structures were selected with no NOE violations >0.5 Å, no dihedral angle violations >5°, and no violations in the covalent geometry. The final quality of the selected ensemble was checked using PROCHECK_NMR (31).
Differential Scanning Calorimetry—Differential scanning calorimetry measurements were recorded on the full-length LP2086-B01 and the recombinant C-terminal fragment 107-261, at concentrations of 14 μm, in 20 mm phosphate buffer, 150 mm NaCl, pH 7.0.
Flow Cytometry—Monoclonal antibodies used in flow cytometry experiments were obtained from mice immunized with rLP2086 subfamily A or B. Bacterial cells were grown to an A650 of 0.45-0.55 and then fixed in 1% paraformaldehyde for 10 min. The bacteria were then plated and washed in 1% bovine serum albumin/phosphate-buffered saline. Following the addition of primary antibody, cells were resuspended and incubated on ice for 30 min. Mouse IgG and MN86-994-11 were used as negative and positive control, respectively. After two washes in 1% bovine serum albumin/phosphate-buffered saline, biotinylated goat anti-mouse IgG was added to the cells and incubated on ice in the dark for 30 min. Cells were washed twice, resuspended in streptavidin-phycoerythrin, and incubated on ice in the dark for an additional 30 min. After two washes in 1% bovine serum albumin/phosphate-buffered saline, the cells were resuspended in 1% paraformaldehyde. Fluorescence intensity was recorded on a BD Biosciences LSR II flow cytometer, and data were analyzed with FlowJo software. The mean fluorescence intensity of the phycoerythrin channel was determined for each sample after gating on bacterial cells.
Peptide Competition—25 dodecapeptides overlapping by 10 amino acids derived from the LP2086-B01 sequence were synthesized (JPT Peptide Technologies). Each peptide was incubated with either MN86-440-18 or MN86-1075-6 antibodies for 30 min on ice. As a negative control, the antibodies were also incubated with an irrelevant cytomegalovirus peptide. LP2086-B01-expressing bacterial cells were then added to the peptide/antibody mixture and stained as reported above.
RESULTS
Solution Structure of the Nonlipidated LP2086 Variant B01—The NMR structure of rP2086-B01 was determined using 3323 homonuclear distances derived from 15N- and 13C-edited NOESY experiments. Comparison of the rP2086-B01 structure with the Protein Data Bank and the DALI server found no structural similarities suggesting a previously unidentified protein fold. rP2086-B01 is composed of 18 β-strands and two short α-helices arranged into two domains separated by a 15-residue linker (supplemental Fig. 1A). The ribbon diagram of a representative structure, the ensemble of the 20 lowest energy structures, and structural statistics are displayed in Fig. 1, A-D, and Table 1, respectively. The N-terminal domain (residues 1-139) consists of a “taco-shaped” antiparallel β-sheet, which folds on itself, enclosing a core of hydrophobic side chains. The β-sheet opens between strands β1 and β10, forming a groove, with three arginines (Arg134, Arg136, and Arg86) protecting the hydrophobic core from solvent exposure (Fig. 2A). Strands β3 and β4 are connected by a type II tight turn (residues 64-67) and are separated by a short amphipathic helix (α1). The C-terminal domain (residues 155-261) consists of an eight-stranded antiparallel β-barrel enclosing a hydrophobic core. The two domains are connected by a 15-residue linker (residues 140-154), which contains a short helical turn (α2) and crosses one end of the β-barrel, forming hydrophobic contacts through Phe147.
FIGURE 1.

Structure, electrostatic surface, and superposition of the backbone heavy atoms for the 20 lowest energy structures of LP2086-B01. A, ribbon representation of LP2086-B01 structure. B, overlay of the structured residues for the C-domain; r.m.s. deviation 0.61 Å (the structured regions span the following residues: β11, 155-164; β12, 171-176; β13, 181-188; β14, 197-206; β15, 212-220; β16, 224-233; β17, 239-249; β18, 252-261. The hydrophobic core in the C-domain comprises residues Tyr158, Tyr174, Tyr203, Tyr228, Ala156, Tyr158, Ala162, Leu172, Tyr174, Ile176, Phe178, Ile187, Val197, Leu199, Val201, Tyr203, Ala212, Ile214, Val218, Tyr220, Tyr228, Leu230, Ile232, Val240, Ala244, Val246, Ile255, Leu257, Ala259). C, overlay of the structured residues for the N-domain; r.m.s. deviation 0.77 Å (the structured regions span the following residues: α1, 21-28; β1, 39-41; β2, 51-55; β3, 59-63; β4, 67-69; β5, 81-87; β6, 97-105; β7, 111-116; β8, 119-121; β9, 128-131; β10, 133-139. The hydrophobic core in the N-domain comprises residues Val104, Leu115, Leu69, Leu54, Leu52, Leu42, Leu40, Leu26, and Phe102, Phe84, Phe82, Tyr63). D, overlay of the structured residues for both domains excluding the interdomain linker; r.m.s. deviation 0.96 Å. b-d, the unfolded N-terminal chain (residues 1-16) has been excluded for purposes of clarity. E, electrostatic surface representation for the entire LP2086-B01. Blue, positively charged residues; red, negatively charged residues; light gray, uncharged residues.
