

Abstract
Tazobactam, sulbactam, and clavulanic acid are the only β-lactamase inhibitors in clinical use. Comparative inhibitory activities of clavulanic acid, sulbactam, and tazobactam against clinically important β-lactamases conclude that tazobactam is superior to both clavulanic acid and sulbactam. Thus far, the majority of explanations for this phenomenon have relied on kinetic studies, which report differences in the ligands’ apparent dissociation constants and number of turnovers before inactivation. Due their innate limitations, these investigations do not examine the identity of intermediates on the reaction pathway and relate them to the efficacy of the inhibitors. In the present study, the reactions between the three inhibitors and SHV-1 β-lactamase have been examined in single crystals using a Raman microscope. The results show that tazobactam forms a predominant population of trans-enamine, a chemically-inert species, with SHV-1 while clavulanate and sulbactam form a mixture of trans-enamine and two labile species, the cis-enamine and imine. The same reactions are then reexamined using a deacylation-deficient variant, SHV E166A, that has been used to trap acyl-enzyme intermediates for X-ray crystallographic analysis. Our Raman data show that significant differences exist between the wild-type and SHV E166A acyl-enzyme populations. Namely, compared to SHV-1, sulbactam shows significantly smaller populations of cis-enamine and imine in the E166A variant while clavulanate exists almost exclusively as trans-enamine in the E166A active site. Using clavulanate as an example, we also show that Raman crystallography can provide novel information on the presence of multiple conformers or tautomers for intermediates within a complex reaction pathway. These insights caution against the interpretation of experimental data obtained with deacylation-deficient β-lactamases to make mechanistic conclusions about inhibitors within the enzyme.
Keywords: SHV-1, E166A, Raman spectroscopy, Raman crystallography, sulbactam, tazobactam, clavulanic acid, mechanism-based inhibitor
INTRODUCTION
β-lactamase production is the most common mechanism by which Gram-negative bacteria become resistant to β-lactam antibiotics such as penicillins and cephalosporins.1, 2 TEM-1 and SHV-1 share 68% sequence identity and are the most commonly encountered class A β-lactamases in Gram-negative bacteria.3 Up to 90% of ampicillin resistance in E. coli is due to the production of TEM-1, whereas SHV-1 is responsible for up to 20% of the plasmid-mediated ampicillin resistance in K. pneumonia.1 Thus, in present day clinical practice, bacterial infections are often treated with two drugs, one such as penicillin to inhibit bacterial cell wall growth and the second (see, for example, Chart 1) to inhibit the β-lactamases produced by the bacteria, thereby blocking penicillin hydrolysis.2 Currently, four classes of β-lactamase enzymes have been described. The Ambler class A, C and D enzymes are serine hydrolases with the fourth class being metallo-enzymes.4–7 The serine β-lactamases share a mechanism whereby their active site serine attacks the carbonyl carbon of the β-lactam ring of the inhibitors shown in Chart 1 to form an acyl enzyme complex (Scheme I). Subsequent 5-membered ring opening occurs and products can be removed from the active site in a deacylation step that regenerates the active enzyme.8
Chart 1.

Schematic diagram of the mechanism-based inhibitors clavulanate, sulbactam, and tazobactam.
Scheme 1.

Proposed reaction scheme for sulbactam in class A β-lactamases. A similar reaction mechanism is accepted for β-lactamase inhibitors tazobactam and clavulanate.
