

Abstract
Since the completion of the genome sequence of Saccharomyces cerevisiae in 19961,2, there has been an exponential increase in complete genome sequences accompanied by great advances in our understanding of genome evolution. Although little is known about the natural and life histories of yeasts in the wild, there are an increasing number of studies looking at ecological and geographic distributions3,4, population structure5-8, and sexual versus asexual reproduction9,10. Less well understood at the whole genome level are the evolutionary processes acting within populations and species leading to adaptation to different environments, phenotypic differences and reproductive isolation. Here we present one- to four-fold or more coverage of the genome sequences of over seventy isolates of the baker's yeast, S. cerevisiae, and its closest relative, S. paradoxus. We examine variation in gene content, SNPs, indels, copy numbers and transposable elements. We find that phenotypic variation broadly correlates with global genome-wide phylogenetic relationships. Interestingly, S. paradoxus populations are well delineated along geographic boundaries while the variation among worldwide S. cerevisiae isolates shows less differentiation and is comparable to a single S. paradoxus population. Rather than one or two domestication events leading to the extant baker's yeasts, the population structure of S. cerevisiae consists of a few well-defined geographically isolated lineages and many different mosaics of these lineages, supporting the idea that human influence provided the opportunity for cross-breeding and production of new combinations of pre-existing variation.
The baker's yeast, S. cerevisiae, has had a long association with human activity11, leading to the idea that use in fermentation lead to domestication. Two domestication events have been suggested, one for sake strains and one for wine12. In contrast, its closest relative, S. paradoxus, has never been associated with human activity and is found globally, sometimes in the same locations as S. cerevisiae3,4. A preliminary comparison within the Saccharomyces sensu stricto group exhibited extensive variation between S. paradoxus populations on different continents but limited variation among S. cerevisiae isolates and no correlation with geographic location8.
Here we report nearly complete genome sequences of S. cerevisiae and S. paradoxus from a large variety of sources and locations (Table S1 and S2). The S. cerevisiae strains included the reference S288c plus other lab, pathogenic, baking, wine, food spoilage, natural fermentation, sake, probiotic and plant isolates. The S. paradoxus isolates were mostly from oak tree bark from the three recognised populations6,8,13 as well as Siberia, Hawaii and the previously designated S. cariocanus14. There is overlap in the general geographic sources of isolates from both species. The majority of strains were sequenced using Sanger sequencing on ABI3730s. For some strains sequence was obtained using the Illumina Genetic Analyser (IGA). Most strains were covered to a depth of 1-4X with a few covered more extensively (Table S3). The sequence reads, assemblies, alignments, a BLAST tool and a genome browser are all publicly available15.
We identified 235127 high-quality SNPs and 14051 indels in the S. cerevisiae nuclear genome, and 623287 SNPs and 25267 indels in S. paradoxus. Our S288c sequence differs from the reference genome by 498 high-quality unambiguous SNPs (Fig. S1). For 480 SNPs our S288c sequence is supported by other strains whereas the reference has no support, while for 18 SNPs the reference sequence is supported by other strains while ours is not. Many of the former are likely to represent errors in the reference sequence (Table S4). The reference sequence for the type strain of S. paradoxus16 was not complete and so we sequenced the type strain CBS432 to 4.3X coverage with ABI and 80X with IGA.
Sequence surveys allow novel sequences not found in the reference genome to be identified. The proportions of unplaced reads for each strain are shown in Table S2. We found 38 new hypothetical ORFs in these sequences that are likely to be real. These ORFs are present in more than one strain (Fig. S2), with some specific to a single lineage, such as the SGRP hypothetical protein 5 (see SI) in the West African lineage, which contains a conserved methyltransferase domain. Much of the unplaced material is subtelomeric. This is in contrast to a genome-wide analysis of copy number based on the numbers of reads of each strain aligning to each gene in the reference sequence, which showed very little significant copy number variation (CNV) outside the rDNA region (see SI).
Neighbour-joining (NJ) phylogenetic trees based on pairwise SNP differences in the alignments were generated (Fig. 1 and S3). The S. paradoxus strains fall into the three previously described populations, plus one isolate from Hawaii. Most of the SNPs in S. paradoxus are private polymorphisms within each population, resulting in a clear separation of the three populations17 (Fig. 2A). The European population was sampled extensively, which provided a picture of within-population structure (Fig. 1B).