TABLE 1.
Experimental constraints and structural statistics for the 20 lowest energy structures of LP2086-B01
| Parameters | Values |
|---|---|
| Experimental restraintsa | |
| Total NOEs | 3323 |
| Intraresidue NOEs | 1210 |
| Interresidue NOEs | |
| Sequential | 933 |
| Medium range | 296 |
| Long range | 884 |
| Hydrogen bonds | 88 |
| Dihedral angles (TALOS) | 208 |
| r.m.s. deviations of residues in folded regionsb | |
| Overall backbone | 0.96 Å |
| N-terminal (residues 22-139) | 0.77 Å |
| C-terminal (residues 155-261) | 0.61 Å |
| Ramachandran statistics | |
| Residues in most favored regions | 62.1% |
| Residues in additionally allowed regions | 31.0% |
| Residues in generously allowed regions | 6.7% |
| Residues in disallowed regions | 0.3% |
Hydrogen bonds were identified from residues that did not exchange with 1H from a perdeuterated sample and from 2H2O exchange experiments. Two constraints were used for each hydrogen bond (dHN-O ≤ 2.2 Å dN-O ≤ 3.3 Å).
r.m.s. deviations are calculated from overlap of 20 final structures. None of the final structures has NOE violations exceeding 0.5 Å and dihedral angle violations above 5°.
FIGURE 2.

N-domain groove and interdomain hydrophobic patches. A, groove formed between strands β1 and β10 on the N-domain of LP2086-B01. Several charged residues protect the hydrophobic core from solvent exposure. B, electrostatic representation of the N- and C-domains separately showing the hydrophobic patches involved in the interaction between the two domains.
The surface of rP2086-B01 (Fig. 1E) is largely populated by charged residues except for two hydrophobic patches (Fig. 2B) located on the N-terminal domain and the C-terminal domain and a patch of five serines on the β-barrel. Long range NOEs and paramagnetic quenching experiments (3-carboxyproxyl and MnCl2) (24) indicate that the two hydrophobic patches are responsible for the interaction between the two domains. A few long range NOEs observed between the two domains lock the interdomain orientation. However, the low intensity of these constraints suggests that the relative orientation between the two domains experiences librational motions. Unlike the other arginines, the side-chain HNε of Arg159 survives quenching with high concentrations of paramagnetic reagents and retains its intensity in 60% D2O, suggesting that Arg159 is protected and probably hydrogen-bonded to the neighboring side chain of Thr157.
A structure comparison between the full-length rP2086-B01 and the C-terminal fragment of the sequence homolog GNA1870 from Neisserial strain MC58, determined previously by NMR (32), reveals that the two proteins have comparable folds at the C-terminal domains with an r.m.s. deviation of 2.5 Å for 73 structured Cα pairs. Differences are observed in the orientation of the short helix His144-Phe147 in the interdomain linker. Although in both cases the helix sits on an open end of the β-barrel, in GNA1870 it adopts a different orientation. This can be attributed to the truncation at residue 106 of the GNA1870 sequence, which unfolds the remaining portion of the N-terminal domain.
Different Backbone Flexibility of the rP2086-B01 Domains—Rates of solvent exchange with D2O are compatible with backbone hydrogen bonds of different strength for the two domains. Several residues at the C-terminal β-barrel on a perdeuterated sample did not exchange back with water during the protein purification, indicating that the β-barrel of the protein is structured around a network of rigid hydrogen bonds. Conversely, for the N-terminal domain, backbone amide protons exchange upon dialysis in 60% D2O, indicating a higher degree of backbone flexibility. The presence of two domains with different thermal stabilities is also confirmed by differential scanning calorimetry experiments, which show two thermal transitions at 59 and 82 °C for the N- and C-terminal domain, respectively (supplemental Fig. 2).