Sulbactam, tazobactam, and clavulanic acid (Chart 1), prevent the degradation of β- lactam molecules by inhibiting serine β-lactamases. As shown in Scheme I, in addition to a final hydrolysis step and regeneration of the active enzyme, these competitive inhibitors can be converted to long-lived or irreversible inhibitors in the active site of the enzyme. For the serine-based enzymes, formation of a Michaelis complex positions the inhibitor’s lactam carbonyl in the oxyanion hole, which involves hydrogen bonding to two backbone amides and significant enzyme-induced C=O polarization.9 This effectively increases the contribution of the C+-O− resonance form to the C=O bond and facilitates nucleophilic attack by an active-site serine on the lactam carbonyl. Following formation of the acyl-enzyme, a second ring opening, the oxazolidinium ring in the case of clavulanic acid, or the thiazolidinium ring for tazobactam or sulbactam, results in an imine (species 1, Scheme I). The imine is thought to be a key intermediate in that it undergoes a number of chemical transformations:
The imine lacks conjugation with the carbonyl of the acyl-enzyme, which makes the serine ester more susceptible to hydrolysis than the tautomeric enamine. Hydrolysis frees the enzyme from inhibition and imine of the acid product may further hydrolyze to malonic semialdehyde. In a parallel pathway, initial hydrolysis of the imine will an acyl enzyme malonic semialdehyde whose subsequent hydrolysis also liberates free malonic semialdehyde (species 5, Scheme 1).
Secondly, attack on the C5 position of the imine by a second nucleophilic amino acid side chain, S130 in SHV and TEM, forms a cross-linked species in the active site that can degrade to an acrylate-like species irreversibly (species 4) bound to the enzyme. The covalent modification of the S130 has been observed by UV spectroscopy,10 isoelectric focusing,11 mass spectrometry,12, 13 and X-ray crystallography.14 Nonetheless, recent investigations by Raman crystallography showed that sulbactam forms only minimal amounts of irreversible acrylate-enzyme with SHV-1 β-lactamase.15 Because this species does not accumulate on a physiologically relevant time-scale, it is unlikely to contribute to overall inhibition of the enzyme.
Lastly, a series of proton transfers can occur in which the imine acyl-enzyme tautomerizes to yield a more stable cis- or trans-enamine (species 2, 3, Scheme I). These intermediates have been detected by UV spectroscopy and are associated with an absorption band at ~280 nm.10, 16 The enzyme-bound species are stable enough to be recovered using isoelectric focusing and high-performance liquid chromatography;11, 16 but, attempts to clearly discriminate between the cis- or trans-enamine in a crystal structure of SHV-1 inhibited with tazobactam were unsuccessful.14 However, recent combined Raman and X-ray studies have confirmed the presence of substantial trans-enamine populations (species 2) in the E166A (deacylation deficient) SHV-1 enzyme.17–19
Previous Raman data showed that all three inhibitors formed a predominant population of the trans-enamine species using the deacylation deficient mutant, E166A.17–19 Class A β-lactamases utilize an invariant glutamic acid residue, E166, to activate a water molecule for nucleophilic attack on the acyl-enzyme.20, 21 An aliphatic residue, such as alanine, cannot activate this water molecule and prevents regeneration of active enzyme by blocking hydrolysis of species 1, 2, and 3.22–24 For the E166A mutant, the accumulation of reaction intermediates in the active site, particularly the trans-enamine species, facilitates their detection by Raman difference spectroscopy and analysis by X-ray crystallography.17–19 However, characterization of the reaction between wild-type SHV-1 and mechanism-based inhibitors proves challenging due to an increased deacylation rate constant, decreased acyl-enzyme populations, and the presence of reaction intermediates other than the trans-enamine. Monitoring the nature of the intermediates, their relative populations, and their time courses of formation and disappearance provides insight into the key steps in β-lactamase inhibition and the effect of enzyme mutations on the formation of these intermediates.25, 26 TEM and SHV enzymes often contain single point mutations that confer resistance to β-lactamase inhibitors.2, 3, 27 In the TEM family, clinical mutants containing M69I, -L, and -V, R244S, and S130G mutations have been reported. In SHV, clinical mutants containing the M69I and S130G mutations have been isolated. Because resistance is directly related to the composition of intermediates formed between inhibitor and β-lactamase, a detailed study of these reaction mechanisms is warranted.
