

Abstract
A medium throughput approach is used to rapidly identify membrane proteins from a eukaryotic organism that are most amenable to expression in amounts and quality adequate to support structure determination. The goal was to expand knowledge of new membrane protein structures based on proteome-wide coverage. In the first phase membrane proteins from the budding yeast Saccharomyces cerevisiae were selected for homologous expression in S. cerevisiae, a system that can be adapted to expression of membrane proteins from other eukaryotes. We performed medium-scale expression and solubilization tests on 351 rationally selected membrane proteins from the budding yeast Saccharomyces cerevisiae. These targets are inclusive of all annotated and unannotated membrane protein families within the organism’s membrane proteome. 272 targets were expressed and of these 234 solubilized in the detergent n-dodecyl-β-D-maltopyranoside. Furthermore, we report the identity of a subset of targets that were purified to homogeneity to facilitate structure determinations. The extensibility of this approach is demonstrated with the expression of ten human integral membrane proteins from the solute carrier superfamily (SLC). This discovery-oriented pipeline provides an efficient way to select proteins from particular membrane protein classes, families, or organisms that may be more suited to structure analysis than others.
Keywords: Discovery-oriented screen, Membrane Protein Structure, Structural Genomics, Eukaryotic Integral Membrane Protein, Saccharomyces cerevisiae
Introduction
Integral membrane proteins (IMP) comprise the channels, transporters, receptors, and enzymes that mediate the flow of information and materials between extracellular and intracellular milieus. Underscoring their importance and relevance is that approximately 60% of currently available therapeutics interact with one or more membrane proteins 1. Studying membrane proteins has proven to be experimentally daunting. This is evident by the observation that to date there are only approximately 100 unique α–helical membrane protein structures within the Protein Data Bank (PDB) 2 (http://blanco.biomol.uci.edu/Membrane_Proteins_xtal.html), accounting for less then 0.25% of all known structures. The hurdles include obtaining sufficient levels of expression or overexpression, detergent extraction from the membrane, and purification.
The majority of currently available eukaryotic structures were purified from natural sources where the target of interest was endogenously expressed at relatively high levels within a specific and readily available tissue. These provisos rarely exist for eukaryotic targets, as required for a general approach to target particular human or pathogenic membrane proteins of importance to human health. Thus, alternative means of generating material must be developed. Only thirteen heterologously expressed eukaryotic integral membrane protein structures have been published so far 3; 4; 5; 6; 7; 8; 9; 10; 11; 12; 13; 14; 15. The first of these was determined in 2005. These structures are generally the result of “family-oriented” approaches - protracted operose feats guided by the pursuit of a particular functional class or family of protein 16. Archetypal examples are the β2-adrenergic receptor 4; 17; 18, the Kv1.2 potassium channel 11, and the Plasmodium glycerol transporter PfAQP 14. To address the barriers and increase the probability of success, we sought to develop a way of selecting membrane proteins from particular membrane protein classes, families, or organisms that may be more suited to structure analysis than others. Using this strategy, we first report an approach to “discovery-oriented” selection of more tractable targets that begins with genomic data and, using predetermined constraints to define an empirical pipeline, that advances selected IMPs through the pipeline based on success at each stage. The objective of this approach is to identify and prioritize targets based on selected criteria, in this case expression level, detergent solubilization and molecular homogeneity characteristics seen on size exclusion chromatography (SEC). Such a discovery-oriented approach is based on the premise that target selection, be is species for a single protein, or choice among the membrane proteome is often vital to successful integral membrane protein structure determination. While this borrows from the concepts used by structural genomics initiatives 16; 19, it begins with a functional focus, namely onto integral membrane proteins that transmit signals or materials across membranes. It also borrows from the notion of broad coverage to find single candidates that might transcend a ‘high barrier’ to success.
The simplest application of this approach to eukaryotic integral membrane proteins is within a system previously demonstrated to be amenable to protein production for structural studies, the budding yeast Saccharomyces cerevisiae 20; 21; 22. Seven of the thirteen currently available eukaryotic integral membrane protein structures expressed heterologously were produced in some form of yeast 4; 6; 8; 9; 10; 11; 13. In addition, S. cerevisiae is an appropriate choice for these studies because it allows for high-throughput cloning and expression via episomal expression plasmids, selection, post-translational modifications, proper membrane targeting and insertion machinery, and an easy platform for downstream functional studies 20; 21. Thus, we sought a pipeline approach for the identification and validation of S. cerevisiae eukaryotic integral membrane protein overexperssion via episomal plasmids within S. cerevisiae. The eventual goal is for a system generally applicable to any eukaryotic set of membrane proteins.
To determine the extensibility of this approach, we expressed and solubilized ten human integral membrane proteins from the SLC superfamily and identified the most appropriate targets for further investigation. The advantage of such a broad screen approach to addressing the problems associated with eukaryotic integral membrane protein structure determination is the rapid and cost-effective identification and prioritization of targets for subsequent scale-up and crystallization trials. Vetting of these targets can occur rapidly by strict screening according to predefined criteria for progression, producing rapid returns. Once identified, an ‘inverse-funnel’ approach can be pursued where one works to obtain pure, homogenous, stable and monodisperse samples prior to crystallization (employing methods such as vapor diffusion, microbatch, microfluidics, and lipidic mesophases). The current paucity of eukaryotic integral membrane protein structures, coupled to the difficulty of success with any single nominated membrane protein warrants this approach; a ratiocinative selection of a large group of targets to move through a single predetermined empirical pipeline with strict standards. Since only thirteen heterologously expressed eukaryotic membrane protein structures have been determined, with the first in 2005, any return of structural information is at this time biologically significant.
Results and Discussion
We developed a medium throughput pipeline to expedite the timeline and reduce the cost of identifying targets amenable to large-scale purification, crystallization and functional characterization. Membrane proteins from the yeast S. cerevisiae were screened for maximal coverage of protein families, which led to a selected group of 384 integral membrane proteins that cover all IMP protein families within the organism with some redundancy. The 384 IMPs were cloned, transformed into S. cerevisiae, and grown in medium-scale (500 ml culture volume) cultures for expression, membrane preparation and solubilization trials. This resulted in 234 IMPs that express in our yeast system, as indicated by signal on a western blot, which could be solubilized (>50%) with a detergent (n-dodecyl-β-D-maltopyranoside, DDM) amenable to crystallization trials. 61 of these targets, from the first 96 (one quarter of the 384), were further grown in large-scale (3 L culture volume) and evaluated based upon post immobilized metal affinity chromatography (IMAC) expression level and quality of size exclusion characteristics. This resulted in twenty-three IMPs with relatively high expression level, soluble in DDM, and fully resident within the included volume on a size-exclusion column (Supplementary Table). These data suggest that 25% of all yeast eukaryotic integral membrane protein targets reach the necessary criteria for a very high probability of success in crystallization for structure determination.