Fig.1. Saccharomyces phylogenomics.

NJ trees based on SNP differences of a, S. cerevisiae and S. paradoxus strains sequenced in this project, using S. mikatae, S. kudriavzevii and S. bayanus as outgroups; b, Close-up of the European S. paradoxus, with UK isolates highlighted in violet; c, S. cerevisiae strains with clean lineages highlighted in grey, with colour indicating source (name) and geographic origin (dots).
Fig. 2. Saccharomyces population structure.
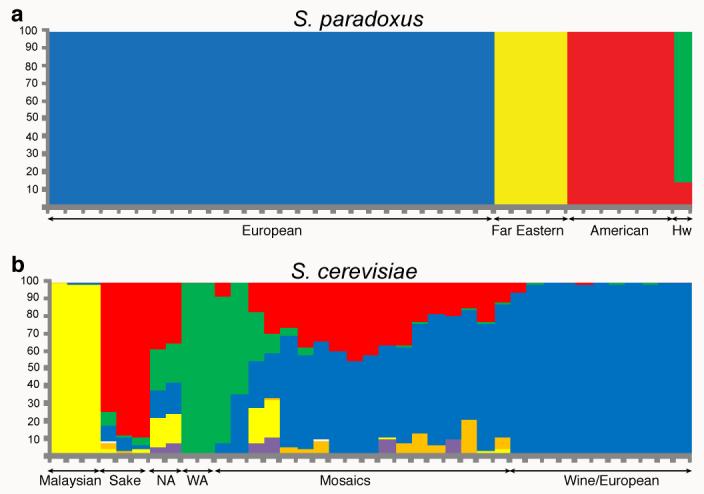
a, Inference of population structure using STRUCTURE on S. paradoxus (markers: 7544 SNPs with >30 strains passing neighbourhood quality standard, NQS), assuming K=6 subpopulations and correlated allele frequencies, linkage model based on marker distances in basepairs, 15000 iteration burn in, and 5000 iterations of sampling. Each mark on the x axis represents one strain, and the blocks of colour represent the fraction of the genetic material in each strain assigned to each cluster.
b, As a, but for S. cerevisiae (markers: 3413 SNPs with >30 strains passing NQS)
The S. cerevisiae population structure is more complex. There are five lineages that exhibit the same phylogenetic relationship across their entire genomes, which we consider to be ‘clean’ non-mosaic lineages (Fig. 1C). These are strains from Malaysia, West Africa, sake and related fermentations (Sake), North America, and a large cluster of mixed sources containing many European and wine strains (Wine/European). The remaining strains are on long branches between the Wine/European cluster and the other four clean lineages. While some lineages correspond to geographic origin, such as those from North America and Malaysia, many closely related strains are from widely separated locations. This mixed architecture could be due to human traffic in yeast strains and subsequent recombination between them. Analysis with STRUCTURE is consistent with separate populations for the West African, Malaysian, Sake and Wine/European lineages (Fig. 2B). The North American isolates share some polymorphisms with all four separate populations while the rest of the strains share polymorphisms with the European lineage and at least one other population. Analysis of SNP distributions (Table S5) is consistent with the NJ tree phylogeny (Fig. 1C) and the STRUCTURE analysis (Fig 2B). Each clean lineage is monomorphic for the majority of segregating sites while the mosaics are polymorphic for the majority of sites.
Phylogenetic trees constructed for individual chromosomes or smaller segments (Fig. S4) demonstrate the mosaic nature of these genomes, as do segmental comparisons (Fig. S5). For example, the laboratory strains SK1 and Y55 appear to be the result of recent crosses between the West African lineage and the European lineage (Fig. S5B). Similarly, W303 is a recent cross between the reference S288c lineage and one or more other lineages. Different segments of the mosaics fall into different locations in the NJ tree (Fig. S4). The recently sequenced clinical derivative YJM78918 is another example. This complex population structure of S. cerevisiae is seen in a similar study reported in this issue19 and is consistent with five well-delineated lineages, two of which contain isolates used in fermentation industries12, plus a number of recombinant strains, many of which are also used for fermentation. Phenotypic profiling (see below), and analyses of rDNA repeat unit variation (Fig. S6) and Ty element abundance (Fig. S7 and Table S6) produce results consistent with this overall picture of the S. cerevisiae population structure.