Heteronuclear steady-state 15N NOE experiments are in agreement with the D2O exchange behavior showing two ranges of fluctuation of the backbone dynamics for the C-terminal and the N-terminal domains (supplemental Fig. 1B). With the exception of one loop in the β-barrel (residues 248-252), the C-terminal domain has a narrow range of NOE relaxation values, with an average NOE above 0.8, indicative of a dynamically restricted backbone motion. For the N-terminal domain, on the other hand, 15N NOE relaxations fluctuate over a broader range. Although the regions enclosing the hydrophobic core have a degree of backbone rigidity comparable with the β-barrel, most of the N-terminal domain has heteronuclear 15N NOE values below 0.7, indicating higher backbone mobility. In the interdomain linker, only two residues, Val154 and Asp153, have high backbone mobility. The most rigid residue in the linker is Phe147, as expected from the contacts observed between this aromatic ring and the β-barrel.
Lipidated LP2086-B01 Is Anchored to the Micelle Surface—The N-terminal Cys residue of the mature bacterial lipoprotein is covalently modified with a tripalmitoyl-S-Cys moiety (Pam3Cys), which serves as an anchor to the bacterial membrane. To characterize the topology of the lipidated protein with respect to the membrane bilayer, we first determined the structure of the nonlipidated recombinant rP2086-B01 and then compared it with a three-dimensional 15N-edited NOESY for the lipidated version rLP2086-B01, in 30 mm Zwittergent 3-12. Under these two conditions, the pattern of NOE peaks for each residue is identical in both number and intensity of the NOE cross-peaks, confirming that the protein fold is not affected by the presence of the micelle and the lipid anchor. We titrated the lipidated rLP2086-B01 with the paramagnetic reagent 5-doxyl stearic acid, which reconstitutes into the micelle, inducing selective paramagnetic broadening of residues bound to the micellar surface (33, 34). With the exception of Cys1 and Gly2 (supplemental Fig. 3), the spectrum remained unaffected by the presence of the paramagnetic probe, confirming that the folded portion of rLP2086-B01 does not interact with the surface of the micelle. As final evidence that rLP2086-B01 does not interact with the detergent either in its monomeric or micellar form, we titrated Zwittergent 3-12 to the nonlipidated rP2086-B01, from concentrations below the critical micellar concentration to concentrations above the critical micellar concentration (2.5 μm to 30.9 mm). No change in the 1H-15N HSQC was observed, confirming our earlier findings that the protein fold is not affected by the presence of the detergent.
Our NMR data for the lipidated protein in micellar solution are consistent with a model of LP2086-B01 bound to the micelle through its N-terminal lipid chain, with no direct contacts between the two folded domains and the surface of the micelle.
Structural Conservation across the LP2086 Family—Based on their primary sequence, proteins of the LP2086 family were divided into two subfamilies, A and B, with a pairwise percentage identity of 83-99% within each subfamily and 60-75% between the two subfamilies (6).4 The NMR structure and sequence alignment of 172 natural LP2086 sequence variants4 suggest conserved structural features common to most of the LP2086 family members (supplemental Fig. 4). Residues forming the hydrophobic cores and the contact patches between the two domains are invariant (with the exception of R159H and T161K), suggesting that most of the LP2086 variants may consist of two domains enclosing hydrophobic cores.
An insertion of three charged residues, KDN, at position 68 of the N-terminal domain is observed in 50% of subfamily A variants and in only seven variants of subfamily B (magenta residues in supplemental Fig. 4). This position corresponds to the beginning of strand β4, close to the rigid type II turn. Additionally, one residue, Lys152 in the interdomain linker of subfamily B, is missing in every subfamily A variant.4
As expected, the Cys in position 1 is present in every LP2086 variant in order to link the Pam3Cys group required to anchor the protein to the bacterial membrane. Interestingly, a Gly-Serrich region at the N terminus is highly conserved across the two subfamilies, although members of the LP2086 family have different lengths of the N-terminal chain, possibly due to the need to accommodate the variation in length and composition of the LOS in the outer membrane.
Mapping Monoclonal Antibody Binding Sites to the N-terminal Domain—Whole cell enzyme-linked immunosorbent assay and flow cytometry show that polyclonal antibodies raised against individual LP2086 variants are largely subfamily-specific. In order to identify the structural epitopes responsible for inducing family and subfamily-specific antibodies, we selected two mAbs from our collection with binding affinity for multiple LP2086 variants. MN86-1075-6 was selected due to its broad reactivity with most of the variants from both subfamilies A and B. MN86-440-18, a subfamily B-specific monoclonal antibody, was selected, since it binds most of the subfamily B variants, but it is insensitive to all subfamily A proteins in flow cytometry experiments. For MN86-1075-6, sequence alignment of 172 unique naturally occurring LP2086 variants4 and competitive flow cytometry with overlapping linear dodecapeptides identified one linear epitope on the N-terminal domain containing loop Glu89-Gln93 (between strands β5 and β6) (Fig. 3A). Within this loop, each variant recognized by MN86-1075-6 contains the sequence EVDGQ (supplemental Fig. 4).