Pipeline Development and Overview
The objective was to streamline the screening aspect and prioritize IMP targets for intensive characterization. The methods and protocols were largely developed a priori and not varied while targets IMPs progressed through the pipeline. This contrasts with the more usual route where multiple tags, expression plasmids, detergents, and purification schemes are varied to pursue specific membrane proteins or protein families 20; 23. The pipeline is divided into three general categories – target selection, expression plasmid construction and target prioritization (Figure 1). Within each category a minimalist approach was pursued to both expedite the time and decrease costs associated with identifying targets amenable to subsequent studies. Generally, one expression plasmid and associated affinity tags were used for cloning with sequencing information for cloned targets obtained only if the target expressed, solubilized in DDM and eluted in the included volume in SEC (using one buffer condition). DDM was chosen as the only detergent for solubilization screening as it was shown previously by multiple groups to be a good performer in solubilizing eukaryotic IMPs, and is often also amenable for crystallization trials 20; 22. In addition, DDM generally solubilizes proteins that can be solubilized in n-octyl-β-D-glucopyranoside (OG), currently the most commonly utilized detergent in generating structures of integral membrane proteins, thereby reducing the number of proteins that need to be initially screened for crystallization in OG 22. No salvage pathways were utilized for any stage of the process so, for example, 351 out of 384 targets attempted were cloned in the first pass and the failed sequences were not pursued. A more inclusive detergent solubilization and SEC buffer screen would be informative starting points for an expanded pipeline. The stringency of methods utilized within this approach derives from the understanding that efforts required to turn identified targets into actual structures will drastically increase during the crystallization phase.
Figure 1.

Pipeline for extensive prioritization phase. Prioritization of targets based on expression level, detergent solubility and size-exclusion profile were determined using the above pipeline. This pipeline is divided into three phases: target selection (pink), plasmid construction (green) and prioritization (blue). Arrows trace the path through the pipeline with black arrows being followed within a level and red arrows denoting the transition between phases. All cloned targets progressed through small-scale prioritization. Targets that expressed and were soluble in the detergent DDM also progressed through the large-scale prioritization step.
Target Membrane Protein Selection
We describe the target selection in detail in a companion publication 24. S. cerevisiae protein sequences were collected from the Saccharomyces Genome Database (http://www.yeastgenome.org) (SGD). 621 of the total 6600 protein sequences were predicted by the TMHMM program 25 to have three or more transmembrane helices (TMH) (targets, Figure 2). This cutoff was chosen to focus on integral membrane proteins rather than monotopic or membrane associated proteins that may only have membrane anchoring helices or signal peptides. We realize such a restriction ignores some important classes of integral membrane proteins, such as one- or two-crossing proteins that oligomerize to form channels, but signal peptide prediction algorithms are currently not robust enough to accurately assign eukaryotic targets with two TMHs 26.
Figure 2.
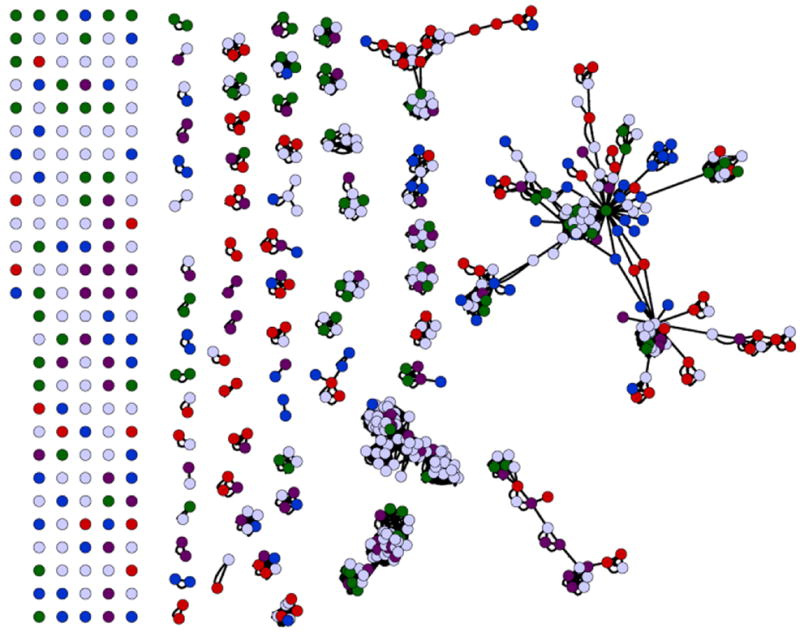
Genome-wide membrane protein target selection from S. cerevisiae for maximum Pfam coverage. The number of integral membrane proteins within yeast that have three or more TMH totals 621. Each protein is represented as a circle. These are clustered according to Pfam families. Colored circles represent the targets chosen for the expression of the first group of 384 membrane proteins, selected for maximal coverage of the Pfam families. As a result of clustering, there are 131 unannotated Pfam proteins, and 84 singletons that were all selected for expression (represented on the left side of the figure). 81 Pfam families have two or more members indicated by the clusters (center to right). In each case two members were chosen for remaining Pfam families. Seven additional proteins were selected from the larger clusters (right), to complete the target set of 384 membrane proteins. These proteins were divided into four sets of 96 tagets each (represented by red, blue, purple or green circles). Proximity within Pfams represents the sequence conservation between family members gleaned from use of multiple sequence alignment methods.
There are 162 unique Pfam membrane protein families in yeast, 79 of these being represented by a single sequence. For the remaining 83 annotated families, two sequences were selected from each to improve the probability of successful advancement of an IMP to structure determination for each family. Of the 621 membrane proteins with three or more TMHs in S. cerevisiae, 131 could not be annotated with a Pfam identifier. Of these sequences, 16 were in two unannotated clusters of 8 sequences each, 62 matched no other sequences, six sequences fell in two clusters of three unannotated sequences each and 14 fell into seven clusters of two sequences each. Thus, we selected a total of 384 targets (4 × 96 to facilitate cloning in 96-well format) from the S. cerevisiae membrane proteome providing complete coverage of all annotated and unannotated IMP families within the organism. In addition, the families are now rationally categorized to facilitate rapid homolog selection from other organisms downstream for expansion around targets performing well within the pipeline.