Here we present steady-state Raman difference spectra of acyl-enzyme derived intermediates in single crystals of the β-lactamase enzyme. The spectra are paired with extensive quantum mechanical calculations on models for enzyme-bound intermediates that permit characterization of inhibitor degradation products, such as the imine and cis- and trans-enamine species. Two enzymes are used to acquire spectra of the acyl-enzymes: wild-type SHV-1 and SHV E166A, a deacylation-deficient laboratory mutant. X-ray structures of the trans-enamine acyl-enzyme of SHV E166A and the inhibitors tazobactam, sulbactam, and clavulanate have served as good starting points for the rational design of new inhibitors.18, 19, 28 However, by comparing the difference spectra obtained with SHV-1 and E166A, this report will show that significant differences exist between the wild-type and E166A acyl-enzyme populations. Consequently, strategic drug design must take into account the identity and composition of reaction intermediates formed with the wild-type enzyme. Laboratory mutants, such as E166A, that bias the reaction pathway in order to trap an acyl-enzyme may provide information on intermediates that are not dominant for the wild-type enzyme and therefore may not be a reliable basis for structure-based drug design.
MATERIALS AND METHODS
Inhibitors
Sodium clavulanate (Smith-Kline-Beecham), sulbactam (Pfizer), and tazobactam (Wyeth Pharmaceuticals) were gifts of the respective companies. 6,6-Dideuterosulbactam was a kind gift from John D. Buynak. A detailed synthesis is provided in ref. 15. Stock solutions (20 mM) in 2 mM HEPES buffer (pH 7.0) were prepared for use with the protein crystals.
Protein Isolation and Purification
The SHV-1 and E166A β-lactamase enzymes were isolated and purified as previously described with an additional HPLC purification step performed using a Sephadex Hi Load 26/60 column (Pharmacia, Uppsala, Sweden) and elution with phosphate-buffered saline (pH 7.4).3, 17, 27
Crystallization
SHV-1 and E166A were concentrated to 5 mg/mL in 2 mM HEPES buffer (pH 7.0) for crystallization by the vapor diffusion method using the protocol of Kuzin et al.3 Briefly, the 10 µL sitting protein drop [2 mg/mL, 0.56 mM Cymal-6 detergent (Hampton Research, Laguna Niguel, CA), 15% poly(ethylene glycol) 6000 (Hampton Research, Laguna Niguel, CA), 50 mM HEPES buffer, pH 7.0] was placed over a 0.50 mL reservoir solution containing 30% poly(ethylene glycol) and 100 mM HEPES buffer. The enzyme crystallized in approximately 1 week.
Raman Crystallography
The Raman microscope system has been described previously.29, 30 Using a cryoloop, crystals, typically 300 µm × 300 µm × 300 µm in size, were transferred from the mother liquor solution to a 4 µL drop of 2 mM HEPES. A 647 nm, 80 mW Kr+ laser beam (Innova 70 C, Coherent, Palo Alto, CA) was focused on the protein crystals in a 4 µL hanging drop using the 20× objective of the Raman microscope. During data collection, spectra were acquired for 10 s and 10 accumulations were averaged for each time point. After obtaining spectra of the apo β-lactamase protein crystals, inhibitors were added to the drop to achieve a final volume of 5 µL and a final inhibitor concentration of 5 mM. Spectra were then acquired every 2–3 min. after addition of an inhibitor. To obtain difference spectra, an apo β-lactamase spectrum was subtracted from the protein: inhibitor spectra at varying time intervals following addition of inhibitor. HEPES buffers in D2O were prepared using nondeuterated HEPES and titrated with NaOD to pD 6.55 (pD = pH + 0.45).31 Stock solutions of inhibitors were prepared in the deuterated 2 mM HEPES buffer (pD 7.0). Protein crystals, prepared as described above, were added to a 4 µL drop of 2 mM deuterated HEPES buffer (pD 7.0). Data collecting and processing were performed using HoloGRAMS and GRAMS/AI 7 software (ThermoGalactic, Inc., Salem, NH). Raman spectra of the inhibitors that were used were obtained under similar conditions. Spectra were obtained for 4 µL drops of 100 mM inhibitor solutions prepared in 100 mM HEPES (pH 7.0). Low temperature spectra were collected at 8 °C. To achieve this, the crystal was set up on a coverslip resting on a metal chamber that was connected to a low-temperature thermostatically controlled bath containing a 1:1 mixture of water and glycerin.
