

Abstract
Prion diseases are associated with the accumulation of an abnormal form of the host‐coded prion protein (PrP). It is postulated that different tertiary or quaternary structures of infectious PrP provide the information necessary to code for strain properties. We show here that different light microscopic types of abnormal PrP (PrPd) accumulation found in each of 10 sheep scrapie cases correspond ultrastructurally with abnormal endocytosis, increased endo‐lysosomes, microfolding of plasma membranes, extracellular PrPd release and intercellular PrPd transfer of neurons and/or glia. The same accumulation patterns of PrPd and associated subcellular lesions were present in each of two scrapie strains present, but they were present in different proportions. The observations suggest that different trafficking pathways of PrPd are influenced by strain and cell type and that a single prion strain causes several PrPd–protein interactions at the cell membrane. These results imply that strains may contain or result in production of multiple isoforms of PrPd.
Keywords: endocytosis, prion disease, scrapie, stroin
INTRODUCTION
Scrapie of sheep is the prototype of a group of fatal neurological diseases called transmissible spongiform encephalopathies (TSEs) or prion diseases that include bovine spongiform encephalopathy (BSE) of cattle and Creutzfeldt–Jakob disease of man. The cause of the TSEs or prion diseases is still a matter of some debate, but most investigators now favor the prion hypothesis 40, 41, which postulates that the cause of prion diseases is an abnormal form of a host‐coded, cell surface, sialoglycoprotein called prion protein (PrP). For many years, a major challenge to the prion hypothesis has been to explain how an abnormal protein could code for the existence of numerous, stable, well‐characterized rodent scrapie strains. According to this hypothesis, strain‐specific information is encoded within the conformation of different tertiary or quaternary structures of an infectious isoform of PrP 41, 46.
Murine scrapie strains are characterized according to their relative incubation periods in different inbred mouse lines, different patterns of pathology and immunochemical forms of abnormal PrP accumulation 3, 43. A smaller number of experimental sheep scrapie strains have been recognized by their relative incubation times in different PrP genotypes (18). Biochemical methods are not readily able to distinguish between natural scrapie sources, although two experimental strains, CH1641 and BSE, have distinctive molecular phenotypes 17, 26; however, both experimental and naturally occurring scrapie sources may be distinguished by pathological phenotypes (14). The specific parameters of the pathological phenotype that permit differentiation of strains differs for sheep and mice: different murine strains show marked variation in vacuolation and neuroanatomic targeting (4), but sheep scrapie strains and sources mainly affect the same brain areas and neurons (14). Following immunohistochemical labeling, individual sheep strains and sources show highly diverse morphological types of disease‐associated PrP (PrPd) accumulation (14), whereas cloned murine strains have fewer types of PrPd accumulation. Although the PrP genotype may affect the magnitude of PrPd accumulation in sheep, the patterns and types of PrPd accumulation are not influenced by breed or route of inoculation 15, 35. From studies of the proportions of different types of PrPd accumulation (the PrPd profile), we have proposed that the pathology phenotype of sheep strains are partly the result of differences in the relative affinity of different sheep scrapie agents for different cell types and by differences in the cellular processing of PrPd (15). In the present study, we have sought to determine the nature of these putative processing or trafficking pathways of PrPd in different sheep scrapie sources.
In previous electron microscopy immunogold studies of rodent scrapie, we have shown that PrPd accumulates in the extracellular space and at the plasma membrane of neurons, glial cells and follicular dendritic cells 21, 25, 37. The degree to which the plasma membrane and extracellular forms show a tendency to aggregate and form fibrils differs between strains and tissues, but no differences in the nature of the changes in neurons of different brain areas were found. In contrast, variation in neuronal pathology both within and between neuroanatomic sites may be found for different sheep scrapie sources 7, 8. To determine whether there are different processing pathways for PrPd sheep scrapie strains, we first determined the in vivo sites of accumulation and processing of PrPd in different naturally occurring sheep scrapie sources and then investigated possible molecular mechanisms for some of these changes in brain and in lymphoid tissues (the latter is reported separately in reference 36). Our results suggest that varying proportions of several functional conformers are present in the brains of different sheep scrapie strains.
MATERIALS AND METHODS
The sheep included in the study and the methods of tissue fixation have been described previously (7). Briefly, ten clinically affected sheep with natural scrapie and four uninfected control animals from New Zealand were perfused under deep surgical anaesthesia with mixed aldehydes and post‐fixed in osmium tetroxide. PrP genotypes and farms of origin are listed in Table 1. The polymorphisms at codons 136, 154 and 171 in the ovine PrP gene are indicated with the appropriate amino acid single‐letter code.
Table 1.
Sheep genotypes, farm of origin and presence or absence of selected light and electron microscopy lesions. Abbreviation: PrP = prion protein; WMAS = white matter astrocyte associated PrPd.
| Sheep genotype | Farm | Light microscopy | Electron microscopy* | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Vascular plaques | Stellate | WMAS | PrPd + ve neuronal/dendrites | PrPd + ve pits and membrane changes | spiral inclusions (PrPd + or − ve) | PrPd + ve astrocytic membrane proliferation | PrPd + ve lysosomes | PrPd − ve reactive astrocytosis | ||
| R881 ARQ/ARQ | A | − | +++ | ++ | +++ | +++ | + | + | ++ | +++ |
| R883 ARQ/ARQ | A | − | ++ | ++ | +++ | +++ | + | + | ++ | +++ |
| R884 ARQ/AHQ | A | − | ++ | ++ | +++ | +++ | + | ++ | +++ | +++ |
| R882 VRQ/ARQ | A | − | ++ | ++ | +++ | +++ | + | ++ | ++ | ++ |
| R789 ARH/ARH | B | − | ++ | ++ | +++ | +++ | + | + | +++ | +++ |
| R816 VRQ/ARQ | C | + | + | ++ | +++ | ++ | +/− | +++ | + | + |
| R817 VRQ/ARQ | C | + | + | + | +++ | + | − | +++ | + | + |
| R790 VRQ/ARH | D | + | + | + | +++ | + | − | +++ | + | + |
| R646 VRQ/VRQ | E | + | ++ | + | +++ | +/− | +/− | +++ | +/− | ++ |
| R796 VRQ/VRQ | F | + | + | + | +++ | + | − | +++ | +/− | + |
| R617 ARR/ARR | G | − | − | − | − | − | − | − | − | − |
| R618 ARR/ARR | G | − | − | − | − | − | − | − | − | − |
| R647 ARQ/ARR | G | − | − | − | − | − | − | − | − | − |
| R682 ARQ/AHQ | G | − | − | − | − | − | − | − | − | − |
Electron microscopy lesions were ordered as negative, −; sparse, +/−; few, +; moderate, ++; widespread, +++; +ve, positive; −ve, negative.