Expression Plasmid Construction
As part of efforts to facilitate multi-system membrane protein expression, the Membrane Protein Expression Center (MPEC.ucsf.edu) created a number of Ligase Independent Cloning (LIC)-compatible expression vectors with different affinity tags, aimed at purification or improving solubility (available upon request). The different host systems include Escherichia coli, yeast S. cerevisiae, yeast Pichia pastoris and HEK296S cells. The LIC vectors have been designed for high-throughput target construction with various tags or fusion proteins from the same PCR product of each target gene. For the current application, all genes were inserted into a S. cerevisiae LIC expression plasmid based on the yeast two-micrometer (2 μ) plasmid. This naturally occurring extrachromosomal DNA plasmid within S. cerevisiae replicates under strict cell cycle control and serves as the backbone for most episomal methods within yeast 27; 28; 29; 30; 31; 32. This MPEC LIC based plasmid, termed “83ν”, contains an N-terminal FLAG tag followed by a 3C protease cleavage site and a C-terminal 10XHis tag preceded by a thrombin protease cleavage site. The complete coding sequence for this plasmid is included within the Supplementary Protocol.
We opted for the galactose inducible GAL1 promotor over a highly constitutive promoter such as TEF2 based on previous data showing higher expression under GAL1 20. Additionally, cell toxicity is common with the overexpression of many IMPs, suggesting tight control of induction is favorable within the current system 33. All genes were cloned from genomic S. cerevisiae DNA (Promega) using LIC in a high-throughput 96-well format as described in Materials and Methods (see also Supplementary Protocol). Following cloning, inserts were validated through colony PCR and double digestion. Sequencing of inserts was not done at this stage. To reduce cost and time only targets performing well within this pipeline were sequenced upon completion. In retrospect, all constructs sequenced following quality assessment via SEC contained no mutations. At this time, we successfully cloned 351 out of 384 targets in the initial pass (91% success rate), with a throughput of up to 192 clones per week. All cloning was done in 96-well format producing four target sets referred to as SC-1, SC-2, SC-3 and SC-4 (Figure 3).
Figure 3.

Schematic overview of discovery-oriented pipeline. The 384 targets were divided into four sets of 96 targets each (SC-1, SC-2, SC-3 and SC-4). Each tier shows the number of targets that were successfully passed through the specified stage of the pipeline (cloning, expression, solubilization and size exclusion) with the percent success rate relative to the previous tier. SC-1 targets which expressed and solubilized in DDM were then scaled-up to size exclusion to determine if the protein was present within the included volume of the column. 31 proteins, of 61 attempted, were shown to be present within the included volume at a level of 50% or greater. Of these, 23 proteins were fully included under the conditions tested. Five of these targets were pushed into the intensive production phase and determined to be readily purifiable and stable (“HS”) (Figure 5).
Prioritization - Test Expression and Solubilization
Samples can be rapidly screened for expression using smaller volumes, typically one to five ml’s for membrane proteins, yet our experience is that this comes at a cost of heightened variability and increased false negatives and sporadic false positives. Therefore we progressed immediately to 500 ml culture volumes to test expression and detergent solubilization for each of the 351 cloned constructs (Materials and Methods). Qualitative expression levels for each membrane protein was determined by western blot from the before spin sample prior to detergent solubilization. Each target was given a score of one through four depending on the amount of western signal from each blot (Supplementary Table). Concurrently, we performed small-scale (300 μl) solubilization trials for each target using DDM and a fifteen-fold dilution of resuspended membranes. A target was deemed soluble in DDM if greater then 50% of the western signal was retained in the supernatant following a high-speed spin of the solubilized membranes (one-hour solubilization at 4 °C). Using this combined approach, we identified 272 targets, out of 351, with positive expression based on the presence of western signal using total membrane fractions. Of these, 234 were observed to be soluble in DDM (> 50%), producing a 61% success rate for the identification of targets that express and are soluble relative to our starting set of 384 membrane proteins. These were then ranked based on qualitative level of expression and detergent solubilization.
Previous demonstrations have correlated protein properties with expression and solubilization profiles for integral membrane proteins 22; 34; 35 23. Significant correlations have implicated protein molecular weight, overall hydrophobicity, hydrophobicity of TMHs, isoelectric point, native expression level, and percentage of charged and polar residues within the TMHs. Only 4% of IMPs with a molecular weight greater than 100 kDa express in the “high” category compared to approximately 23% for other lower molecular weight ranges (Figure 4A). This relationship does not extend to the number of TMHs in each protein, as we observe a consistent level of expression with increasing number of TMHs (Figure 4B). Increasing hydrophobicity, as measured by a GRAVY score 36, for each candidate IMP also correlates negatively with overall expression (Figure 4C) for both IMPs and soluble proteins 22; 37. We expected a positive correlation between the percentage of hydrophobic residues (WFLIVMY) within TMHs and expression level, with proteins containing > 70% hydrophobic amino acids within their TMH region expressing at a higher level 22. However, only six proteins within the current dataset of 272 proteins contain TMHs that are > 70% hydrophobic. To increase the dataset size we used a lower hydrophobicity cutoff of 60% (Figure 4D) resulting in 140, out of 272, targets with TMH regions that are > 60% hydrophobic. Within this dataset we do not observe a positive correlation between expression and hydrophobicity of TMHs, with 21% of targets above or below the 60% threshold expressing within the high category (> 1 mg protein/L of culture). This disparity is likely the result of the datasets used within the two studies. The previous work included putative integral membrane proteins with one or two transmembrane helices 22, IMPs excluded here. Indeed, IMPs within this study tend to have more aromatic and charged TMHs. 206 of the 272 targets have transmembrane regions with > 16% aromatic residues (WFY) with no statistically significant correlation between this aromaticity and expression.
Figure 4.

Protein expression profiles relative to protein size and hydrophobicity. Bar graphs for each panel represent the total number of proteins within the respective bins, as enumerated on the right-axis (“# of Proteins”). Line plots represent the percent of targets expressing greater then one mg of protein per liter of culture within that bin (“% High Expressing”) and correspond to the left axis. Panels correspond to the following: A) molecular weight for each target divided into bins of 20 kDa each, B) number of transmembrane helices, C) overall hydrophobicity of the protein as indicated by a GRAVY score 36 and D) percent of residues within the predicted transmembrane region for each target which are hydrophobic (WFLIVMY).