Calculations
Ab initio quantum mechanical calculations were performed to predict the Raman spectra of compounds A-G (see Table 1, Table 3, and Table 4) using Gaussian 03.32 Calculations were performed at the DFT level using the 6–31+G(d) basis set. DFT calculations were performed with Becke’s three-parameter hybrid method using the correlation functional of Lee, Yang, and Parr (B3LYP), with 20% HF exact exchange mixing. A standard scaling factor of 0.961 was applied to the calculated values.33
Table 1.
Comparison of the theoretical and experimental O=C-C=C-NH- stretching frequencies of a model trans-enamine acyl-enzyme and isotopologues.
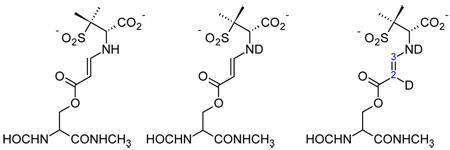 | |||
|---|---|---|---|
| A trans-enamine (-NH-) |
B trans-enamine (-ND-) |
C trans-enamine (-ND-, D at C6) |
|
|
Theoretical O=C-C=C-N- stretch (cm−1) |
1594 | 1590 | 1579 |
|
Experimental O=C-C=C-N- stretch (cm−1) |
1595 | - | 1581 |
Table 3.
Comparison of the theoretical and experimental O=C-C=C-NH- stretching frequencies of a model cis-enamine acyl-enzyme and isotopologue.
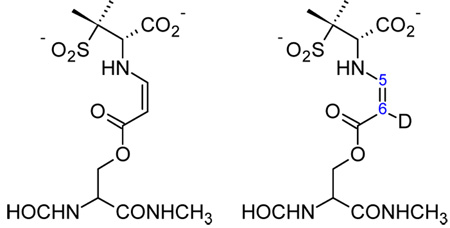 | ||
|---|---|---|
| D cis-enamine (s-cis) |
E cis-enamine (D at C6) |
|
|
Theoretical O=C-C=C-N-stretch (cm−1) |
1585 | 1579 |
|
Experimental O=C-C=C-N-stretch (cm−1) |
1588 | 1581 |
Table 4.
Comparison of the theoretical and experimental C=NH+ stretching frequencies of two tautomeric imines of clavulanate.
 | ||
|---|---|---|
| F protonated imine |
G protonated imine |
|
|
Theoretical C=NH+ stretch (cm−1) |
1663 | 1648 |
|
Experimental C=NH+ stretch (cm−1) |
1658 | 1649 |
RESULTS AND DISCUSSION
Raman Spectra of the Inhibitors
The Raman spectra of tazobactam, sulbactam and clavulanate are shown in Figure 1. The weak band near 1775 cm−1 corresponds to the stretch of the carbonyl in the β-lactam ring. Nucleophilic attack on the lactam carbonyl by the active-site S70 produces acyl-enzymes that result in the disappearance of the lactam carbonyl stretching feature. This indicates breakage of the C-N bond in the lactam ring, and gives rise to a broad, weak carbonyl band between 1720 and 1740 cm−1. The latter feature is often too diffuse to be seen in the Raman spectrum. Following formation of the initial acyl-enzyme, opening of the thiazolidine ring, after departure of the sulfinate group at C5, results in the formation of an imine. In the Raman spectrum, this second ring opening leads to disappearance of the C-S stretch seen near 630 cm−1 in the parent spectra of tazobactam and sulbactam. The imine rapidly partitions amongst three competing pathways: 1) hydrolysis, which regenerates active enzyme, 2) formation of a S130-bound vinyl carboxylic acid species, or 3) rearrangement to a cis- or trans-enamine acyl-enzyme.