FIGURE 3.

Flow cytometry analysis and peptide binding competition to LP2086-B01 bacterial cells. A, binding of the broad-spectrum monoclonal MN86-1075-6 raised against LP2086-B01 is inhibited by a peptide spanning sequence Phe82-Glu101. B, subfamily B-specific monoclonal MN86-440-18 raised against 8529 has reactivity for LP2086-B01 meningococcal cells but is not competed by a peptide spanning the sequence Phe82-Glu101. C, monoclonal antibodies MN86-813 and MN86-626-15, which cross-react with rP2086 from both subfamilies, do not recognize LP2086-B01 on the bacterial surface. Mouse IgG and the broad-spectrum monoclonal MN86-994-11 are used as negative and positive control, respectively.
Antigen-antibody interactions occurring with micromolar affinities can be studied by NMR by titrating the unlabeled mAb to the 15N-labeled antigen and mapping residues experiencing above average line broadening to the binding interface (25, 26). Unlike other antibodies that bind with nanomolar affinity, MN86-440-18 binds to rP2086-B01 with a KD of 1.6 μm. We identified the structural regions affected by the subfamily B-specific monoclonal MN86-440-18 by observing the effects of binding a Fab fragment derived from MN86-440-18 through differential line broadening experiments and calculating values of Δ as reported under “Materials and Methods.” Residues that are most strongly involved in binding interactions will exhibit the higher Δ. Most of the residues affected by the binding are located on the N-terminal domain (Fig. 4A) and appear to overlap with the same residues contained in the linear epitope recognized by the broadly reactive antibody MN86-1075-6. At a 10:1 antigen/Fab ratio, the most affected residues are Leu94, Glu89, and Gly50 (Δ= 2.41 ± 0.11, 1.24 ± 0.06, and 1.32 ± 0.13), which form a small patch on the neighboring strands β2, β5, and β6 (Fig. 4B). In their vicinity, the type II turn, the hydrophobic face of helix α1, and the flanking strands β3 and β4 are also broadened (Δ in the range of 0.31 ± 0.08 for Tyr63 to 0.85 ± 0.05 for Gly66). At higher concentration of Fab (antigen/Fab ratio of 5:1), identical results are obtained with the addition of residues Glu124 and His125 (0.70 ± 0.04 and 1.29 ± 0.11) on the neighboring loop between strands β8 and β9. MN86-440-18 does not compete with a peptide spanning sequence Phe82 to Glu101 in competitive flow cytometry experiments, confirming that it requires a structural binding epitope comprising non-contiguous residues on neighboring loops (Fig. 3B). Interestingly, residues at the contact interface between the two domains are also broadened, indicating that the interdomain interaction is affected by the binding. In particular, Val104, Leu112, and Thr113 on the N-domain and Gly160 and Val240 on the C-domain experience above average line broadening and are part of the contact faces between the two domains. On the interdomain linker, Ser146 and His144 are the most affected residues together with Glu208 and Gly234 on the nearby loops. Although most of the residues in the C-domain experience below average line broadenings, Thr175 and Asp198 on the opposite side of the β-barrel have values of Δ of 1.18 ± 0.05 and 0.69 ± 0.07, suggesting that the binding may allosterically affect remote parts of the protein.
FIGURE 4.

Binding epitope of MN86-440-18 displayed on the surface of LP2086-B01. A, at an antigen/Fab ratio of 10:1, the most affected residues are Leu94, Glu89, and Gly50. These residues form a small patch on one face of the N-domain. At higher concentrations of antibody (antigen/Fab ratio of 5:1), residues Glu124 and His125 on the nearby loop are also affected substantially. The reported values are the summation over a total of nine experiments conducted on three identical samples. Positive values of Δ (red) and negative values of Δ (green) correspond to residues experiencing above average and below average line broadening upon binding. Residues in white either have a value of Δ between the cut-offs of -1.0 and 1.0 or are overlapped. B, the position of the KDN, a common insertion in the A subfamily, is indicated. In this region of the protein, strands β3 and β4, which separate the type II turn, and the hydrophobic face of α1 between the two strands experience above average line broadening upon binding with the Fab fragment 440-18.