Prioritization – Quality Assessment based on Size Exclusion Chromatography
A key component of the discovery-oriented approach to identifying and prioritizing IMPs for downstream studies is obtaining size exclusion profiles that assess protein stability and integrity. 61 targets within the first list of 96 cloned genes (64%) (SC-1) were demonstrated to express and were solubilized using our single chosen detergent DDM (Figure 3). The range of expression for 60 of these 61 targets ranged from 0.5 mg to 5.8 mg of protein per liter of culture as determined from post-IMAC elutions. The remaining target, YPL087W, expressed at 0.3 mg protein per liter of culture (Supplementary Table). These targets were advanced to chromatographic analysis, as they were the first to be cloned, and contained the most diversity of selected protein families within S. cerevisiae. This first list of 96 targets is composed almost entirely of singletons where the selected proteins are the only representatives of the selected Pfam families within the S. cerevisiae genome.
For each of these 61 targets, three liters of yeast culture were grown, producing, on average, approximately 100 grams of wet cells and fifteen grams of wet membranes. We found during the course of these experiments that it was essential to desalt the sample after running through the IMAC column and prior to SEC to prevent protein precipitation or aggregation, as revealed by a resulting shift into the void volume of the target (data not shown). To expedite this step, we elected to screen only one buffer condition which, based upon experience, we projected to be a safe compromise for the majority of targets: 20 mM TRIS-HCl pH 7.4 RT, 200 mM NaCl, 1 mM DDM and 10% glycerol. Thus, we identified 31 of 61 targets (i.e. 51%) which are > 50% resident within the included volume of a size exclusion column, 23 (38%) of which are fully included and of high quality for downstream functional and crystallization screening (Supplementary Table). This corresponds to 24% retention of targets through the extensive phase of our pipeline even while applying relatively strict and limited criteria for target progression (single detergent, single SEC buffer, etc.). This rate of target retention for eukaryotic proteins (24%) corresponds to the 25% obtained for globular prokaryotic membrane proteins in a recent systems-oriented screen 23.
Several of the selected targets have progressed into an “intensive” phase, involving protein specific purification and characterization protocols. The objective of this phase is to obtain well-characterized and pure protein for structure determination. Of the 31 targets mentioned above, six have been moved into production mode with large-scale growths and purification trials. Each of these IMP targets were sharp included peaks on SEC following IMAC purification and tag cleavage (Supplementary Protocol). Representative SEC profiles and SDS-PAGE gels for five of these targets are shown in Figure 5. In addition, three of these IMP targets have now been shown to crystallize; diffraction data for one of these extending to 3 Å resolution has been obtained (Figure 6), however it is generally the case that resolution is progressively improved within the same crystal form by ‘micellar tuning’ during purification.
Figure 5.

Representative set of eukaryotic integral membrane proteins identified within this screen. Each target is fully cleaved and injected on a Superdex 200 size exclusion column (approximately 24 ml bed volume flowing at 0.33 ml/minute) on day one (red) and day fourteen (blue) following storage at 4°C. Q14542 was reinjected on day eight. Each target has a symmetrical, unnormalized, peak with the ordinate being absorbance at 280 nm wavelength (in milli-absorbance units) and abscissa being column time (in minutes). In addition, a Coomassie stained SDS-PAGE gel is shown for each sample to demonstrate purity. In each case the primary band corresponds to the correct molecular weight for the specified target. The Precision Plus Protein Standard (BIO-RAD) standard is included with the corresponding molecular weights (top to bottom in kD): 250, 150, 100, 75, 50, 37, 25, 20, 15 and 10. A UniProt ID identifies each target.
Figure 6.

Crystal of a S. cerevisiae integral membrane protein transporter obtained from a sparse matrix grid screen that diffracts to 3 Å resolution. This target was identified as a result of our discovery-oriented screen and was one of the first two targets for which crystallization screens were performed. Diameter of the circle is 100 microns. Imagine obtained from ALS beamline 8.3.1.
Signal Peptide Processing
Inclusion of dual tags at N- and at –C termini within this expression system was designed to provide empirical insights into the presence or absence of a signal peptide relative to the calculated D-scores for each target 26. The D-score is a statistical probability that a given sequence contains a signal peptide; D-scores > 0.43 indicate a signal peptide is likely present within the target sequence. Currently, signal peptide prediction methodologies are more robust and accurate in identifying signal peptides within prokaryotic protein sequences when compared with eukaryotic sequences. Surprisingly, of the 99 targets within our target list predicted to have a signal peptide (D-score > 0.43), only nine targets are negative for N-terminal anti-FLAG signal and positive for C-terminal anti-His signal on a western blot as would be expected if the signal sequence had been cleaved off by the signal peptidase. For these nine targets, the D-score ranges from 0.44 to 0.86. Seven targets with D-scores between 0.03 and 0.36 also appear to have signal peptides based on our empirical data (Supplementary Table), indicating that D-scores < 0.43 do not preclude the presence of a signal peptide. These include a thiamine transporter (SGD accession code YOR192C), polyamine transporter (YOR273C) and lysophospholipid acyltransferase (YOR175C). However, for the vast majority (~80%) of targets predicted to contain a signal peptide, there is clear N-terminal anti-FLAG signal on western blots. This may be a consequence of incorrect signal peptide processing by the signal peptidase due to the presence of the N-terminal tag that precedes the signal sequence, perhaps by too great a distance. As these targets are expressed and purified one can best ascertain the presence of a cleavable signal peptide through mass spectrometry or Edman degradation.