Figure 1.

Raman spectra of sulbactam (top), tazobactam (middle), and clavulanate (bottom). The β-lactam carbonyl stretch near 1775 cm−1 is common to the inhibitors and disappears as a result of acylation.
SHV-1 Reacting with Tazobactam Predominantly Forms a trans-Enamine Population
Raman difference spectra from 1200–1800 cm−1 for [SHV-1+tazobactam]-[SHV-1] are shown in Figure 2. The region from 1550–1800 cm−1 is critical to our discussion and contains many bands largely due to stretching modes from a number of acyl-enzyme species; these are described in detail below. In addition, the low intensity feature near 1678 cm−1 is assigned to protein amide I modes due to minor secondary structural changes occurring upon acylation. For the native crystals, the amide I profile, 1620–1700 cm−1, contains contributions at different positions from modes due to α-helices, β-sheets, and unordered regions of polypeptide. When β-strand features appear in the difference spectra, such as the Raman lines near 1680 cm−1 in Figure 2, it indicates a ligand-induced conformational change involving β-structure.34 However, these conformational changes are small: when the intensity at 1680 cm−1 is normalized to the parent amide I band in the apo spectrum, less than 5 percent of the overall amide I band intensity moves to β-space. The residues giving rise to the β-strand features near 1680 cm−1 are in “Ramachandran β-space” but lack the inter-strand hydrogen bonds found in β-sheet. Other major modes include 1460 cm−1, due to protein and acyl group C-H deformations and a peak near 1380 cm−1 due to the symmetric –CO2−1 stretch of tazobactam. The band near 1290 cm−1 is due to the substrate’s triazole moiety, while the 1240 cm−1 feature probably contains contributions from –N-H deformation modes of the acyl group and amide III motions of the protein. The broad features centered at 1500 cm−1 are experimental artifacts due to light scattering from the material of the crystal mounting cell.
Figure 2.

Steady-state Raman difference spectra of the acyl-enzyme derived intermediates from tazobactam with SHV-1 in a) H2O at room temperature, b) H2O at 8 °C, and c) D2O at room temperature.
For tazobactam in the crystal complex, the Raman difference spectra in the double bond stretching region reveal the formation of an intermediate band at 1596 cm−1 in H2O and 1580 cm−1 in D2O (Figure 2a and c, respectively). Based on previous studies with the deacylation deficient variant, SHV E166A, this feature is assigned to the trans-enamine.17 The conjugated O=C-C=C-NH- fragment gives rise to a fairly intense Raman band that responds to NH/ND exchange in D2O. The calculated stretching frequencies for a trans-enamine model compound A and mono- and di-deuterated analogues B and C are shown in Table 1. The calculated stretching frequency for trans-enamine A (1594 cm−1) agrees with the experimental result (1596 cm−1); however, replacing the amine hydrogen of A with deuterium (as in B) does not reproduce the isotope shift observed on NH/ND exchange in Fig. 2c (Table 1). The difference between the experimental and observed isotope shifts suggests that another deuterium from the solvent is incorporated into the enamine skeleton. The imine is formed immediately after the second ring opening and rapidly equilibrates with the cis- and trans-enamine. As illustrated in Scheme 2, deuterium incorporation occurs at C6 after multiple tautomerization events between the enamine and imine species. If a deuterium is included in model compound C at C6, the calculated stretching frequency (1581 cm−1) now agrees with the experimental value (1580 cm−1). If deuterium incorporation at C6 proceeded slowly in D2O, then the trans-enamine peak would shift from 1596 cm−1 to 1580 cm−1 as a function of time. However, time dependence over a period of 1 to 60 min. was not observed which suggests that tautomerization is rapid and deuterium incorporation at C6 is complete before 1 minute. Based on QM calculations, the band observed at 1630 cm−1 is assigned to the antisymmetric –CO2−1 stretch coupled to the O=C-C=C-NH- stretch of the enamine backbone. As illustrated by the Raman frequency at 1612 cm−1 (Figure 2c), this mode also responds to NH/ND exchange due to significant contribution from –NH– bending motions of the trans-enamine. The shift from 1630 to 1612 cm−1 is faithfully reproduced in the calculations. Lastly, the presence of the trans-enamine is also supported by a band at 969 cm−1, which is assigned to the trans HC=CH wag of the enamine (data not shown).