Has the entire sequence space of S. cerevisiae been sampled? It is clear that segments from many of the mosaic strains are not related to any of the five clean lineages and are probably derived from yet to be determined or no longer existing lineages. A quarter (24%) of SNPs are found only in the mosaics (Table S5), which provides a measure of the unsampled S. cerevisiae species space.
Sequence variability was quantified using the average pairwise divergence within a population (θπ) and the proportion of polymorphic sites (θS)10. We estimated these parameters for various populations (Table S7). Both θπ and θS are about 0.001 in the UK population of S. paradoxus. The Wine/European cluster of S. cerevisiae has approximately the same level of diversity. In both the global and Wine/European samples of S. cerevisiae Tajima's D20 is significantly negative, indicating an excess of singleton polymorphisms, which may be a consequence of our sampling strategy. By contrast, the UK sample of S. paradoxus from a single population has a positive Tajima's D, though not significantly, indicating a relative abundance of mid-frequency polymorphisms. Linkage disequilibrium differs between samples (Fig. 3A). For S. paradoxus linkage disequilibrium declines smoothly with distance, decaying to half its maximum value at about 9kb, as previously reported10. For both S. cerevisiae samples the linkage disequilibrium decays much faster, with a half maximum at 3kb or less. This implies more recombination in S. cerevisiae, perhaps due to more opportunities for strains to mate and recombine.
Fig. 3. Population genomics: variation and selection.
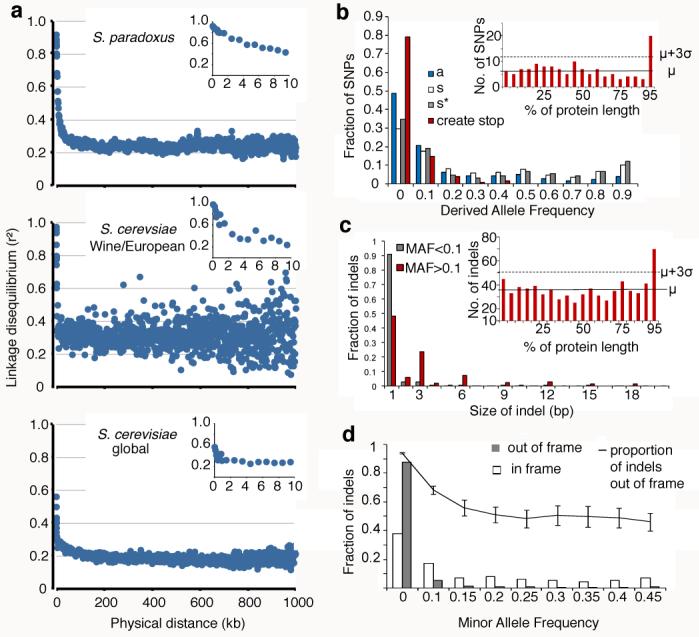
a, Linkage disequilibrium as a function of distance averaged over 1kb. Insets show the decline in linkage disequilibrium over the first 10kb. Details shown in Table S7.
b, Derived allele frequencies of SNPs in coding regions. Amino acid changing SNPs (‘a’) show an excess of low frequencies compared to synonymous SNPs (‘s’). Synonymous SNPs in genes with strong codon bias (‘s*’) are in excess at low and high frequencies. SNPs that create stop codons (‘create stop’) show skew to low frequencies. Inset is the number of mutations occurring over the length of the protein, exceeding three standard deviations from the mean in the C-terminus.
c, Distribution of sizes of indel polymorphisms in coding regions. High frequency indels (>10%, red) more often occur in multiples of 3 than low frequency indels (grey). Inset is as for b.
d, Frequency distribution of indels in coding regions. Out of frame indels (grey) show excess at low frequencies relative to in frame indels (unfilled). The proportion of out of frame indels decreases as frequency increases. Error bars represent the standard error of the proportion.