The type and location of expression tags on IMPs can have a dramatic effect on not only protein expression but function as well. In a previous study of P-type ATPases N-terminally tagged constructs expressed at a higher level and tended to be functional relative to their C-terminally tagged counterparts 23. Others suggest that proper membrane insertion, stability and function of IMPs was not adversely affected by C-terminal tags 38; 39; 40. Indeed, a recent work with C-terminally tagged GFP constructs resulted in the expression of all 43 candidate IMPs 20. Dual tags within the current study were designed to provide insights into this important issue relating to overexpression of IMPs for structure determination. Our desire, a priori, was that a comparison of target D-scores with the presence/absence of anti-FLAG western signal would provide novel empirical insights into the presence of signal peptides for selected targets. Unfortunately, as evidenced by the high abundance of FLAG signal in our results, it remains inconclusive. This high basal level of anti-FLAG signal may, in part, be the result of unprocessed signal peptide. To test this we cloned and expressed the AmtB ammonia channel, a polytopic membrane protein with a validated N-terminal signal sequence 41; 42, into the same LIC cassette with flanking tags. The resulting western blot of membrane fractions showed approximately 10% of the expressed protein retained an uncleaved upstream FLAG tag (data not shown). Future discovery-oriented screens may benefit from a streamlined construct containing only C-terminally poly-histidine tagged proteins, though this may come at a cost of capturing fewer targets within the broad screen. Alternatively, the presence of N-terminally charged residues may also help facilitate membrane insertion, as demonstrated by recent studies on prokaryotic integral membrane protein expression 23 and orientation 43
Expression of Human Integral Membrane Proteins
To determine if this approach is applicable to a higher eukaryotic system, we selected ten human integral membrane protein transporters from the SLC superfamily for S. cerevisiae expression trials (Supplementary Table). We were able to express all ten targets within our yeast system at levels of 0.3 to 1.0 mg of protein per liter of culture. These levels are considered to be medium to high for human integral membrane protein overexpression in yeast. Seven of these targets, were > 50% soluble in DDM and correctly targeted to the membrane fraction. Four of them are completely extracted from the membrane with DDM.
Four members of this family: hENT1 (SLC29A1), hENT2 (SLC29A2), hCNT1 (SLC28A1) and hCNT2 (SLC28A3) were scaled up to twelve liters of culture volume (Materials and Methods; Supplementary Protocol). All were shown to be included in SEC when solubilized in DDM, except hCNT2 (data not shown for hENT1, hCNT1 and hCNT2). Furthermore, hENT1 and hENT2 were pushed forward for full-scale purification using IMAC, size-exclusion, cation and anion exchange chromatography. Both of these were purified to homogeneity, run as single peaks in size exclusion and are stable in 1 mM DDM (Q14542 in Figure 5). Expression and purification of these important human membrane transporters, which play a critical role in drug disposition and response 44, will greatly facilitate their structural and functional characterization. The effect of genetic polymorphisms in these transporters could also be predicted and functionally studied in vitro. Thus, these results demonstrate the feasibility of using S. cerevisiae within a discovery-oriented pipeline for the overexpression of human integral membrane proteins for downstream structural studies. Indeed, previous data using GFP fusion constructs found similar expression levels for a small group of human integral membrane proteins overexpressed in S. cerevisiae 20. Once targets are identified they can be integrated into a P. pastoris system to further increase protein yield.
Conclusions
An efficient screen for eukaryotic IMPs that can be advanced to structure determination could best be described as an “hourglass” which is divided into two phases: extensive and intensive. The extensive (funnel) phase starts very broadly with an organism’s membrane proteome, narrows in onto specific targets which appear amenable to downstream studies and ranks them based on expression level, detergent solubility and size exclusion characteristics. The list resulting from this phase can be described as the bottleneck within the hourglass. The intensive (inverse funnel) phase focuses on developing robust purification, concentration, crystallization and functional characterization protocols for specific membrane protein targets that do progress through the bottleneck criteria. Within the context of this approach one is only concerned with specific membrane protein identity within the refined intensive phase of the pipeline. This approach is designed to inject additional capture of targets at the front end (target selection, cloning and prioritization) to attenuate laborious efforts on the intensive purification end when pursuing pure, homogenous, stable, and monodisperse protein for crystallization.
This study bolsters the utility of S. cerevisiae as a viable system for the overexpression of eukaryotic integral membrane proteins. Using a protease deficient yeast strain with a GAL1 inducible plasmid allows maximal control, and yield, of target overexpression. The ability to clone targets in a high throughput LIC format with episomal expression makes S. cerevisiae a promising system for broad discovery-oriented screens such as this. Functional complementation and utilization of the extensive Yeast Knockout Collection 45 allows one to also rapidly characterize the function and phenotype of a specific membrane protein. The extensive phase of a discovery-oriented pipeline (Figure 1) intentionally sets aside protein function, along with numerous other criteria, in lieu of strict empirical standards for identifying viable targets for downstream purification and crystallization. Function would be pursued in the intensive phase of the pipeline to gain insights into protein function within the larger biological context.
We implement this approach by rationally selecting 384 S. cerevisiae integral membrane proteins covering all of the represented protein families within the organism. From this list we rapidly identified twenty-three targets (out of the first set of 96 targets) that expressed and were fully soluble in DDM and included on a size exclusion column. To facilitate structure determination and functional characterization efforts within the community we identify each of these targets and the remaining 173 DDM soluble expressed targets in a Supplementary Table. The first five of these targets were subsequently demonstrated to be stable within the assigned buffer and easily purified using established protocols (Figure 5). Two of the top targets have been shown to crystallize readily from standard sparse matrix screens (Figure 6) highlighting the benefits of stringently vetting targets during an extensive prioritization phase. A 24% return of identified targets from the original starting subset of 96 targets correlates well with a previously published study of prokaryotic P-type transporters 23. Thus, a streamlined discovery-oriented pipeline can be successfully implemented for the identification and prioritization of eukaryotic integral membrane proteins for downstream crystallization and functional characterization efforts.
Extending the current pipeline to the human membrane proteome would provide insights into human biology. Using ten human integral membrane proteins from the SLC family, we demonstrate the utility of this S. cerevisiae system for episomal heterologous overexpression of human targets, as recently reported elsewhere 20. If we applied the same strict criteria presented by us here to all of the ≥ 3 TMH membrane proteins of the human membrane proteome (3158 proteins) we expect approximately 400 targets to pass the criteria for crystallization trials, assuming a modest return of only 12% (two-fold less than the current study which is based upon our previous experience with the ability of DDM to solubilize human IMPs). There are currently only four structures of human integral membrane proteins solved from heterologously expressed protein 8; 12; 13. The current discovery-oriented approach of screening human IMP expression within yeast can be utilized to identify proteins amenable to structure determination. Such a screen would likely produce significant insights considering the current paucity of structural detail for human IMPs.
Materials and Methods
An explicit experimental protocol is included within the Supplementary Information outlining exactly how the experimental work was employed at the bench. This is designed to facilitate not only the application of our methods to other systems but also utilization of specific methods for other projects (such as the optimized high-throughput LIC-cloning protocol).