Scheme 2.

Proposed reaction scheme for C6 H/D exchange in D2O upon enamine/imine tautomerization.
We assign the band at 1658 cm−1 to a protonated imine species (Fig. 2a). The strongest evidence for this comes from calculations and chemical modifications of the intermediate involving clavulanate (see below). The corresponding Raman peak for tazobactam is assigned to the C=NH+ stretch of the protonated imine by analogy to clavulanate. In D2O, the hydrogen atom attached to the imine nitrogen is expected to exchange with deuterium after repeated deprotonation/protonation cycles. As anticipated, the NH/ND exchange shifts the 1658 cm−1 mode to 1641 cm−1 (Fig. 2c). Calculations on model compounds indicate that the Raman scattering cross-section of the imine may be up to two times greater than the scattering cross-section of the trans-enamine. As such, the strong feature at 1658 cm−1 only represents a minor population of imine acyl-enzymes.
Additional insights regarding the thermodynamics of protein-ligand interactions can be obtained by comparing data collected at low and room temperature. Thus, using a simple home-built cell, the reaction between tazobactam and SHV-1 was followed in a single crystal at 8 °C. In the difference spectra at 8 °C, several ligand-derived band shapes narrow including the symmetric –CO2−1 stretch near 1380 cm−1, the breathing mode of the triazolyl moiety near 1290 cm−1, and the trans-enamine peak near 1596 cm−1 (Fig. 2b). The change is most striking for the trans-enamine feature whose bandwidth at half-height decreases from 34 cm−1 at room temperature to 28 cm−1 at 8 °C, or approximately 20% (Fig. 2a and 3b, respectively). In Raman spectroscopy, broadened band shapes often result from conformational heterogeneity.18 The broader widths of the tazobactam-derived bands at room temperature are ascribed to the existence of a range of close-lying conformations. Essentially, these have the parent trans-enamine structure found in the tazobactam complex; however, dynamic excursions about the observed mean torsional angles leads to line broadening of the main feature near 1596 cm−1. A crystal structure of the SHV E166A/tazobactam complex, revealed the inhibitor covalently bound in the trans-enamine intermediate state with close to 100% occupancy in the active site. The covalently bound tazobactam participates in a number of interactions with the protein: 1) The inhibitor’s carboxylate moiety forms a 2.7 Å hydrogen bond with the Nδ2 atom of N132. 2) The tazobactam triazolyl moiety’s N17 nitrogen atom makes a 2.7 Å hydrogen bond with a water, which is involved in additional interactions with K234 and S130. 3) In addition to this hydrogen bond, the triazolyl moiety is also involved in hydrophobic interactions with Y105.19 It is assumed that the trans-enamine conformation observed by X-ray crystallography is thermodynamically stable and that the aforementioned protein-ligand interactions (e.g. van der Waals, hydrophobic, hydrogen bonds, etc.) contribute to its enthalpic stabilization. Lowering the temperature selects for the lowest energy trans-enamine conformations, i.e. having the optimal protein-ligand interactions. In the Raman spectrum, these contributions are reflected by narrower band shapes, especially for functional groups that participate in these key interactions, such as the carboxylate and triazole moieties.
Figure 3.

Steady-state Raman difference spectra of the acyl-enzyme derived intermediates from sulbactam with SHV-1 (top) and SHV E166A in H2O at room temperature.