After perfusion, brains were removed and two serial 1 mm slices were taken at the level of the obex. One of these slices was used for electron microscopy. The second slice and additional brain slices from representative brain areas were taken and processed into paraffin wax for PrPd profiling 14, 15. Sections were mounted, dried overnight, dewaxed and immersed in 98% formic acid, then washed and autoclaved in 0.2% citric acid for 30 minutes. Endogenous peroxidase was inhibited with freshly prepared 3% hydrogen peroxide and slides were washed and blocked with 1:20 normal horse serum for 60 minutes. All slides were incubated with the rat monoclonal antibody R145. The wax‐embedded medulla tissue sections were also immunolabeled using the technique described previously with the following antibodies: monoclonal anti‐clathrin heavy chain (BD biosciences, Franklin Lakes, NJ, USA), monoclonal anti‐clathrin light chain con1 (Sigma‐Aldrich, Gillingham, UK), monoclonal anti‐dynamin Hudy 1 (Upstate, Dundee, Scotland), monoclonal anti‐dynamin D5 (Santa Cruz, York, UK), monoclonal anti‐amphiphysin VAM‐SV030 and VAM SV031 (Bioquote Ltd., Heidelberg, Germany), and polyclonal anti‐ubiquitin (Dako UK Ltd., Ely, UK). Tissues were immunolabeled using these antibodies both with and without formic acid and autoclave steps.
Tissues for electron microscopy were selected under a dissecting microscope and embedded in araldite. Light microscopic labeling of 1‐µm‐thick plastic‐embedded tissues for PrP was performed as described previously, but tissue sections were first etched using saturated sodium ethoxide immersed in 98% formic acid. Ubiquitin labeling was carried out as described previously; however, both labeling and section quality were improved with the omission of the formic acid step.
For electron microscopy, 55 nm sections were placed on 600‐mesh gold grids and etched in sodium periodate for 60 minutes. Endogenous peroxidase was blocked and sections de‐osmicated with 3% hydrogen peroxide in distilled water for 10 minutes followed by enhancement of antigen expression with 98% formic acid for 10 minutes. Primary antibody (523.7, a gift from Jan Langeveld) at a 1:250 dilution was then applied for 15 h at 27°C and incubated with Auroprobe™ 1 nm colloidal gold (Amersham Ltd., Amersham, UK). Sections were then postfixed with 2.5% gluteraldehyde in phosphate buffered saline (PBS) and labeling enhanced with Goldenhance® (Nanoprobes Ltd.) for 10 minutes. Grids were counterstained with uranyl acetate and lead citrate as described previously. In order to have exact correlation for individual cells and specific PrPd accumulation types, serial 1‐µm‐ and 55‐nm‐thick sections were labeled for PrP by light and electron microscopy, respectively. Ubiquitin labeling was carried out using Dako anti‐ubiquitin® antibody at a 1:50 dilution without formic acid.
Sections stained for PrPd were initially examined blind and the number of PrPd‐positive structures were recorded as negative, sparse, few, moderate or marked. The immunogold labeling made identification of coated‐pit associated changes more conspicuous, reflecting a greater magnitude of scoring, but the trends were identical to those reported by Ersdal et al (7) on the same material.
RESULTS
Light microscopy: two distinct pathological phenotypes indicate two strains
We assembled PrPd profiles on each sheep as previously described 13, 15. Sheep could be divided into two groups according to the presence or absence of vascular plaques and astrocyte‐related types of PrPd accumulation (Table 1). Those in which plaques were absent tended to have higher total PrPd scores (arbitrary units), principally manifested by higher amounts of stellate, perivascular and perivacuolar (white matter astrocyte‐associated) PrPd types. In contrast, sheep with vascular amyloid plaques had lower amounts of the glia‐associated PrPd types. When compared with previous data, these results show that two distinct separate strains or sources of scrapie were present in the 10 infected subjects.
Three brainstem nuclei were selected for electron microscopic examination: the dorsal motor nucleus of the vagal nerve (DMNV), the spinal tract nucleus of the trigeminal nerve and olivary nuclei. Only some of the 13 possible types of PrPd labeling present when whole sheep scrapie brains are examined (15) were present in these sites and are shown in Figure 1.
Figure 1.
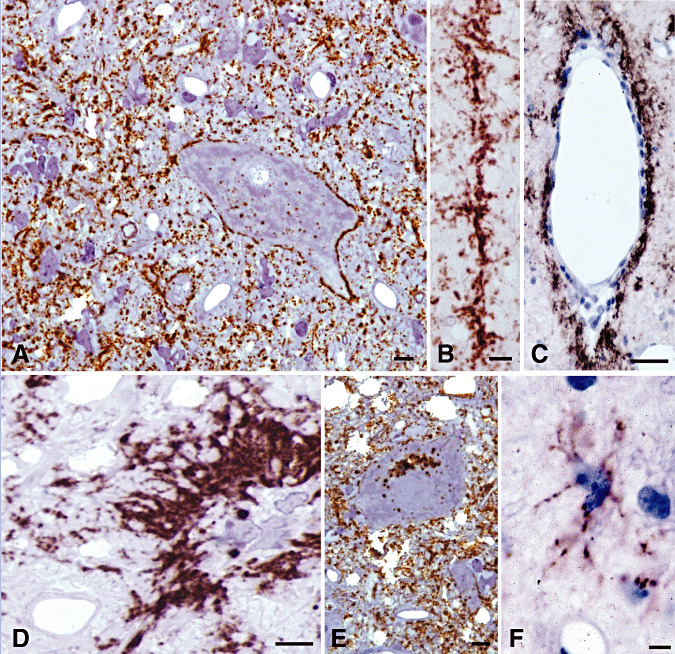
Different types of PrPd accumulation observed in 1‐µm‐thick immunolabeled sections. In the neuropil of the dorsal motor nucleus of the vagal nerve (DMNV), most PrPd accumulated in a diffuse punctate or granular pattern (A,E). Patterns of accumulation that clearly related to neurons included deposits around neuronal perikarya (A) and around neuronal processes as linear accumulations (B). PrPd accumulation was detected within neuronal cytoplasm at all neuroanatomic sites as individual (A) and perinuclear puncta or clusters (E) of ovoidal granular deposits. Other patterns of PrPd accumulation included perivascular (C) and intense coalescing patterns often seen around glial perikarya (D). Intense puncta of labeling, presumed to be intracellular, was often seen adjacent to glial cell nuclei (D). Though not distinguishable against the diffuse or granular neuropil labeling of the DMNV, a distinct pattern associated with the processes of astrocytes was seen elsewhere (F, olivary nucleus). Bar: A, 6 micron; B, 6 micron; C, 20 micron; D, 6 micron; E, 6 micron; F, 6 micron. Immunohistochemistry for PrPd in 0.5–1‐µm‐thick plastic embedded sections using R523 antibody.
Electron microscopy
Neuronal and dendritic plasmalemmal PrPd is associated with abnormal endocytosis and cell membrane transfer
In previous studies of murine scrapie, we have shown that PrPd is found mainly on the cell membranes of neurons and dendrites. The present study also shows that most sheep scrapie PrPd is also found at the cell membranes of neurons and dendrites but not of axons. Cell membrane PrPd was associated with abnormal endocytosis or was transferred to adjacent cell membranes. In contrast to murine scrapie, PrPd was not aggregated into fibrils in the extracellular space.
To determine the relationship between light microscopic types of PrPd and subcellular sites of PrPd accumulation, individual patterns were identified by light microscopy in 1‐µm‐thick immunolabeled sections and then located in serial sections by electron microscopy. The light microscopy patterns of perineuronal, linear and diffuse punctate labeling (Figure 1A,B,E) all correlated at electron microscopy with PrPd located at perikaryonal and dendritic plasma membranes.