High-throughput Ligase Independent Cloning
Except where noted, all cloning methods were performed in 96-well high throughput format. S. cerevisiae genes were amplified from S288C genomic DNA stock (Promega) with synthetic oligonucleotide primers. Each primer sequence contained an additional sequence to engender complementary overhangs for LIC. The modified 2 μ plasmid pRS423 containing a GAL1 promoter was named 83nu and used for all cloning. This modified LIC compatible plasmid contains an N-terminal FLAG epitope, two amino acid spacer followed by a PreScission 3C protease cleavage site while the C-terminus contains a thrombin protease cleavage site, two amino acid spacer with a deca-histidine affinity tag. T4 polymerase-mediated 3′ to 5′ exonuclease reactions were incubated at 25° C for 40 min and heat inactivated at 75° C for 20 minutes. The same reaction was performed on the linearized 83nu vector with dTTP instead of dATP. Annealing reactions were incubated at RT for 15 min after which EDTA was added to start the reaction for 10 min at RT. The annealing reaction between plasmid and amplified gene insert was transformed directly into chemically competent DH5α cells (house stock). Transformants were selected on ampicillin (100 μg/ml) plates and positive clones were identified. The S. cerevisiae strain used for expression was W303-Δpep4 (leu2-3, 112 trp1-1 can1-100 ura3-1 ade2-1 his3-11,15 Δpep4 MATa) and by applying a modified Lithium acetate protocol the target genes were transformed by adding 2 μl miniprep plasmid DNA and incubating at 42 °C for 15 minutes in a heating block. Successful yeast transformants were selected on plates contained synthetic complete medium with histidine drop-out (SC-HIS) after incubating at 30°C for 2–3 days.
Expression and Solubilization Test
351 successfully cloned targets were subjected to an initial test expression and solubilization screen. Growths were performed in 500 ml SC-HIS with 2% glucose as a carbon source. Cultures were induced with 2% galactose following 24 hours at 30°C and 220 rpm in baffled flasks. After overnight induction, cells were harvested by centrifugation at 6,000 × g for five minutes, and resuspended in lysis buffer (50 mM TRIS HCl, pH 7.4RT, 20% glycerol, and 1 mM fresh PMSF). Cells were mechanically lysed on ice with 0.5 mm glass beads in a bead beater. Lysate was spun at 6,000 × g for ten minutes at 4°C. Total membrane fractions were collected by ultracentrifuging supernatant at 138,000 × g for 60 minutes at 4°C. Membranes were resuspended in buffer containing 50 mM TRIS HCl, pH 7.4RT, 200mM NaCl, 10% glycerol, 2 mM fresh PMSF (Buffer A) and a protease inhibitor cocktail. Solubilization screens were conducted in 300 μl total volume mixtures with a fifteen-fold dilution of membranes. Membranes were solubilized for one hour at 4° C in 30 mM DDM, 50 mM TRIS HCl, pH 7.4RT and 100 mM NaCl. This mixture was then spun at 100,000 g for 20 minutes. Before and after spin samples were collected to determine the extent of expression and solubilization from western blots (probing both N-terminal FLAG and C-terminal 10XHis tags).
Large Scale Expression and Purification
The 61 soluble targets of our first set were subjected to larger scale purification and size exclusion chromatography to access the quality of the target within the conditions of assignation. Three liters of culture for each of these targets were grown in SC-HIS as described. Membranes were prepared as described above and then solubilized in 25mM TRIS pH 8.0RT, 100mM Sucrose, 500mM NaCl, 30mM DDM with 15 mM imidazole for one hour at 4° C then spun at 138,000 × g for an additional hour. The supernatant was recovered for incubation with IMAC resin (Ni-NTA, Qiagen). Following a 1.5 hour incubation with IMAC resin on a nutator at 4 °C the protein was purified using steps of 15 mM, 30 mM and finally 300 mM imidazole. Eluted target was immediately exchanged into Buffer A with 1mM DDM using a NAP-10 Sephadex G-25 desalting column. Tags were removed through overnight incubation with 5 U thrombin protease per OD of protein and a five-fold excess of target protein to 3C protease. The cleavage reaction was subsequently purified by reapplication to a column containing benzamidine and Talon resin. Following elution the samples were applied to a Superdex 200 column and further purified in downstream studies. Purity and tag cleavage was verified via SDS-PAGE and western blot analysis.
Supplementary Material
Acknowledgments
We thank Stroud laboratory colleagues: Zachary Newby, David Savage, Franz Gruswitz, Bill Harries and Melissa del Rossario for helpful discussions during the course of this work; Meseret Tessema, Arceli Joves and Lynn Martin for experimental support; Ying Chen for supplying the hENT1 plasmid. We thank Peter Walter for generously providing yeast strains. This work was supported by the N.I.H. Roadmap Center grant P50 GM073210 (to R.M.S.), Specialized Center for the Protein Structure Initiative grant U54 GM074929 (to R.M.S., A.S. and K.G.), and U01 GM61390 (to K.G. and A.S.). F.A.H. is supported by a National Research Service Award from NIGMS (F32 GM078754) and a Sandler Biomedical Research postdoctoral fellowship.