Two Functional Faces on the LP2086-B01 Structure—In an initial sequence analysis, the highest degree of sequence similarity between the two subfamilies was observed on the N-domain, whereas the less conserved C-domain (residues 154-261) was suggested to determine the specificity for the two subfamilies (7). Recently, sequence alignment of 172 naturally occurring LP2086 variants identified two groupings of signature residues that are responsible for the subfamily identity and subdomain identity.4 Mapping these signature residues on the structure shows that the two groups are distributed over two opposite faces of the protein involving both domains (Fig. 5). Many of the subfamily-defining residues are located in one region of the C-terminal β-barrel, supporting the initial hypothesis that the C-domain contributes to the subfamily specificity. However, the presence of subfamily-defining residues on the N-domain indicates that this domain is also involved in defining the two subfamilies. Residues that are specific for either the A or B subfamily are surface-exposed and hydrophilic (with the exception of Leu257, Ile232, Ala259, and Val218, which are embedded inside the β-barrel). On the β-barrel, subfamily-defining residues alternate with the invariant hydrophobic residues orienting the hydrophobic side chains toward the barrel interior (Fig. 5, A and D). Similarly, the side chains of each subdomain-defining residue (with the exception of Tyr63) are exposed on the surface of the protein and are not involved in forming the hydrophobic cores. Subdomain-defining residues form two separate patches on the two domains (green in Fig. 5, E and F). On the N-terminal domain, loops Glu43-Thr51 and Tyr63-Gly66 (separating strands β1-β2 and β3-β4) and residues on the neighboring strand β5 form a large cluster, which includes the type II turn and partly extends into the region affected by the binding with MN86-440-18 (Fig. 5, B and E). On the C-terminal domain, a smaller patch of subfamily-defining residues involves neighboring strands β11-β14 (Fig. 5F).
FIGURE 5.

Mapping of the signature residues as derived4 by aligning 172 known LP2086 sequences. Red, invariant residues; green, subdomain-defining residues; yellow, subfamily-defining residues. A and D, subfamily-defining residues (yellow) populate one face of the protein, involving both domains. Many of these residues are located at the C-domain and alternate the conserved (red) hydrophobic residues that form the β-barrel hydrophobic core. B and C and E and F, on the opposite side of the protein, the subdomain-defining residues (green) form two separate patches on the two domains: on the N-domain, loops Glu43-Thr51 and Tyr63-Gly66 and residues on strands β2, β3, and β5; on the C-domain, neighboring residues on strands β11-β14. A and E, the MN86-1075-6 and MN86-440-18 binding regions are indicated in purple and blue, respectively. The MN86-440-18 recognizes a discontinuous epitope, which extends into regions containing subfamily-defining and subdomain-defining signature residues. MN86-1075-6 maps at a linear sequence comprising the loop Glu89-Gln93 separating strands β5 and β6, whereas MN86-440-18 induces line broadening at residues Leu94, Glu89, Gly50, Glu124, and His125, forming a patch on neighboring strands. In all models, the unfolded N-terminal chain (residues 1-16) has been excluded.
Topology of LP2086 Relative to the Outer Membrane—Proper exposure of the immunogenic determinants to the host immune system is a major factor affecting the immunological properties of outer membrane antigens. Mechanisms of humoral evasion can involve masking of functional epitopes (35), and for meningococcal porins, membrane-bound lipopolysaccharides with different chain lengths affect the accessibility of epitopes that bind porin-specific antibodies (36). Two monoclonals, MN86-626-15 and MN86-813, recognize the linear epitopes Gly21-Leu30 and Gly21-Leu40 in the free rP2086-B01 but do not bind to whole meningococcal cells in flow cytometry experiments, suggesting that the corresponding epitopes are not exposed on the bacterial surface (Fig. 3C). The inability of these antibodies to recognize LP2086 on the bacterial cell suggests a topology in the outer membrane such that the epitopes recognized by these monoclonals are oriented toward the membrane surface (violet residues in Fig. 6). Since the mAbs MN86-440-18 and MN86-1075-6 recognize both the purified antigen and the antigen bound to the bacterial outer membrane, we suggest a model in which LP2086 is anchored to the bacterial outer membrane with the loops Glu89-Leu94 and Asp122-Glu127 projected toward the extracellular space, whereas the recognition sequence of MN86-626-15 and MN86-813 (Gly21-Leu40) is oriented toward the bacterial surface (Fig. 6). This orientation would project the subfamily-defining face and the N-terminal groove toward the extracellular space, making it accessible to the host complement system. Our data in micellar solution indicate that the N-terminal chain of LP2086 is long enough to span the lipooligosaccharide layer. The N-terminal chain (which is unstructured in our model) may be partially folded if it interacts with the LOS or capsule, effectively shortening the chain that tethers the protein to the bacterial surface.