In relation to each of the previously mentioned light microscopic types of PrPd labeling, including the punctate patterns, some PrPd was located precisely at the plasmalemma of neuronal cell bodies and of dendrites with no other changes evident, neither of the adjacent neuropil nor of the neuronal cell body or dendritic cytoplasm (Figure 2A).
Figure 2.
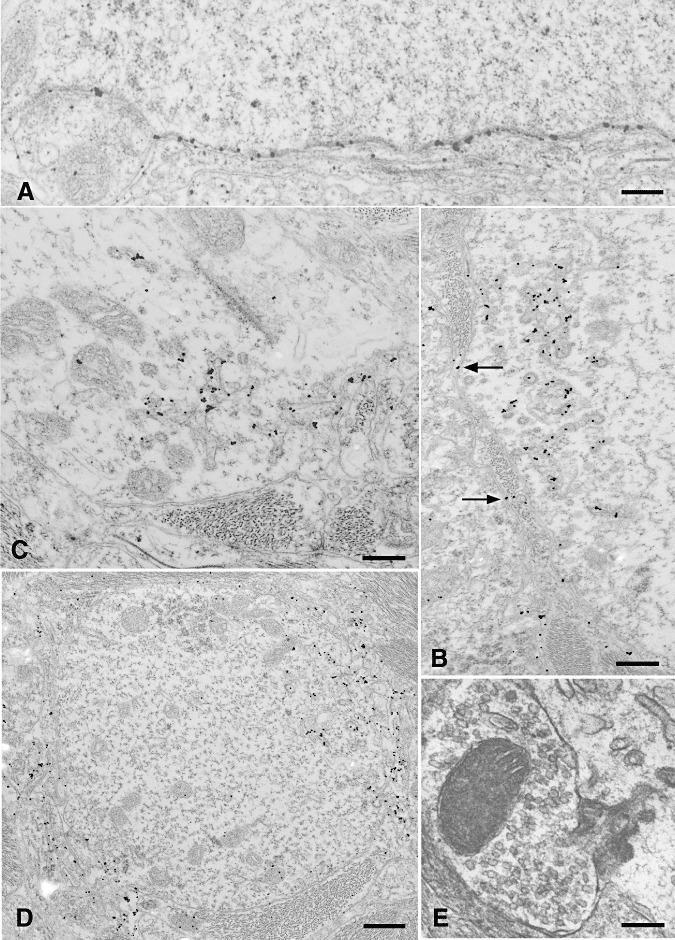
PrPd accumulation was found at the surface of neurons and dendrites corresponding to perineuronal, linear and punctate patterns of labeling at light microscopy. Some PrPd accumulation occurred at the plasma membrane of neurons and dendrites in the absence of any other changes (A). Some membrane accumulation was associated with the formation of excess coated pits and with the transfer to membranes of adjacent processes (B). In appropriate sections, the continuity between coated pits with abnormally long necks and the cell surface could be established (B,C,E). Some of these pits formed fused tubular networks beneath the cell membrane (B,C). In dendrites, PrPd often accumulated at two poles of the same process, and with increasing accumulation PrPd, could be detected on membranes adjacent to the cell (D) (see also arrows at B). Complex dendrite membrane disturbances associated with the formation of abnormal pits incorporated the membranes of adjacent processes; these were usually astrocytic, but rarely, axon terminals were also involved (E). Bar: A, 0.29 micron; B, 0.39 micron; C, 0.30 micron; D, 0.53 micron; E, 0.19 micron. A–D, PrPd immunolabeling using R523 antibody; E, uranyl acetate/lead citrate, not immunolabeled.
A fraction of neuronal and dendritic plasmalemmal PrPd was associated with morphological changes. PrPd was associated with numerous abnormal coated pits and vesicles (Figure 2B,C). In appropriate planes of section, vesicles were connected to the cell surface by tubular, sometimes spiral, invaginations of the cell membrane. PrPd labeling was found within these connecting twisted tubules and in the associated coated structures and pits (Figure 2B,C). Fusion of pits and vesicles resulted in complex subplasmalemmal tubular networks (Figure 2B,C). Often, pits and associated changes were present at two poles of a cross‐sectioned dendrite (Figure 2D); and in longitudinal sections of dendrites, they were present as discrete focal areas distributed discontinuously along a process. Where the entrance to the coated pits was relatively broad, adjacent processes, usually astrocytic but occasionally dendritic, and rarely, axon terminal membranes (Figure 2E), extended into the mouth of the open pits. Close proximity, and rarely, continuity between coated membranes and PrPd‐positive endosomes or lysosomes was occasionally seen. Similar structures were not found on axons.
PrPd was also seen on the plasmalemmas of processes adjacent to PrPd‐positive dendrites and neuronal soma (Figure 2D). The labeling of adjacent processes was indiscriminate and could involve astrocytic processes, axonal structures, occasionally including synaptic boutons (though we never observed involvement of synaptic densities), as well as other dendrites. The surface contours of the dendrite could be very convoluted, some showing finger‐like cytoplasmic extensions or microfolds of the plasmalemma (Figure 2D). The extracellular space between these folded membranes was often increased.
Astrocyte‐associated PrPd is mainly related to plasmalemmal microfolding
When located in serial light and electron microscope preparations, perivascular, coalescing and periastrocytic types of PrPd accumulation (Figure 1C,D,F) were associated with the plasma membrane of astrocytes (Figure 3A). Even presumed early accumulations of PrPd at the plasma membrane of astrocytes were associated with complex membrane changes (Figure 3B–E). In the presence of PrPd at the cell surface, astrocytic process plasmalemmas became markedly irregular with many small microfolds or projections. In areas such as the adjacent reticular formation or the subependymal neuropil which contained closely packed astrocytes, the nature of these finger‐like projections were more obvious (Figure 3C). In other areas, the membranes had a linear form, which on immunolabeling were strongly positive for PrPd. Where PrPd accumulations associated with astrocytes were intense, astrocyte processes were characterized by prominent glial bundles, a markedly disordered neuropil and numerous microglial processes. Such areas correlated with a coalescing pattern of PrPd accumulation at light microscopy (Figure 3F). Rare PrPd‐labeled coated pits were seen but not in association with abnormal membrane invaginations.
Figure 3.