Abbreviations
- DDM
n-Dodecyl-β-D-Maltopyranoside
- IMAC
immobilized metal affinity chromatography
- SEC
size exclusion chromatography
- SGD
Saccharomyces Genome Database
- TMH
transmembrane helices
- LIC
ligase independent cloning
- SLC
solute carrier superfamily
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Overington JP, Al-Lazikani B, Hopkins AL. How many drug targets are there? Nat Rev Drug Discov. 2006;5:993–6. doi: 10.1038/nrd2199. [DOI] [PubMed] [Google Scholar]
- 2.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–42. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Standfuss J, Xie G, Edwards PC, Burghammer M, Oprian DD, Schertler GF. Crystal structure of a thermally stable rhodopsin mutant. J Mol Biol. 2007;372:1179–88. doi: 10.1016/j.jmb.2007.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rasmussen SG, Choi HJ, Rosenbaum DM, Kobilka TS, Thian FS, Edwards PC, Burghammer M, Ratnala VR, Sanishvili R, Fischetti RF, Schertler GF, Weis WI, Kobilka BK. Crystal structure of the human beta2 adrenergic G-protein-coupled receptor. Nature. 2007;450:383–7. doi: 10.1038/nature06325. [DOI] [PubMed] [Google Scholar]
- 5.Nishida M, Cadene M, Chait BT, MacKinnon R. Crystal structure of a Kir3.1-prokaryotic Kir channel chimera. EMBO J. 2007;26:4005–15. doi: 10.1038/sj.emboj.7601828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Long SB, Tao X, Campbell EB, MacKinnon R. Atomic structure of a voltage-dependent K+ channel in a lipid membrane-like environment. Nature. 2007;450:376–82. doi: 10.1038/nature06265. [DOI] [PubMed] [Google Scholar]
- 7.Jasti J, Furukawa H, Gonzales EB, Gouaux E. Structure of acid-sensing ion channel 1 at 1.9 A resolution and low pH. Nature. 2007;449:316–23. doi: 10.1038/nature06163. [DOI] [PubMed] [Google Scholar]
- 8.Ago H, Kanaoka Y, Irikura D, Lam BK, Shimamura T, Austen KF, Miyano M. Crystal structure of a human membrane protein involved in cysteinyl leukotriene biosynthesis. Nature. 2007;448:609–12. doi: 10.1038/nature05936. [DOI] [PubMed] [Google Scholar]
- 9.Pedersen BP, Buch-Pedersen MJ, Morth JP, Palmgren MG, Nissen P. Crystal structure of the plasma membrane proton pump. Nature. 2007;450:1111–4. doi: 10.1038/nature06417. [DOI] [PubMed] [Google Scholar]
- 10.Tornroth-Horsefield S, Wang Y, Hedfalk K, Johanson U, Karlsson M, Tajkhorshid E, Neutze R, Kjellbom P. Structural mechanism of plant aquaporin gating. Nature. 2006;439:688–94. doi: 10.1038/nature04316. [DOI] [PubMed] [Google Scholar]
- 11.Long SB, Campbell EB, Mackinnon R. Crystal structure of a mammalian voltage-dependent Shaker family K+ channel. Science. 2005;309:897–903. doi: 10.1126/science.1116269. [DOI] [PubMed] [Google Scholar]
- 12.Ferguson AD, McKeever BM, Xu S, Wisniewski D, Miller DK, Yamin TT, Spencer RH, Chu L, Ujjainwalla F, Cunningham BR, Evans JF, Becker JW. Crystal structure of inhibitor-bound human 5-lipoxygenase-activating protein. Science. 2007;317:510–2. doi: 10.1126/science.1144346. [DOI] [PubMed] [Google Scholar]
- 13.Horsefield R, Norden K, Fellert M, Backmark A, Tornroth-Horsefield S, Terwisscha van Scheltinga AC, Kvassman J, Kjellbom P, Johanson U, Neutze R. High-resolution x-ray structure of human aquaporin 5. Proc Natl Acad Sci U S A. 2008;105:13327–32. doi: 10.1073/pnas.0801466105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Newby ZE, O’Connell J, 3rd, Robles-Colmenares Y, Khademi S, Miercke LJ, Stroud RM. Crystal structure of the aquaglyceroporin PfAQP from the malarial parasite Plasmodium falciparum. Nat Struct Mol Biol. 2008;15:619–25. doi: 10.1038/nsmb.1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Warne T, Serrano-Vega MJ, Baker JG, Moukhametzianov R, Edwards PC, Henderson R, Leslie AG, Tate CG, Schertler GF. Structure of a beta1-adrenergic G-protein-coupled receptor. Nature. 2008;454:486–91. doi: 10.1038/nature07101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stevens RC. Long live structural biology. Nat Struct Mol Biol. 2004;11:293–5. doi: 10.1038/nsmb0404-293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cherezov V, Rosenbaum DM, Hanson MA, Rasmussen SG, Thian FS, Kobilka TS, Choi HJ, Kuhn P, Weis WI, Kobilka BK, Stevens RC. High-resolution crystal structure of an engineered human beta2-adrenergic G protein-coupled receptor. Science. 2007;318:1258–65. doi: 10.1126/science.1150577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rosenbaum DM, Cherezov V, Hanson MA, Rasmussen SG, Thian FS, Kobilka TS, Choi HJ, Yao XJ, Weis WI, Stevens RC, Kobilka BK. GPCR engineering yields high-resolution structural insights into beta2-adrenergic receptor function. Science. 2007;318:1266–73. doi: 10.1126/science.1150609. [DOI] [PubMed] [Google Scholar]
- 19.DiDonato M, Deacon AM, Klock HE, McMullan D, Lesley SA. A scaleable and integrated crystallization pipeline applied to mining the Thermotoga maritima proteome. J Struct Funct Genomics. 2004;5:133–46. doi: 10.1023/B:JSFG.0000029194.04443.50. [DOI] [PubMed] [Google Scholar]
- 20.Newstead S, Kim H, von Heijne G, Iwata S, Drew D. High-throughput fluorescent-based optimization of eukaryotic membrane protein overexpression and purification in Saccharomyces cerevisiae. Proc Natl Acad Sci U S A. 2007;104:13936–41. doi: 10.1073/pnas.0704546104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bill RM. Yeast--a panacea for the structure-function analysis of membrane proteins? Curr Genet. 2001;40:157–71. doi: 10.1007/s002940100252. [DOI] [PubMed] [Google Scholar]
- 22.White MA, Clark KM, Grayhack EJ, Dumont ME. Characteristics affecting expression and solubilization of yeast membrane proteins. J Mol Biol. 2007;365:621–36. doi: 10.1016/j.jmb.2006.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lewinson O, Lee AT, Rees DC. The funnel approach to the precrystallization production of membrane proteins. J Mol Biol. 2008;377:62–73. doi: 10.1016/j.jmb.2007.12.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kelly L, Pieper U, Eswar N, Hays FA, Li M, Roe-Zurz Z, Kroetz D, Giacomini KM, Stroud RM, Sali A. A taxonomic profile of the membrane protein universe. Genome Biology (pending review) 2008 [Google Scholar]