Patterns of variation can reveal evidence of natural selection. As expected for weakly deleterious mutations, the derived allele frequencies (DAF, SI) for nonsynonymous polymorphism are lower than synonymous polymorphism (Fig. 3B). For polymorphisms with DAF<20%, there were 0.86 amino acid changing polymorphisms for each silent one. In contrast, for those with DAF>20% this ratio was 0.34, indicating that at least 61% (1-0.34/0.86) of the 24418 amino acid changing polymorphisms with DAF<20% are deleterious. Similar calculations (SI) indicate that 27% of non-coding polymorphisms with DAF <20% are deleterious (Fig. S8A). We also performed McDonald-Kreitman tests21 on 1105 genes for which we had enough statistical power. No evidence for positive selection after a multiple testing correction was found (Fig. S8B). These analyses assume that synonymous polymorphisms are neutral. However, we found an excess of polymorphism at both low and high frequency (Fig. 3B) in genes with high codon bias (CAI >0.6). Further analysis (SI) indicates that codon bias in S. cerevisiae is maintained by both purifying and positive selection, as suggested by the mutation-selection-drift model22.
A previous genome-wide study in Arabidopsis23 reported a large number of seemingly highly deleterious alleles. We found 134 mutations that were predicted to introduce stop codons (Fig. 3B), including 5 in genes previously reported to be essential in S288c (SI). These mutations showed a skewed frequency distribution and were enriched in the C-termini (the final 5% of proteins, Fig. 3B inset).
This dataset allowed the consideration of insertions and deletions (Fig. 3C). We identified 3870 indels in the coding regions of the S. cerevisiae population. Of these 731 had minor allele frequency (MAF) greater than 10%. We also found 657 indels (72 with MAF >10%) in genes identified as essential (SI). Indels with MAF >10% predicted to cause frame-shifts were enriched in the C-terminal 5% of the protein (Fig. 3C, inset). The proportion of frame-shift to in-frame indels decreases strongly as a function of MAF (Fig. 3D). For example, at MAF >15% there are 1.0 out-of-frame indels for every in-frame indel, compared to 15.5 at MAF <10%. We estimate that 93% (1-1.0/15.5) of the 2949 out-of-frame indels with MAF <10% are deleterious.
All strains were subjected to high throughput phenotypic analysis under multiple conditions (Fig. 4 and Fig. S9). Growth curves were sampled (>250 time points) over three days and the relevant growth variables: lag (adaptation), rate (slope) and efficiency (maximum density) were extracted24, providing roughly 200 phenotypic traits. The phenotypic variation allowed clustering of strains. There is a high qualitative overlap between the phenotypic clustering and the phylogenies based on SNPs (Fig. 4 and Fig. 1). Also the correlation between genotypic and phenotypic similarity within S. cerevisiae is surprisingly good (Spearman rank test, correlation coefficient = 0.30, p=10−26) given that conventional phenotypic taxonomy generally fails even to resolve the Saccharomyces sensu stricto species. No individual environment determined the overall correlation between genotype and phenotype.
Fig. 4. Saccharomyces phenotype variation.

A selection of growth phenotypes for S. cerevisiae and S. paradoxus strains in different environments and drugs. The complete set of lag, rate and density phenotypes in 67 environments is displayed in Fig. S9. Phenotypes were quantified using high-resolution micro-cultivation measurements of population density. Strain (n=2) doubling time (rate) phenotypes in relation to the S288c derivative BY4741 (n=20) are displayed. Green = poor growth, red = good growth. Hierarchical clustering of phenotypes was performed using a centered Pearson correlation metric and average linkage mapping. Blue = S. paradoxus, pink = S. cerevisiae, grey = S. bayanus isolate CBS7001.