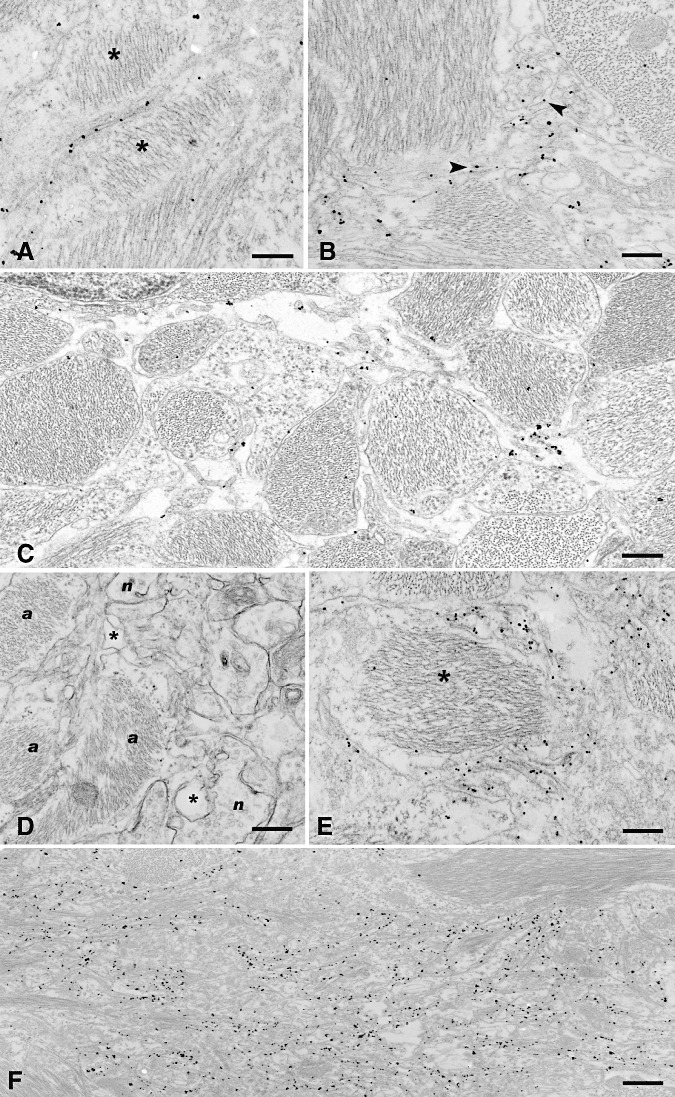
Patterns of accumulation of PrPd in association with astrocytic plasmalemma. As for dendrites, some PrPd accumulation was seen at the plasmalemma with little other change present, but most PrPd was associated with a proliferative membranous change. The simplest pattern of PrPd labeling was present at the plasmalemma as seen between two adjacent astrocytic (stars) processes in (A). In (B), PrPd labeling is seen on the plasma membrane of small cytoplasmic projections (small arrows) arising from astrocytic processes containing abundant glial filaments. In areas where astrocytes form a dense network of processes, the nature of the microfolding of these projections can be more clearly visualized (C) and is seen as PrPd‐labeled filiform cytoplasmic projections originating from and running between glial processes. The complex nature of the membranes around astrocytes is shown in a nonimmunolabeled section in (D); a indicates astrocytic processes, n neuronal processes and star expanded extracellular space and in a PrPd‐labeled section in (E). The association of PrPd with these convoluted membranes is shown around an astrocyte process containing a large bundle of intermediate filaments (star). The coalescing pattern around glial cells such as shown in Figure 1D translates to an area comprising extensive PrPd‐labeled membrane microfolds and convolutions between glial bundle‐containing astrocytic processes (F). Bar: A, 0.27 micron; B, 0.30 micron; C, 0.32 micron; D, 0.44 micron; E, 0.25 micron; F, 0.60 micron. A–C,E,F, PrPd immunolabeling using R523 antibody; D, uranyl acetate/lead citrate, not immunolabeled.
Intracellular PrPd labeling is located in lysosomes
At light microscopy, intracellular labeling was evident in neurons (Figure 1A,E), astrocytes (Figure 1D) and microglia. Electron microscopy showed that PrPd was located within mature and immature lysosomes of neuronal perikarya (Figure 4A,D), occasional dendrites (Figure 4C) and glial cells (Figure 4B). In favorable planes of sections, continuity between subplasmalemmal endosomal or lysosomal structures and abnormal pits invaginating from the cell membrane could be identified, suggesting intra‐endosomal and intralysosomal PrPd was derived from the cell surface PrPd.
Figure 4.
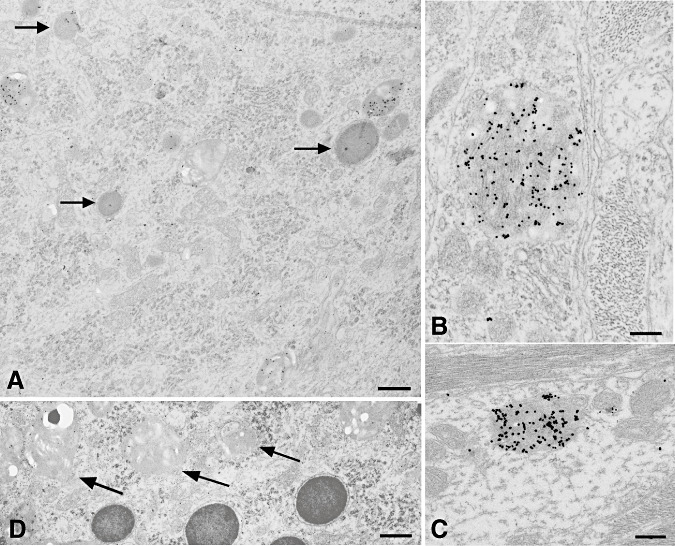
Intralysosomal PrPd accumulation was found in several cell types. In neurons, levels of labeling were higher on immature lysosomes, identified by a less electron dense content, than on mature lysosomes (arrows) (A). Intralysosomal PrPd was also identified in astrocytic and microglial glial cell bodies and processes (B) and in dendrites (C). Immature lysosomes of neurons were generally paler, often vacuolated and irregular in shape (D). Bar: A, 0.88 micron; B, 0.29 micron; C, 0.33 micron; D, 0.81 micron; A–D, PrPd immunolabeling using R523 antibody.
Many neurons contained very frequent lysosomes and endosomal–lysosomal structures, particularly in the perinuclear Golgi zones. To determine whether lysosomes were increased in these sheep, we performed cathepsin immunolabeling at light microscopy and counted endosomal‐lysosomal structures by electron microscopy. No increased light microscopic labeling for cathepsin was observed, but substantially more endosomes–lysosomes were encountered in neuronal perikarya of scrapie‐infected neurons when compared with controls in formal morphometric examination (data not shown).
PrPd is associated with only some scrapie‐specific cytopathological changes
As is common with chronic end‐stage neurological disease, many different lesions are present in brains of clinically sick scrapie‐infected animals. Some of these changes are nonspecific, but a number of changes are pathognomonic or highly indicative of scrapie infection. As partly described previously, some scrapie‐specific lesions were closely associated with PrPd but other changes were not.
Several morphological features have been described in TSE‐affected brains that we consider to be pathognomonic. These are tubulovesicular bodies (31), spiral membrane inclusions of axons previously referred to as spiroplasma (1), highly abnormal coated pits (7) and membrane proliferation or microfolding. Vacuolation is not itself pathognomonic at light microscopy, but the nature of tissue changes that surround and are present within vacuoles appears to be a type of spongiform change unique to the TSEs.
As in previous studies of murine scrapie, the membrane and contents of vacuoles and tubulovesicular bodies were unlabeled 20, 32. None of the major nonspecific degenerative features observed, including cerebellar lamellar bodies (8), clumped synaptic vesicles (2), degenerate processes, dystrophic axons or autophagic vacuoles, were labeled.