- 25.Krogh A, Larsson B, von Heijne G, Sonnhammer EL. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol. 2001;305:567–80. doi: 10.1006/jmbi.2000.4315. [DOI] [PubMed] [Google Scholar]
- 26.Emanuelsson O, Brunak S, von Heijne G, Nielsen H. Locating proteins in the cell using TargetP, SignalP and related tools. Nat Protoc. 2007;2:953–71. doi: 10.1038/nprot.2007.131. [DOI] [PubMed] [Google Scholar]
- 27.Kojo H, Greenberg BD, Sugino A. Yeast 2-micrometer plasmid DNA replication in vitro: origin and direction. Proc Natl Acad Sci U S A. 1981;78:7261–5. doi: 10.1073/pnas.78.12.7261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hartley JL, Donelson JE. Nucleotide sequence of the yeast plasmid. Nature. 1980;286:860–5. doi: 10.1038/286860a0. [DOI] [PubMed] [Google Scholar]
- 29.Hindley J, Phear GA. Sequence of 1019 nucleotides encompassing one of the inverted repeats from the yeast 2 micrometer plasmid. Nucleic Acids Res. 1979;7:361–75. doi: 10.1093/nar/7.2.361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mumberg D, Muller R, Funk M. Regulatable promoters of Saccharomyces cerevisiae: comparison of transcriptional activity and their use for heterologous expression. Nucleic Acids Res. 1994;22:5767–8. doi: 10.1093/nar/22.25.5767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mumberg D, Muller R, Funk M. Yeast vectors for the controlled expression of heterologous proteins in different genetic backgrounds. Gene. 1995;156:119–22. doi: 10.1016/0378-1119(95)00037-7. [DOI] [PubMed] [Google Scholar]
- 32.Sikorski RS, Hieter P. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics. 1989;122:19–27. doi: 10.1093/genetics/122.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Osterberg M, Kim H, Warringer J, Melen K, Blomberg A, von Heijne G. Phenotypic effects of membrane protein overexpression in Saccharomyces cerevisiae. Proc Natl Acad Sci U S A. 2006;103:11148–53. doi: 10.1073/pnas.0604078103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Daley DO, Rapp M, Granseth E, Melen K, Drew D, von Heijne G. Global topology analysis of the Escherichia coli inner membrane proteome. Science. 2005;308:1321–3. doi: 10.1126/science.1109730. [DOI] [PubMed] [Google Scholar]
- 35.Gelperin DM, White MA, Wilkinson ML, Kon Y, Kung LA, Wise KJ, Lopez-Hoyo N, Jiang L, Piccirillo S, Yu H, Gerstein M, Dumont ME, Phizicky EM, Snyder M, Grayhack EJ. Biochemical and genetic analysis of the yeast proteome with a movable ORF collection. Genes Dev. 2005;19:2816–26. doi: 10.1101/gad.1362105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kyte J, Doolittle RF. A simple method for displaying the hydropathic character of a protein. J Mol Biol. 1982;157:105–32. doi: 10.1016/0022-2836(82)90515-0. [DOI] [PubMed] [Google Scholar]
- 37.Goh CS, Lan N, Douglas SM, Wu B, Echols N, Smith A, Milburn D, Montelione GT, Zhao H, Gerstein M. Mining the structural genomics pipeline: identification of protein properties that affect high-throughput experimental analysis. J Mol Biol. 2004;336:115–30. doi: 10.1016/j.jmb.2003.11.053. [DOI] [PubMed] [Google Scholar]
- 38.Gavin AC, Bosche M, Krause R, Grandi P, Marzioch M, Bauer A, Schultz J, Rick JM, Michon AM, Cruciat CM, Remor M, Hofert C, Schelder M, Brajenovic M, Ruffner H, Merino A, Klein K, Hudak M, Dickson D, Rudi T, Gnau V, Bauch A, Bastuck S, Huhse B, Leutwein C, Heurtier MA, Copley RR, Edelmann A, Querfurth E, Rybin V, Drewes G, Raida M, Bouwmeester T, Bork P, Seraphin B, Kuster B, Neubauer G, Superti-Furga G. Functional organization of the yeast proteome by systematic analysis of protein complexes. Nature. 2002;415:141–7. doi: 10.1038/415141a. [DOI] [PubMed] [Google Scholar]
- 39.Ghaemmaghami S, Huh WK, Bower K, Howson RW, Belle A, Dephoure N, O’Shea EK, Weissman JS. Global analysis of protein expression in yeast. Nature. 2003;425:737–41. doi: 10.1038/nature02046. [DOI] [PubMed] [Google Scholar]
- 40.Huh WK, Falvo JV, Gerke LC, Carroll AS, Howson RW, Weissman JS, O’Shea EK. Global analysis of protein localization in budding yeast. Nature. 2003;425:686–91. doi: 10.1038/nature02026. [DOI] [PubMed] [Google Scholar]
- 41.Khademi S, Stroud RM. The Amt/MEP/Rh family: structure of AmtB and the mechanism of ammonia gas conduction. Physiology (Bethesda) 2006;21:419–29. doi: 10.1152/physiol.00051.2005. [DOI] [PubMed] [Google Scholar]
- 42.Gruswitz F, O’Connell J, 3rd, Stroud RM. Inhibitory complex of the transmembrane ammonia channel, AmtB, and the cytosolic regulatory protein, GlnK, at 1.96 A. Proc Natl Acad Sci U S A. 2007;104:42–7. doi: 10.1073/pnas.0609796104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rapp M, Seppala S, Granseth E, von Heijne G. Emulating membrane protein evolution by rational design. Science. 2007;315:1282–4. doi: 10.1126/science.1135406. [DOI] [PubMed] [Google Scholar]
- 44.Leabman MK, Huang CC, DeYoung J, Carlson EJ, Taylor TR, de la Cruz M, Johns SJ, Stryke D, Kawamoto M, Urban TJ, Kroetz DL, Ferrin TE, Clark AG, Risch N, Herskowitz I, Giacomini KM. Natural variation in human membrane transporter genes reveals evolutionary and functional constraints. Proc Natl Acad Sci U S A. 2003;100:5896–901. doi: 10.1073/pnas.0730857100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Winzeler EA, Shoemaker DD, Astromoff A, Liang H, Anderson K, Andre B, Bangham R, Benito R, Boeke JD, Bussey H, Chu AM, Connelly C, Davis K, Dietrich F, Dow SW, El Bakkoury M, Foury F, Friend SH, Gentalen E, Giaever G, Hegemann JH, Jones T, Laub M, Liao H, Liebundguth N, Lockhart DJ, Lucau-Danila A, Lussier M, M’Rabet N, Menard P, Mittmann M, Pai C, Rebischung C, Revuelta JL, Riles L, Roberts CJ, Ross-MacDonald P, Scherens B, Snyder M, Sookhai-Mahadeo S, Storms RK, Veronneau S, Voet M, Volckaert G, Ward TR, Wysocki R, Yen GS, Yu K, Zimmermann K, Philippsen P, Johnston M, Davis RW. Functional characterization of the S. cerevisiae genome by gene deletion and parallel analysis. Science. 1999;285:901–6. doi: 10.1126/science.285.5429.901. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.