The S. paradoxus strains were well separated from the S. cerevisiae strains (Fig. 4), except for the Hawaiian isolate. The phenotypes most clearly (p<10−9) separating the two species were strong S. paradoxus resistance to cycloheximide and sensitivity to paramomycin, heat and copper (Fig. S10A). The S. cerevisiae isolates fell into two groups (Fig. 4). One contains most of the Wine/European, Sake lineages and most of the long-branch recombinants, while the other mainly consists of the North American, Malaysian and African lineages. The main phenotypic characteristic separating these groups is rapid growth (short lag and steep slope in rate, p<10−4) for the Wine/European and mosaics, which could be advantageous for the fermentation processes many of these strains are used for (Fig. S10B). Despite genomic variation, S. paradoxus strains (excluding the Hawaiian isolate) show 38% lower phenotypic variation than S. cerevisiae strains (p=0.002). In S. cerevisiae, the phenotypic variance is as high among the clean lineages as among the mosaic lineages (p=0.78). Hence, the higher phenotypic variance in S. cerevisiae is not driven by outbreeding or domestication per se, but rather suggests that S. cerevisiae occupies a wider diversity of ecological niches than S. paradoxus.
This survey of S. cerevisiae and S. paradoxus population genomics reveals extensive differences in genomic and phenotypic variation despite ecological similarities and will allow rapid fine mapping of the genetic determinants. Is there evidence of domestication in S. cerevisiae as previously debated12? One could interpret our results in two ways. One is a domestication of one or two groups, the Wine/European and Sake strains, with selection for improved fermentation properties. These domesticated groups then gave rise to feral and clinical derivatives as well as being involved in the generation of outcrossed derivatives found in all sources. Alternatively, human activity simply may have utilised existing strains from populations that had appropriate fermentation properties providing the opportunity to outbreed through movement of strains as well as providing a novel disturbed environment. Using domestication to imply “species bred in captivity”25 the strains that best fulfil this definition are the baking isolates as they have clearly arisen from crosses between lineages. Lineages that were selected from captively bred strains would be expected to have lower diversity than other lineages. This is not the case for the Wine/European or Sake lineages, which have similar or greater levels of diversity compared to the other clean lineages or to S. paradoxus populations. This view of human activity simply moving yeast strains around without captive breeding is consistent with analysis of over 600 strains26. Recent findings in the Malaysian rainforest (from which our three Malaysian S. cerevisiae strains were isolated) of chronic intake of alcoholic nectar from bertram palm by wild treeshrews suggest that the association of fermented beverages and primates is ancient and not exclusive to humans27.
Beyond the analyses we have presented here, the sequence data we have obtained for these strains have many other applications, and have already been used both for global28 and gene specific29 studies. With the advent of new sequencing technology it is becoming possible to undertake similar population genomic studies for species with much larger genomes, including human30, enabling a new era of genome wide evolutionary and functional genetics.
Methods Summary
Strains to be sequenced were selected in order to maximise the variety of sources and locations of isolation. Except for laboratory strains, a single meiotic diploid spore was isolated from the original strain to remove any heterozygosity8. DNA was extracted from overnight cultures8 for subsequent sequencing on ABI3730s and an Illumina Genetic Analyser15. Reference-based genome assemblies were created for each strain in a series of steps15. Each read was aligned to the reference genome (S288c or CBS432). As this approach cannot deal with large indels or with sequences not present in the reference genome, we developed an iterative parallel alignment assembling tool, PALAS (see Supplementary Methods), to introduce insertions that were allowed to share material between related strains. Two versions of each strain sequence were produced, a partial assembly derived just from data collected from that strain, and a more complete assembly using an imputation process to infer the most likely sequence of the strain taking into account data from related strains. In both cases confidence estimates are given for each base call. The SNPs obtained were used to generate Neighbour-joining phylogenetic trees15, infer population structure17, estimate sequence divergence10, analyse polymorphisms10. Non-aligned reads (those missing in the reference genome) were searched for potential novel genes. Each strain isolate was subjected to precise phenotyping in 67 experimental conditions using a high-resolution micro-cultivation Bioscreen C (Growth curve Oy, Finland)24. Two consecutive rounds of 48-hour pre-cultivation in SC media were followed by a 72-hour cultivation in stress media. Readings of optical density were taken every 20 minutes. Strains were tested as duplicates (N=2). Growth variables were normalized to the behaviour of the 20 BY4741 replicates.
Details of the methods mentioned above are provided in Supplementary Information.