Spiral membrane inclusions occurred predominantly in axons and synaptic boutons. Continuity with the cell surface was occasionally evident. Unlike dendritic and neuronal membrane invaginations, the coating of the axonal spiral inclusions was electron lucent and lacked buds or branches. Some PrPd labeling was present but to a much more limited extent than that found on the abnormal coated pits. We hypothesize that these forms are specific, predominantly axonal responses to dendrite‐derived PrPd at the axonal plasma‐membrane.
Cell membrane PrPd is endocytosed via a ubiquitinated classical pathway
Clathrin‐coated pits are involved in the endocytosis and recycling of cell membrane proteins including normal PrPc (44). The bizarre extensions and branching of the necks of coated pits led us to consider the possibility that molecular abnormalities of the endocytosis and recycling of PrPd might be involved in these changes. We therefore investigated proteins involved in the formation of coated pits and in their excision from the plasma membrane in several light microscopic studies. Immunohistochemistry of clathrin, dynamin and amphiphysin showed no significant changes (data not shown). However, ubiquitin, which is involved in tagging proteins for proteolysis in endosomal‐lysosomes was increased.
By light microscopy, punctate ubiquitin immunolabeling was present in neuropil of varying intensity in scrapie‐affected sheep but not in controls. For electron microscopy, paired thin sections were labeled for PrPd or ubiquitin to confirm sites of PrPd accumulation on individual sections and then to determine levels of ubiquitin at these and other sites. Conspicuous ubiquitin labeling was associated with abnormal coated pits and subplasmalemmal tubular networks wherever these occurred on dendrites or neuronal cell bodies (Figure 5A). Ubiquitin was also present on the membranes of other cell processes immediately adjacent to these structures (Figure 5A). In paired sections, the PrPd labeling was present in corresponding locations. Some multivesicular bodies in dendrites were labeled strongly for ubiquitin in both controls and scrapie‐affected sheep, though in the latter, these structures were either not labeled for PrPd or showed only slight labeling. Weak labeling for ubiquitin was found on endosomal–lysosomal structures in neuronal perikarya, and rarely, in endosomal‐like structures linked with the spiral membrane inclusions (Figure 5D).
Figure 5.
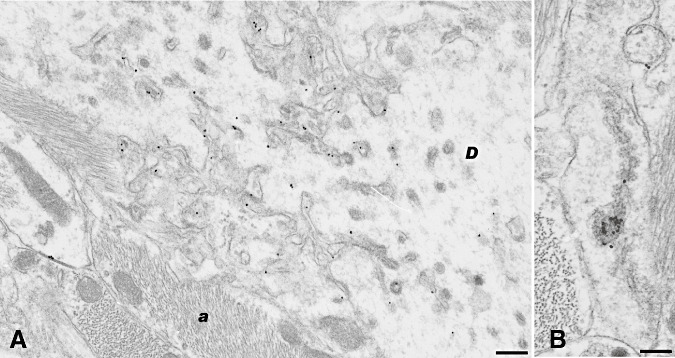
Ubiquitin labeling. Conspicuous ubiquitin labeling was present on abnormal coated pits at the cell membrane of dendrites (D) and on coated pits and vesicles and on the subplasmalemmal fused tubular complexes. It was also present on membranes of cell processes [in this case astrocytic processes (A)] adjacent to such areas. Variable; sometimes high levels of ubiquitin labeling were found on multivesicular bodies and on endosomal‐lysosomal structures, shown here attached to spiral inclusions (B). Bar: A, 0.37 micron; B, 0.24 micron; immunogold for ubiquitin.
Effect of scrapie strain on subcellular lesions and PrPd accumulation
The same range of morphological features and immunolabeling was seen in each of the sheep examined, but the frequency of changes differed markedly between individuals. When sheep were allocated to strain type, two distinct groups were evident. Thus, the two scrapie strains present differed according to discrete differences in the proportions of lesions present (Table 1).
At the level of the DMNV or olivary nuclei, there were few differences in the light microscopic patterns of PrPd accumulation when each of the 10 scrapie‐affected sheep was examined. A semiquantitative analysis of electron microscopy lesions present in the DMNV and in olivary nuclei was performed on coded grids. When the magnitude of selected features are tabulated (Table 1), the 10 scrapie‐affected sheep can be divided into two distinct groups (Table 1) that correspond with the strains identified by light microscopic PrPd profiling. The first group showed marked formation of PrPd‐associated abnormal coated pits and associated membrane irregularity and invaginations, increased lysosomes in neurons (mostly PrPd positive) and had more readily detectable spiral membrane inclusions. The five remaining sheep also had each of these features but in much less abundance. However, this second group of sheep had marked PrPd‐associated proliferative and microfolding membrane changes in astrocytes. These changes are morphologically distinct from classical reactive astrocytosis. Thus, when the light microscopy strain typing approaches and the electron microscopy changes are considered together, the same groupings are present and show that interanimal differences in PrPd‐related pathological changes differ only in magnitude and not in nature. Nevertheless, these magnitude changes segregate according to the strain of infection.
DISCUSSION
The present study informs on the in vivo cellular and subcellular location of PrPd and its relationships to subcellular morphological changes. From these observations, we can infer the sites of conversion of PrPc to PrPd and the inter and intracellular trafficking of PrPd in two strains of sheep scrapie.
PrPc to PrPd conversion occurs at the cell membrane
In agreement with conventional murine scrapie models that show lysosomal, cell membrane and extracellular PrPd accumulations 20, 21, 22, this study shows that PrPd of scrapie‐affected sheep is also associated with endosomes, lysosomes and plasma membranes of neurons and glia. In previous studies of sheep scrapie and experimental ovine BSE, we have shown that intracellular PrPd accumulations are N‐terminally truncated 23, 24, but all other neuropil types of PrPd accumulations are full length. When compared with these data, the present results suggest that PrPd first accumulates at the cell surface of neurons and dendrites from where it is either released or internalized and then digested. In vitro pulse‐chase experiments first suggested that the transformation of normal to abnormal PrP probably takes place at the cell surface or at some later stage in the cell cycle (5). Our results therefore agree with this data insofar as the first site of the cell cycle at which inferred full‐length PrPd is visualized is at the cell membrane.
PrPd is internalized by an abnormal classical endocytotic mechanism
Consistent with other glycosylphosphatidylinositol (GPI)‐anchored proteins, some studies suggest that PrPc is internalized by one of the noncoated pit mechanisms for raft‐associated proteins 27, 39, but others show that PrPc is internalized from N2a cells via coated pits (42). In N2a cells, PrPc resides within the lipid‐rich fragments of the membrane but leaves these regions to be endocytosed within nonraft regions by a clathrin‐coated mechanism (44). As GPI‐anchored proteins do not have a transmembrane domain, they cannot directly interact with clathrin‐associated proteins to elicit endocytotic vesicle formation; basic residues within the N terminus of PrPc are necessary to link between it and other membrane ligands that possess cytoplasmic domains recognized by clathrin (44). The present study suggests that PrPd on neuronal cell membranes is also internalized via a coated‐pit mechanism.