Lastly, the triazole moiety of tazobactam serves as an internal intensity standard to assess the relative contribution of a particular intermediate to the total acyl-enzyme population. This is accomplished by determining the signal ratio of the main triazole mode at 1290 cm−1 to the respective enamine or imine mode. For example, a crystal structure of SHV E166A with tazobactam revealed a covalent bound trans-enamine intermediate with close to 100 percent occupancy.19 In the Raman spectrum of the tazobactam/SHV E166A complex, the ratio of intensities of the trans-enamine peak to the triazole peak (I1595/I1290) is 1.60 (Supporting Figure 1). In the difference spectrum between the wild-type enzyme and tazobactam, the I1595/I1290 is 1.25 (Fig 3a). This signifies that the wild-type enzyme forms slightly less of the trans-enamine intermediate with tazobactam than E166A, which forms a stoichiometric trans-enamine intermediate. The remainder of the acyl-enzyme population for the wild-type enzyme is attributed to the imine. As predicted from the X-ray data, the 1658 cm−1 band of the imine is absent in the E166A difference spectrum with tazobactam (Supporting Figure 1). We conclude that with wild-type SHV-1 tazobactam forms an acyl-enzyme population dominated by the trans-enamine but which does have a minor contribution from the imine. The major peaks in the spectrum are assigned in Table 2.
Table 2.
Raman peak assignments for the major protein and acyl-enzyme bands in the tazobactam/SHV-1 difference spectra shown in Figure 2.
| Vibration | Frequency in H2O (cm−1) | Frequency in D2O (cm−1) |
|---|---|---|
| Amide I β-sheet | 1679 | 1678 |
| v(C=NH+) of imine | 1658 | 1641 |
|
vas(CO2−) coupled to v(O=C-C=C-NH-) |
1629 | 1612 (sh) |
|
v(O=C-C=C-NH-) of trans-enamine |
1596 | 1580 |
| Artifact | 1500 (br) | - |
| vs(CO2−) | 1372 | 1378 |
| triazolyl | 1288 | 1291 |
|
-NH-deformation of ligand and/or protein |
1238 | - |
| trans HC=CH wag | 969 | 930 |
SHV-1 Reacting with Sulbactam Forms a Mixture of Acyl-Enzymes
Sulbactam is another inhibitor of serine β-lactamases and is normally partnered with the common penicillin-based antibiotic ampicillin. In a previous account, it was suggested that sulbactam was a poorer inactivator than tazobactam because it formed less trans-enamine with the deacylation-deficient E166A enzyme.17 The Raman spectra in this section confirm this but now show that wild-type SHV-1 forms even greater populations cis-enamine and imine.
Difference spectra between sulbactam and SHV-1 (top) or SHV E166A (bottom) are shown in Figure 3. With the wild-type enzyme, it is immediately obvious that sulbactam, unlike tazobactam, does not form a predominant population of trans-enamine. Compared to tazobactam, the conjugated O=C-C=C-NH- stretch of the trans-enamine at 1606 cm−1 is up-shifted by 10 cm−1. While the parent trans-enamine structure of sulbactam is chemically identical to tazobactam, the torsional angles in the O=C-C=C-NH- fragment are slightly different from those for the tazo-derived fragment, accounting, in part, for the differences in the position of the stretching feature.18 A peak of similar intensity at 1588 cm−1 is assigned to the O=C-C=C-NH- stretch of the cis-enamine. The most compelling evidence for this assignment comes from combined X-ray and Raman crystallographic studies on the intermediates formed between SHV S130G, an inhibitor resistant β-lactamase, and tazobactam. By X-ray, electron density consistent with a cis-enamine was found in the active site.35 When the same complex was subject to Raman analysis, a prominent peak at 1585 cm−1 was present in the difference spectrum (Supporting Figure 1). While the agreement between the X-ray and Raman data alone allows us to confidently assign the 1585 cm−1 signature to the C=C stretch of the cis-enamine, the assignment is additionally supported by two other factors: a) in simple model olefins, the cis-isomer