FIGURE 6.

Proposed model representing LP2086-B01 in the bacterial membrane. The flexible linker between residues 1 and 19 allows the protein to span the LOS layer of the outer membrane. Subfamily-defining residues (yellow) populate one region on one face of the protein oriented toward the extracellular space. In violet, the linear epitopes for the monoclonals MN86-626-15 and MN86-813 are oriented toward the membrane surface and are not accessible by these mAbs in flow cytometry experiments. In green, the binding epitopes for the monoclonals MN86-440-18 and MN86-1075-6 are also exposed to the extracellular space. A single LOS molecule is indicated in red in the model. The carbohydrate torsional angles have been manually set to achieve maximum extension of the headgroup.
DISCUSSION
The potential of a bivalent LP2086-based vaccine that offers broad protection against serogroup B meningococci has led to the initiation of clinical trials, which are currently in phase 2. The solution structure of rLP2086-B01 reveals a novel fold and provides interesting insights into the immunogenic properties of the LP2086 outer membrane protein family. The presence of highly conserved residues in the hydrophobic cores and the interdomain contacts suggests structural features common across both LP2086 subfamilies. Subfamily-defining and subdomain-defining signature residues populate opposite sides of the structure, revealing two functional faces of the protein that span both domains. Our model suggests that the subfamily-defining face and the N-domain groove are oriented toward the extracellular space, where they are the targets of antibodies that initiate the complement-mediated bacteriolysis in a subfamily-specific manner. In this context, the subfamily-defining surface may be structurally conserved across the LP2086 variants.
Two broad-spectrum mAbs target overlapping regions on the N-domain, suggesting an antigenic determinant exposed to the extracellular space. The subfamily B-specific monoclonal MN86-440-18 is directed to a conformational epitope that includes both subfamily-defining residues and subdomain-defining residues, explaining its specificity for the subfamily B variants. A second antibody, MN86-1075-6, with reactivity to most of the variants from both LP2086 subfamilies, binds at a linear epitope on the same loop targeted by MN86-440-18 recognizing the intervening sequence Glu89-Gln93. Unlike MN86-440-18, the binding epitope of MN86-1075-6 does not contain subdomain signature residues, and it is competed by the Phe82-Glu101 peptide (Fig. 3, A and B), explaining its broader reactivity to LP2086 variants belonging to both subfamilies. The nature of these binding epitopes is consistent with previous structural analysis of antigen-antibody complexes, which reveal contact surfaces limited to as few as three critical residues, located on flexible loops, that contribute to most of the change in the binding free energy (20, 37-40). These residues are generally surrounded by other residues that are energetically less important for the interaction. Not surprisingly, the binding epitopes are located on flexible regions of LP2086-B01 (supplemental Fig. 1B), suggesting that the binding with the mAb proceeds through an induced fit mechanism, which requires a structural rearrangement of the epitope residues.
The NMR structure of rLP2086-B01 in micellar solution provides an initial model to elucidate the architecture of this family of proteins in the bacterial outer membrane. The specific interaction of LP2086 with the capsule, LOS, and other outer membrane proteins is an area of future research. Future studies are also required to link the antigen topology to its role in modulating the host-pathogen interaction via binding with human factor H.
Supplementary Material
Acknowledgments
We thank Christopher Levins, Lidia Mosyak, and Will Somers for useful discussion on the manuscript. We also thank Ronald Kriz, Laura Lin, Mark Stahl, Wayne Stochaj, Amy Tam, and May Tam for help with the expression of isotopically labeled protein.
The atomic coordinates and structure factors (code 2KDY) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
The nucleotide sequence(s) reported in this paper has been submitted to the GenBank™/EBI Data Bank with accession number(s) AY330406, AY330361, AY330357, AY330398, AY330362, AY330373, and FJ184157.
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. 1-4.
Footnotes
The abbreviations used are: rLP2086, lipidated recombinant LP2086; rP2086, nonlipidated recombinant LP2086; Pam3Cys, N-α-palmitoyl-S-[2,3-bis(palmitoyloxy)-(2RS)-propyl]-l-cysteine; 3-carboxy-proxyl, 3-carboxy-2,2,5,5-tetramethyl-1-pyrolidinyloxy; LOS, lipooligosaccharides; NOE, nuclear Overhauser effect; NOESY, NOE spectroscopy; HSQC, heteronuclear single quantum coherence; mAb, monoclonal antibody; Zwittergent 3-12, 3-(dodecyldimethylammonio)-propanesulfonate; TROSY, transverse relaxation optimized spectroscopy.