The bizarre tubules connecting coated pits to the cell surface suggest that the excision of the PrPd‐containing coated pits from the plasma membrane of scrapie‐infected neurons occurs inefficiently. We hypothesized that PrPd might interfere with the excision of pits from the cell surface and attempted to identify potential molecular defects using antibodies directed at proteins involved in vesicle excision, but we were unable to find any consistent changes in the abundance of amphiphysin, dynamin or clathrin (data not shown). We therefore propose that PrPd alters endocytosis by interfering with one of the vesicle excision proteins, which then elicits excess ubiquitin amplification prior to endosomal degradation of such complexes. Similar features are found in the scrapie‐infected TG3 PrP0/0 mouse, which generates PrPd from astrocytes and lack neuronal PrPc (25). This suggests that astrocyte‐derived PrPd can be transferred to neuronal membranes and drive abnormalities of endocytosis of abnormal PrPd. Abnormal endocytosis may therefore be an acquired toxic change of PrPd and may not require any alteration in cytoplasmic expression of proteins associated with clathrin‐mediated endocytosis. In the present study, astrocytes of scrapie‐infected sheep showed PrPd internalization by normal coated pits. We have also shown, in lymphoid tissues of the same sheep described here, that abnormal endocytotic mechanisms are present in macrophages internalizing PrPd. However, the macrophages internalize the PrPd via a noncoated (probably caveolin) mediated mechanism (36). Together, these observations suggest that membrane processing of PrPd is independent of PrPc expression and is influenced by cell type.
Scrapie‐associated alterations of the ubiquitin–proteosomal system are reported 6, 28, 47, with one study proposing that neurodegeneration may be caused by abnormally trafficked PrPc (34). In these scrapie‐infected sheep, PrPd and ubiquitin were present together at the cell surface, in abnormal coated pits, in fused subplasmalemmal tubular networks and in a proportion of lysosomes but not in association with the endoplasmic reticulum or the cytosol adjacent to Nissl or Golgi bodies, suggesting that ubiquitin‐associated trafficking of PrPd does not involve the proteosome. Rather, these results suggest that PrPc conversion or PrPd accumulation signals to a cytosolic protein that can instigate the clathrin endocytotic process, either directly—as a consequence of an abnormal location within the membrane—or more probably by complexing with another ligand as previously suggested (42). This protein complex is not recycled but directed to the endosomal system for degradation by ubiquitin signaling, which appears to increase cellular catabolic activity as shown by increased lysosomes. Astrocyte membrane‐associated PrPd was not detectably endocytosed with ubiquitin. Thus, mechanisms by which astrocytes and neurons deal with cell membrane‐associated PrPd are different.
Intercellular transfer of PrPd
Scrapie‐infected cells in vitro demonstrate release of PrPd and/or infectivity into the culture medium (9), and we have previously shown that PrPd may also be released from the surface of scrapie‐infected mouse neurons and dendrites in vivo (21). Exosomes are 50–90‐nm‐diameter vesicles of endosomal origins that are released into the extracellular environment and can transfer PrPc from neuroblastoma cells to PrP null cell lines (33). We were not able to identify exosome‐like structures in routine preparations in this or in previous rodent studies. To eliminate the possibility that small numbers of exosomes might by chance escape detection in two‐dimensional membrane representations seen at electron microscopy, we performed serial reconstructions of putative PrPd‐containing membrane fragments but failed to detect any exosomes (data not shown).
Previous studies have shown that GPI‐anchored proteins, including PrP, may be transferred between membranes, sometimes as a complex of molecules 19, 33. Our observations suggest that a fraction of PrPd that reaches the cell membrane is then transferred to other membranes of adjacent processes. This transfer is indiscriminate and independent of cell type and also occurs in the lymphoid tissues of these sheep (36). As the TG3 PrP0/0 mouse also demonstrates PrPd‐associated pathology on PrP null neurons (25), we can also infer that the transfer of PrPd between cells is independent of PrPc expression. Because both ubiquitin and PrPd are seen on membranes adjacent to a scrapie‐infected PrPd‐releasing cell, we surmise that PrPd is moved between membranes as a multimeric protein complex.
PrPd largely accumulates on dendritic rather than axonal membranes
Most brain PrPc appears to be located in the neuropil (10), and previous light microscopic studies have sought to relate the diffuse neuropil accumulation of PrPd to synapses (29). Some studies have suggested a preferential synaptic membrane concentration of PrPc 11, 38, but others show generalized localization on plasmalemmas 16, 30, 36. In these latter studies, PrPc was found to follow standard biosynthetic pathways for GPI‐anchored proteins, with PrPc expressed in Golgi bodies and recycled through late endosomes after reaching the cell membrane.
At the cell surface, the GPI anchor and N‐terminal domains direct PrPc to detergent‐resistant membrane (DRM) fractions (45). However, when the DRM lipid content is experimentally altered, PrPc is redistributed from axons to dendrites (12). In the present study, as in previous studies of murine scrapie, we show that PrPd accumulations in sheep are primarily related not to axons or to synapses but to dendrites. The observed preference for PrPd accumulation and release from dendrites may therefore be related to altered trafficking of the abnormal protein within the membrane or to an altered distribution of PrPc and PrPd following changes in the raft domains of axonal and dendritic membranes.
Scrapie strains or sources differ in the proportion of functional PrPd conformers
When the 10 scrapie‐infected sheep investigated in the present study were segregated according to strain or by their electron microscopy cytopathology features (Table 1), they segregate into the same two groups. The results therefore suggest that there are strain‐directed cytopathological effects of scrapie infection. However, the differences in cytopathological changes observed between the two strain groups were not qualitative but quantitative; that is, the same PrPd‐associated ultrastructural pathology was recognized for the two strains but in different proportions. This is in agreement with previous light microscopic immunohistochemical observations, suggesting that different strains may target some cell populations rather than others, giving rise to, for example, a more prominent neuron‐associated rather than glia‐associated morphological pattern of PrPd deposition, and that infection with different strains may lead to a different processing of PrPd, which is translated into, for example, more abundant intracellular rather than cell membrane aggregates (14).
Different cell types, such as neurons, glial cells and follicular dendritic cells (36), can exhibit different cytopathological changes when infected with the same strain of TSE agent; this is most likely caused by biological differences between these cell types. However, diversity of ultrastructural morphological changes can also be found when a single strain of TSE agent infects a particular cell type. Thus, a neuron can internalize cell membrane PrPd via abnormal coated pits, show PrPd‐associated plasma membrane microfolds and can release membrane PrPd to the extracellular space or transfer it to the membranes of adjacent cells. These different processing mechanisms result in the diversity of ultrastructural changes described, and the fact that these vary quantitatively, not qualitatively, in infections by different strains can be interpreted as follows: PrPd originating from infection with one particular strain interacts mainly with one cell membrane protein resulting in a main processing pathway and pathological change, and less so with other cell membrane proteins, resulting in different, secondary trafficking pathways and less pronounced cytopathology features. In this scenario, PrPd produced following infections by different strains would have distinct relative affinities for different cell membrane proteins, resulting in diverse proportions of processing mechanisms and pathological changes. It is conceivable that for this to happen, PrPd from a single strain would consist of several tertiary or quaternary isoforms, each of which would interact with a different cell membrane protein; different proportions of those isoforms in different TSE strains would result in relatively diverse trafficking mechanisms and different proportions of essentially the same cytopathological changes, as observed. Either through interaction with other cell membrane proteins or on its own, this notion of TSE strains being composed of several PrPd isoforms in varying proportions—rather than consisting solely of single isoforms—appears to fit better with the consistency of pathological changes observed for particular strains. If prion strains generated only single isoforms, the diversity of processing mechanisms and the different resulting cytopathological changes would be best explained as random events that would not lead to consistent pathological phenotypes for different strains.