Supplementary Material
‘Supplementary Information accompanies the paper on www.nature.com/nature.
Acknowledgements
We thank all the members of the Sanger sequencing production teams for generating the sequence data. We thank members of the Durbin and Louis laboratories and Conrad Nieduszynski for comments and suggestions, to L. Kruglyak and J. Schacherer for sharing their unpublished manuscript and to Ryan Ames and Simon Lovell for sharing unpublished results. We also thank the British Council and Chinese Academy of Sciences for providing the opportunity to conceive and develop this project. Research at The Wellcome Trust Sanger Institute (D.M.C., A.M.M., L.P, M.J., M.A.Q., I.G., S.S., F.S. and R.D.) is supported by The Wellcome Trust. G.L., D.B.H.B., E.B. and E.J.L. were supported by The Wellcome Trust, The Royal Society and the BBSRC. S.A.J., R.P.D. and I.N.R. were supported by the BBSRC. A.B. and J.W. were supported by the Swedish Research Council and the Swedish Foundation for Strategic Research. A.B., V.K. were supported by NERC and I.J.T. by The Wellcome Trust. D.B. was supported by NERC. A.M.M. was supported by Canada Foundation for Innovation. M.J.T.O. and A.V. were supported by NSF, NIH and a Hertz fellowship.
References
- 1.Goffeau A, et al. Life with 6000 genes. Science. 1996;274:546, 563–7. doi: 10.1126/science.274.5287.546. [DOI] [PubMed] [Google Scholar]
- 2.Mewes HW, et al. Overview of the yeast genome. Nature. 1997;387:7–65. doi: 10.1038/42755. [DOI] [PubMed] [Google Scholar]
- 3.Sampaio JP, Goncalves P. Natural populations of Saccharomyces kudriavzevii in Portugal are associated with oak bark and are sympatric with S. cerevisiae and S. paradoxus. Appl Environ Microbiol. 2008;74:2144–52. doi: 10.1128/AEM.02396-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sniegowski PD, Dombrowski PG, Fingerman E. Saccharomyces cerevisiae and Saccharomyces paradoxus coexist in a natural woodland site in North America and display different levels of reproductive isolation from European conspecifics. FEMS Yeast Res. 2002;1:299–306. doi: 10.1111/j.1567-1364.2002.tb00048.x. [DOI] [PubMed] [Google Scholar]
- 5.Aa E, Townsend JP, Adams RI, Nielsen KM, Taylor JW. Population structure and gene evolution in Saccharomyces cerevisiae. FEMS Yeast Res. 2006;6:702–15. doi: 10.1111/j.1567-1364.2006.00059.x. [DOI] [PubMed] [Google Scholar]
- 6.Koufopanou V, Hughes J, Bell G, Burt A. The spatial scale of genetic differentiation in a model organism: the wild yeast Saccharomyces paradoxus. Philos Trans R Soc Lond B Biol Sci. 2006 doi: 10.1098/rstb.2006.1922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kuehne HA, Murphy HA, Francis CA, Sniegowski PD. Allopatric divergence, secondary contact, and genetic isolation in wild yeast populations. Curr Biol. 2007;17:407–11. doi: 10.1016/j.cub.2006.12.047. [DOI] [PubMed] [Google Scholar]
- 8.Liti G, Barton DB, Louis EJ. Sequence diversity, reproductive isolation and species concepts in Saccharomyces. Genetics. 2006;174:839–50. doi: 10.1534/genetics.106.062166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ruderfer DM, Pratt SC, Seidel HS, Kruglyak L. Population genomic analysis of outcrossing and recombination in yeast. Nat Genet. 2006;38:1077–81. doi: 10.1038/ng1859. [DOI] [PubMed] [Google Scholar]
- 10.Tsai IJ, Bensasson D, Burt A, Koufopanou V. Population genomics of the wild yeast Saccharomyces paradoxus: Quantifying the life cycle. Proc Natl Acad Sci U S A. 2008;105:4957–62. doi: 10.1073/pnas.0707314105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pretorius IS. Tailoring wine yeast for the new millennium: novel approaches to the ancient art of winemaking. Yeast. 2000;16:675–729. doi: 10.1002/1097-0061(20000615)16:8<675::AID-YEA585>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 12.Fay JC, Benavides JA. Evidence for domesticated and wild populations of Saccharomyces cerevisiae. PLoS Genet. 2005;1:66–71. doi: 10.1371/journal.pgen.0010005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liti G, Peruffo A, James SA, Roberts IN, Louis EJ. Inferences of evolutionary relationships from a population survey of LTR-retrotransposons and telomeric-associated sequences in the Saccharomyces sensu stricto complex. Yeast. 2005;22:177–92. doi: 10.1002/yea.1200. [DOI] [PubMed] [Google Scholar]