E. Murphy, L. Andrew, K. L. Lee, D. A. Dilts, L. Nunez, P. S. Fink, K. Ambrose, R. Borrow, J. Findlow, M. K. Taha, A. E. Deghmane, P. Kriz, M. Musilek, J. Kalmusova, D. A. Caugant, T. Alvestad, L. W. Mayer, C. T. Sacchi, X. Wang, D. Martin, A. von Gottberg, M. du Plessis, K. P. Klugman, A. S. Anderson, K. U. Jansen, G. W. Zlotnick, and S. K. Hoiseth, manuscript in preparation.
References
- 1.Morley, S., and Pollard, A. (2002) Vaccine 20 666-687 [DOI] [PubMed] [Google Scholar]
- 2.Rosenstein, N., Perkins, B., Stephens, D., Popovic, T., and Hughes, J. (2001) N. Engl. J. Med. 344 1378-1388 [DOI] [PubMed] [Google Scholar]
- 3.Nassif, X. (2000) Science 287 1767-1768 [DOI] [PubMed] [Google Scholar]
- 4.Jódar, L., Feavers, I. M., Salisbury, D., and Granoff, D. M. (2002) Lancet 359 1499-1508 [DOI] [PubMed] [Google Scholar]
- 5.Poolman, J. T. (1995) Infect. Agents Dis. 4 13-28 [PubMed] [Google Scholar]
- 6.Fletcher, L. D., Bernfield, L., Barniak, V., Farley, J. E., Howell, A., Knauf, M., Ooi, P., Smith, R. P., Weise, P., Wetherell, M., Xie, X., Zagursky, R., Zhang, Y., and Zlotnick, G. W. (2004) Infect. Immun. 72 2088-2100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pillai, S., Howell, A., Alexander, K., Bentley, E. B., Jiang, H. Q., Ambrose, K., Zhu, D., and Zlotnick, G. (2005) Vaccine 23 2206-2209 [DOI] [PubMed] [Google Scholar]
- 8.Giuliani, M. M., Adu-Bobie, J., Comanducci, M., Aricò, B., Savino, S., Santini, L., Brunelli, B., Bambini, S., Biolchi, A., Capecchi, B., Cartocci, E., Ciucchi, L., Di Marcello, F., Ferlicca, F., Galli, B., Luzzi, E., Masignani, V., Serruto, D., Veggi, D., Contorni, M., Morandi, M., Bartalesi, A., Cinotti, V., Mannucci, D., Titta, F., Ovidi, E., Welsch, J., Granoff, D., Rappuoli, R., and Pizza, M. (2006) Proc. Natl. Acad. Sci. U. S. A. 103 10834-10839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Masignani, V., Comanducci, M., Giuliani, M. M., Bambini, S., Adu-Bobie, J., Arico, B., Brunelli, B., A, A. P., L, L. S., Savino, S., Serruto, D., Litt, D., Kroll, S., Welsch, J. A., Granoff, D. M., R, R. R., and Pizza, M. (2003) J. Exp. Med. 197 789-799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhu, D., Zhang, Y., Barniak, V., Bernfield, L., Howell, A., and Zlotnick, G. (2005) Infect. Immun. 73 6838-6845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhu, D., Barniak, V., Zhang, Y., Green, B., and Zlotnick, G. (2006) Vaccine 24 5420-5425 [DOI] [PubMed] [Google Scholar]
- 12.Lex, A., Wiesmüller, K. H., Jung, G., and Bessler, W. G. (1986) J. Immunol. 137 2676-2681 [PubMed] [Google Scholar]
- 13.Madico, G., Welsch, J. A., Lewis, L. A., McNaughton, A., Perlman, D. H., Costello, C. E., Ngampasutadol, J., Vogel, U., Granoff, D. M., and Ram, S. (2006) J. Immunol. 177 501-510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Metzler, W., Wittekind, M., Goldfarb, V., Mueller, L., and II, B. T. F. (1996) J. Am. Chem. Soc. 118 6800-6801 [Google Scholar]
- 15.Crespi, H. L., Rosenberg, R. M., and Katz, J. J. (1968) Science 161 795-796 [DOI] [PubMed] [Google Scholar]
- 16.Markley, J. L., Putter, I., and Jardetzky, O. (1968) Science 161 1249-1251 [DOI] [PubMed] [Google Scholar]
- 17.Nietlispach, D. (2005) J. Biomol. NMR 31 161-166 [DOI] [PubMed] [Google Scholar]
- 18.Pervushin, K., Riek, R., Wider, G., and Wüthrich, K. (1997) Proc. Natl. Acad. Sci. 94 12366-12371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vuister, G. W., and Bax, A. (1993) J. Am. Chem. Soc. 115 7772-7777 [Google Scholar]
- 20.Decanniere, K., Transue, T., Desmyter, A., Maes, D., Muyldermans, S., and Wyns, L. (2001) J. Mol. Biol. 313 473-478 [DOI] [PubMed] [Google Scholar]
- 21.Delaglio, F., Grzesiek, S., Vuister, G. W., Zhu, G., Pfeifer, J., and Bax, A. (1995) J. Biomol. NMR 6 277-293 [DOI] [PubMed] [Google Scholar]
- 22.Goddard, T. D., and Kneller, D. G.