The scrapie‐infected TG3 PrP0/0 mouse shows the same PrPd‐associated neuronal coated pit and membrane changes (25) as found in sheep. Despite the astrocytic origin of infection in this transgenic model, these mice did not show the same level of astrocytic plasma membrane proliferation of the sheep. This indicates that the responses of both astrocytes and neurons are also directed by specific properties of PrPd and not simply by the presence of infectivity in different cell types. It further suggests that it is the nature of the PrPd isoforms that dictates the abnormal cytopathology and inferred trafficking and not the converse. In general, rodent scrapie infections release more PrPd to the extracellular space and internalize less than sheep sources; nevertheless, the nature of the tissue changes and sites of PrPd localization are similar for rodent and sheep scrapie strains. We therefore suggest that every TSE or prion infection produces a range of PrPd isoforms, each of which produces different cytopathological effects. The relationship between different forms of PrPd and infectivity, and the significance of certain prion disease‐specific, PrP‐negative cytological changes such as tubulovesicular structures have yet to be established.
ACKNOWLEDGMENTS
We are grateful to Jim Hope for critical reading of the manuscript and to Janey Witz for morphometric studies of lysosomes. The study was supported by DEFRA in project Se1940.
The authors have nothing to disclose; there were no conflicts of interest.
Re‐use of this article is permitted in accordance with the Creative Commons Deed, Attribution 2.5, which does not permit commercial exploitation.
REFERENCES
- 1. Bastian FO, Foster JW (2001) Spiroplasma sp. 16s rDNA in Creutzfeldt–Jakob disease and scrapie as shown by PCR and DNA sequence analysis. J Neuropathol Exp Neurol 60:613–620. [DOI] [PubMed] [Google Scholar]
- 2. Bouzamond Bernstein E, Hopkins SD, Spilman P, Uyehara Lock J, Deering C, Safar J, et al (2004) The neurodegeneration sequence in prion diseases: evidence from functional, morphological and ultrastructural studies of the GABAergic system. J Neuropathol Exp Neurol 63:882–899. [DOI] [PubMed] [Google Scholar]
- 3. Bruce ME (2003) TSE strain variation. Br Med Bull 66:99–108. [DOI] [PubMed] [Google Scholar]
- 4. Bruce ME, McBride PA, Farquhar CF (1989) Precise targeting of the pathology of the sialoglycoprotein, PrP, and vacuolar degeneration in mouse scrapie. Neurosci Lett 102:1–6. [DOI] [PubMed] [Google Scholar]
- 5. Caughey B, Raymond GJ (1991) The scrapie‐associated form of PrP is made from a cell surface precursor that is both protease‐ and phospholipase‐sensitive. J Biol Chem 266:18217–18223. [PubMed] [Google Scholar]
- 6. Drisaldi B, Stewart RS, Adles C, Stewart LR, Quaglio E, Biasini E, et al (2003) Mutant PrP is delayed in its exit from the endoplasmic reticulum, but neither wild‐type nor mutant PrP undergoes retrotranslocation prior to proteasomal degradation. J Biol Chem 278:21732–21743. [DOI] [PubMed] [Google Scholar]
- 7. Ersdal C, Simmons MM, Goodsir C, Martin S, Jeffrey M (2003) Sub‐cellular pathology of scrapie: coated pits are increased in PrP codon 136 alanine homozygous scrapie‐affected sheep. Acta Neuropathol (Berl) 106:17–28. [DOI] [PubMed] [Google Scholar]
- 8. Ersdal C, Simmons MM, Gonzalez L, Goodsir CM, Martin S, Jeffrey M (2004) Relationships between ultrastructural scrapie pathology and patterns of abnormal prion protein accumulation. Acta Neuropathol 107:428–438. [DOI] [PubMed] [Google Scholar]
- 9. Fevrier B, Vilette D, Archer F, Loew D, Faigle W, Vidal M, et al (2004) Cells release prions in association with exosomes. Proc Natl Acad Sci USA 101:9683–9688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ford MJ, Burton LJ, Li H, Graham CH, Frobert Y, Grassi J, et al (2002) A marked disparity between the expression of prion protein and its message by neurones of the CNS. Neuroscience 111:533–551. [DOI] [PubMed] [Google Scholar]
- 11. Fournier JG, Escaighaye F, Grigoriev V (2000) Ultrastructural localization of prion proteins: physiological and pathological implications. Microsc Res Tech 50:76–88. [DOI] [PubMed] [Google Scholar]
- 12. Galvan C, Camoletto PG, Dotti CG, Aguzzi A, Ledesma MD (2005) Proper axonal distribution of PrP(C) depends on cholesterol– sphingomyelin‐enriched membrane domains and is developmentally regulated in hippocampal neurons. Mol Cell Neurosci 30:304–315. [DOI] [PubMed] [Google Scholar]
- 13. Gonzalez L, Martin S, BegaraMcGorum I, Hunter N, Houston F, Simmons M, Jeffrey M (2002) Effects of agent strain and host genotype on PrP accumulation in the brain of sheep naturally and experimentally affected with scrapie. J Comp Pathol 126:17–29. [DOI] [PubMed] [Google Scholar]
- 14. Gonzalez L, Martin S, Jeffrey M (2003) Distinct profiles of PrPd immunoreactivity in the brain of scrapie‐and BSE‐infected sheep: implications for differential cell targeting and PrP processing. J Gen Virol 84:1339–1350. [DOI] [PubMed] [Google Scholar]
- 15. Gonzalez L, Martin S, Houston FE, Hunter N, Reid HW, Bellworthy SJ, Jeffrey M (2005) Phenotype of disease‐associated PrP accumulation in the brain of bovine spongiform encephalopathy experimentally infected sheep. J Gen Virol 86:827–838. [DOI] [PubMed] [Google Scholar]
- 16. Herms J, Tings T, Gall S, Madlung A, Giese A, Siebert H, et al (1999) Evidence of presynaptic location and function of the prion protein. J Neurosci 19:8866–8875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hope J, Wood SCER, Birkett CR, Chong A, Bruce ME, Cairns D, et al (1999) Molecular analysis of ovine prion protein identifies similarities between BSE and an experimental isolate of natural scrapie, CH1641. J Gen Virol 80:1–4. [DOI] [PubMed] [Google Scholar]
- 18. Hunter N (1997) PrP genetics in sheep and the implications for scrapie and BSE. Trends Microbiol 5:331–334. [DOI] [PubMed] [Google Scholar]
- 19. Ilangumaran S, Robinson PJ, Hoessli DC (1996) Transfer of exogenous glycosylphos‐phatidylinositol (GPI)‐linked molecules to plasma membranes. Trends Cell Biol 6:163–167. [DOI] [PubMed] [Google Scholar]
- 20. Jeffrey M, Goodsir CM, Bruce ME, McBride PA, Scott JR, Halliday WG (1992) Infection specific prion protein (PrP) accumulates on neuronal plasmalemma in scrapie infected mice. Neurosci Lett 147:106–109. [DOI] [PubMed] [Google Scholar]