- 14.Naumov GI, James SA, Naumova ES, Louis EJ, Roberts IN. Three new species in the Saccharomyces sensu stricto complex: Saccharomyces cariocanus, Saccharomyces kudriavzevii and Saccharomyces mikatae. Int J Syst Evol Microbiol. 2000;50(Pt 5):1931–42. doi: 10.1099/00207713-50-5-1931. [DOI] [PubMed] [Google Scholar]
- 15.Carter DM. Saccharomyces Genome Resequencing Project. 2005. http://www.sanger.ac.uk/Teams/Team118/sgrp/
- 16.Kellis M, Patterson N, Endrizzi M, Birren B, Lander ES. Sequencing and comparison of yeast species to identify genes and regulatory elements. Nature. 2003;423:241–54. doi: 10.1038/nature01644. [DOI] [PubMed] [Google Scholar]
- 17.Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155:945–59. doi: 10.1093/genetics/155.2.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wei W, et al. Genome sequencing and comparative analysis of Saccharomyces cerevisiae strain YJM789. Proc Natl Acad Sci U S A. 2007;104:12825–30. doi: 10.1073/pnas.0701291104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schacherer J, et al. This issue.
- 20.Tajima F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 1989;123:585–95. doi: 10.1093/genetics/123.3.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McDonald JH, Kreitman M. Adaptive protein evolution at the Adh locus in Drosophila. Nature. 1991;351:652–4. doi: 10.1038/351652a0. [DOI] [PubMed] [Google Scholar]
- 22.Bulmer M. The selection-mutation-drift theory of synonymous codon usage. Genetics. 1991;129:897–907. doi: 10.1093/genetics/129.3.897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Clark RM, et al. Common sequence polymorphisms shaping genetic diversity in Arabidopsis thaliana. Science. 2007;317:338–42. doi: 10.1126/science.1138632. [DOI] [PubMed] [Google Scholar]
- 24.Warringer J, Blomberg A. Automated screening in environmental arrays allows analysis of quantitative phenotypic profiles in Saccharomyces cerevisiae. Yeast. 2003;20:53–67. doi: 10.1002/yea.931. [DOI] [PubMed] [Google Scholar]
- 25.Diamond J. Evolution, consequences and future of plant and animal domestication. Nature. 2002;418:700–7. doi: 10.1038/nature01019. [DOI] [PubMed] [Google Scholar]
- 26.Legras JL, Merdinoglu D, Cornuet JM, Karst F. Bread, beer and wine: Saccharomyces cerevisiae diversity reflects human history. Mol Ecol. 2007;16:2091–102. doi: 10.1111/j.1365-294X.2007.03266.x. [DOI] [PubMed] [Google Scholar]
- 27.Wiens F, et al. Chronic intake of fermented floral nectar by wild treeshrews. Proc Natl Acad Sci U S A. 2008;105:10426–31. doi: 10.1073/pnas.0801628105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mancera E, Bourgon R, Brozzi A, Huber W, Steinmetz LM. High-resolution mapping of meiotic crossovers and non-crossovers in yeast. Nature. 2008;454:479–85. doi: 10.1038/nature07135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Demogines A, Wong A, Aquadro C, Alani E. Incompatibilities involving yeast mismatch repair genes: a role for genetic modifiers and implications for disease penetrance and variation in genomic mutation rates. PLoS Genet. 2008;4:e1000103. doi: 10.1371/journal.pgen.1000103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Siva N. 1000 Genomes project. Nat Biotechnol. 2008;26:256. doi: 10.1038/nbt0308-256b. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.