- 23.DeLano, W. L. (2000) PyMol, Delano Scientific, San Carlos, CA [DOI] [PubMed]
- 24.Deschamps, M. L., Pilka, E. S., Potts, J. R., Campbell, I. D., and Boyd, J. (2005) J. Biomol. NMR 31 155-160 [DOI] [PubMed] [Google Scholar]
- 25.Walters, K., Gassner, G., Lippard, S., and Wagner, G. (1999) Proc. Natl. Acad. Sci. 96 7877-7882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Matsuo, H., Walters, K., Teruya, K., Tanaka, T., Gassner, G., Lippard, S., Kyogoku, Y., and Wagner, G. (1999) J. Am. Chem. Soc. 121 9903-9904 [Google Scholar]
- 27.Hwang, P. M., Choy, W.-Y., Lo, E. I., Chen, L., Forman-Kay, J. D., Raetz, C. R., Privé, G. G., Bishop, R. E., and Kay, L. E. (2002) Proc. Natl. Acad. Sci. U. S. A. 99 13560-13565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wuthrich, K., Billeter, M., and Braun, W. (1983) J. Mol. Biol. 169 949-961 [DOI] [PubMed] [Google Scholar]
- 29.Cornilescu, G., Delaglio, F., and Bax, A. (1999) J. Biomol. NMR 13 289-302 [DOI] [PubMed] [Google Scholar]
- 30.Schwieters, C., Kuszewski, J., Tjandra, N., and Clore, G. (2003) J. Magn. Reson. 160 66-74 [DOI] [PubMed] [Google Scholar]
- 31.Laskowski, R., Rullman, J., MacArthur, M., Kaptein, R., and Thornton, J. (1996) J. Biomol. NMR 8 477-486 [DOI] [PubMed] [Google Scholar]
- 32.Cantini, F., Savino, S., Scarselli, M., Masignani, V., Pizza, M., Romagnoli, G., Swennen, E., Veggi, D., Banci, L., and Rappuoli, R. (2006) J. Biol. Chem. 281 7220-7227 [DOI] [PubMed] [Google Scholar]
- 33.Buck-Koehntop, B. A., Mascioni, A., Buffy, J. J., and Veglia, G. (2005) J. Mol. Biol. 354 652-665 [DOI] [PubMed] [Google Scholar]
- 34.Hilty, C., Wider, G., Fernández, C., and Wüthrich, K. (2004) Chembiochem 5 467-473 [DOI] [PubMed] [Google Scholar]
- 35.Zhou, T., Xu, L., Dey, B., Hessell, A., Van Ryk, D., Xiang, S., Yang, X., Zhang, M., Zwick, M., Arthos, J., Burton, D., Dimitrov, D., Sodroski, J., Wyatt, R., Nabel, G., and Kwong, P. (2007) Nature 445 732-737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bentley, A. T., and Klebba, P. E. (1988) J. Bacteriol. 170 1063-1068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sharon, M., Kessler, N., Levy, R., Zolla-Pazner, S., Görlach, M., and Anglister, J. (2003) Structure 11 225-236 [DOI] [PubMed] [Google Scholar]
- 38.Rosen, O., Chill, J., Sharon, M., Kessler, N., Mester, B., Zolla-Pazner, S., and Anglister, J. (2005) Biochemistry 44 7250-7258 [DOI] [PubMed] [Google Scholar]
- 39.Sundberg, E., and Mariuzza, R. (2002) Adv. Protein Chem. 61 119-160 [DOI] [PubMed] [Google Scholar]
- 40.Li, Y., Urrutia, M., Smith-Gill, S., and Mariuzza, R. (2003) Biochemistry 42 11-22 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.