- 21. Jeffrey M, Goodsir CM, Bruce M, McBride PA, Scott JR, Halliday WG (1994) Correlative light and electron microscopy studies of PrP localisation in 87V scrapie. Brain Res 656:329–343. [DOI] [PubMed] [Google Scholar]
- 22. Jeffrey M, Halliday WG, Bell J, Johnston AR, MacLeod NK, Ingham C, et al (2000) Synapse loss associated with abnormal PrP precedes neuronal degeneration in the scrapie‐infected murine hippocampus. Neuropathol Appl Neurobiol 26:41–54. [DOI] [PubMed] [Google Scholar]
- 23. Jeffrey M, Martin S, Gonzalez L, Ryder SJ, Bellworthy SJ, Jackman R (2001) Differential diagnosis of infections with the bovine spongiform encephalopathy (BSE) and scrapie agents in sheep. J Comp Pathol 125:271–284. [DOI] [PubMed] [Google Scholar]
- 24. Jeffrey M, Martin S, Gonzalez L (2003) Cell‐associated variants of disease‐specific prion protein immunolabelling are found in different sources of sheep transmissible spongiform encephalopathy. J Gen Virol 84:1033–1046. [DOI] [PubMed] [Google Scholar]
- 25. Jeffrey M, Goodsir CM, Race RE, Chesebro B (2004) Scrapie‐specific neuronal lesions are independent of neuronal PrP expression. Ann Neurol 55:781–792. [DOI] [PubMed] [Google Scholar]
- 26. Jeffrey M, Gonzalez L, Chong A, Foster J, Goldmann W, Hunter N, Martin S (2006) Ovine infection with the agents of scrapie (CH1641 isolate) and bovine spongiform encephalopathy: Immunochemical similarities can be resolved by immunohistochemistry. J Comp Pathol 134:17–29. [DOI] [PubMed] [Google Scholar]
- 27. Kaneko K, Vey M, Scott M, Pilkuhn S, Cohen FE, Prusiner SB (1997) COOH‐terminal sequence of the cellular prion protein directs subcellular trafficking and controls conversion into the scrapie isoform. Proc Natl Acad Sci USA 94:2333–2338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kang SC, Brown DR, Whiteman M, Li RL, Pan T, Perry G, et al (2004) Prion protein is ubiquitinated after developing protease resistance in the brains of scrapie‐infected mice. J Pathol 203:603–608. [DOI] [PubMed] [Google Scholar]
- 29. Kovacs GG, Preusser M, Strohschneider M, Budka H (2005) Subcellular localization of disease‐associated prion protein in the human brain. Am J Pathol 166:287–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Laine J, Marc ME, Sy MS, Axelrad H (2001) Cellular and subcellular morphological localization of normal prion protein in rodent cerebellum. Eur J Neurosci 14:47–56. [DOI] [PubMed] [Google Scholar]
- 31. Liberski PP, Budka H, Yanagihara R, Gibbs CJ, Gajdusek DC (1993) Tubulovesicular structures. In: Light and Electron, Microscopic Neuropathology of Slow Virus Disorders. Liberski P (ed), pp. 373–392. CRC Press: Boca Raton. [Google Scholar]
- 32. Liberski PP, Jeffrey M, Goodsir C (1996) Immunogold electron microscopy studies of tubulovesicular structures: they are not composed of prion protein (PrP). In: Transmissible Subacute Spongiform Encephalopathies: Prion Diseases. Court L, Dodet B (eds), pp. 137–142. Elsevier Editions Scientifiques: Paris, France. [Google Scholar]
- 33. Liu T, Li RL, Pan T, Liu DC, Petersen RB, Wong BS, et al (2002) Intercellular transfer of the cellular prion protein. J Biol Chem 277:47671–47678. [DOI] [PubMed] [Google Scholar]
- 34. Ma JY, Wollmann R, Lindquist S (2002) Neurotoxicity and neurodegeneration when PrP accumulates in the cytosol. Science 298:1781–1785. [DOI] [PubMed] [Google Scholar]
- 35. Martin S, Gonzalez L, Chong A, Houston FE, Hunter N, Jeffrey M (2005) Immunohistochemical characteristics of disease‐associated PrP are not altered by host genotype or route of inoculation following infection of sheep with bovine spongiform encephalopathy. J Gen Virol 86:839–848. [DOI] [PubMed] [Google Scholar]
- 36. McGovern G, Jeffrey M (2007) Scrapie‐specific pathology of sheep lymphoid tissues. Plos One 2:e1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. McGovern G, Brown KL, Bruce ME, Jeffrey M (2004) Murine scrapie infection causes an abnormal germinal centre reaction in the spleen. J Comp Pathol 130:181–194. [DOI] [PubMed] [Google Scholar]
- 38. Moya KL, Hassig R, Creminon C, Laffont I, Di Giamberardino L (2004) Enhanced detection and retrograde axonal transport of PrPc in peripheral nerve. J Neurochem 88:155–160. [DOI] [PubMed] [Google Scholar]
- 39. Peters PJ, Mironov A Jr, Peretz D, Van Donselaar E, Leclerc E, Erpel S, et al (2003) Trafficking of prion proteins through a caveolae‐mediated endosomal pathway. J Cell Biol 162:703–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Prusiner SB (1982) Novel proteinaceous infectious particles cause scrapie. Science 216:136–144. [DOI] [PubMed] [Google Scholar]
- 41. Prusiner SB (1999) An introduction to prion biology and diseases. In: Prion Biology and Diseases. Prusiner SB (ed), pp. 1–66. Spring Harbor Laboratory Press: New York. [Google Scholar]
- 42. Shyng S‐L, Heuser JE, Harris DA (1994) A glycolipid‐anchored prion protein is endocytosed via clathrin‐coated pits. Cell Biol 125:1239–1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Somerville RA, Chong A, Mulqueen OU, Birkett CR, Wood SCER, Hope J, et al (1997) Biochemical typing of scrapie strains. Scientific correspondence. Nature 386:564. [DOI] [PubMed] [Google Scholar]
- 44. Sunyach C, Jen A, Deng J, Fitzgerald KT, Frobert Y, Grassi J, et al (2003) The mechanism of internalization of glycosylphosphatidylinositol‐anchored prion protein. EMBO J 22:3591–3601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Taraboulos A, Scott M, Semenov A, Avraham D, Laszlo L, Prusiner SB (1995) Cholesterol depletion and modification of COOH‐terminal targeting sequence of the prion protein inhibit formation of the scrapie isoform. J Cell Biol 129:121–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wille H, Michelitsch MD, Guenebaut V, Supattapone S, Serban A, Cohen FE, et al (2002) Structural studies of the scrapie prion protein by electron crystallography. Proc Natl Acad Sci USA 99:3563–3568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Yedidia Y, Horonchik L, Tzaban S, Yanai A, Taraboulos A (2001) Proteasomes and ubiquitin are involved in the turnover of the wild‐type prion protein. EMBO J 20:5383–5391. [DOI] [PMC free article] [PubMed] [Google Scholar]