has its C=C stretch about 20 cm−1 below the signature for trans36 and b) calculations for the cis-enamine, as in compound D (Table 3), reveal good agreement for the major feature near 1588 cm−1. Thus, for wild-type SHV-1, the main assignments in the double bond region are i) 1588 cm−1 cis-enamine, ii) 1606 cm−1 trans-enamine, iii) 1629 cm−1 asymmetric –CO2−1 stretch coupled to enamine skeletal stretch, iv) 1654 cm−1 protonated imine. Therefore, unlike tazobactam, significant populations of imine and cis-enamine co-exist with the trans-enamine. In the difference spectra with E166A, the Raman data show that sulbactam forms a trans-enamine acyl-enzyme that exhibits a relatively intense band near 1603 cm−1 due to a stretching motion of the O=C-C=C-NH. The asymmetry of this main band profile suggests the presence of a feature near 1587 cm−1, which likely results from the O=C-C=C-NH stretch of a small population of cis-enamine (Fig. 3, bottom). The cis/trans-enamine vibrations couple with the asymmetric –CO2 stretch to give an unresolved medium intensity band near 1630 cm−1. Lastly, the 1655 cm−1 band is assigned to the C=NH+ stretch of the imine acyl-enzyme. The assignment of the trans-enamine’s major Raman mode is reported in ref. 17; however, in the present report, an upgraded Raman cell was used to collect the spectra. This latest cell results in an increased signal-to-noise ratio and better resolution of the complex double-bond stretching region. As a result, we are able to confidently assign vibrations of the cis-enamine and imine and extend our previous work. Thus, with sulbactam, both E166A and SHV-1 show evidence for trans- and cis-enamine and an imine population. However, for E166A, the trans-enamine population appears to predominate.
Further insight is provided by following the reaction with 6,6-dideuterosulbactam. The difference spectrum of 6,6-dideuterosulbactam reacting with SHV-1 after 15 min. is shown in Figure 4a. The spectrum is similar to that obtained with sulbactam; however, the positions of key peaks in the double bond region are down-shifted due to isotopic substitution at C6. Additionally, the data (shown at 15, 60, and 90 min. in Fig. 4a, b, and c, respectively) are time- dependent due to the effect of isotope scrambling with the water solvent. We assign the feature at 1581 cm−1 to a deuterated cis-enamine (E, Table 3). In the vibrational spectrum obtained with unlabeled sulbactam, the cis O=C-C=C-NH- group gives rise to a strong feature near 1588 cm−1 (Figure 3). Thus, we anticipated that this stretch down-shifts to 1581 cm−1 when deuterated at C6. QM calculations on deuterated cis-enamine E support this assignment and predict a strong stretching vibration near 1579 cm−1 (Table 3). As was the case with tazobactam, D/H exchange with the solvent results in protium incorporation at C6 after multiple tautomerization events between the enamine and imine species (Scheme 2). This phenomenon is apparent when we compare the 6,6-dideuterosulbactam spectra at 15 and 60 min. (Fig. 4a and 5b, respectively). In these spectra, D/H exchange results in movement of the cis-enamine’s O=C-C=C-NH- stretch from 1581 cm−1 at 15 minutes to 1586 cm−1 at 60 minutes. Note that the cis-enamine frequency at 60 and 90 minutes (Fig. 4b and 5c) with dideuterosulbactam is the same as that obtained with unlabeled sulbactam. This is further confirmation that exchange at C6, although slow, is complete. The peak for the trans-enamine is unresolved in the 1600 cm−1 region at 15 min. (Fig. 4a) but gains intensity throughout the reaction. At 60 minutes, the intensity of the trans form is equal to the cis form and at 90 minutes, the trans stretch at 1602 cm−1 is the dominant feature of the spectrum and appears at initially the same position as that for isotopically unlabeled sulbactam (Fig. 3a).
Figure 4.

Partial Raman difference spectra of the acyl-enzyme derived intermediates from dideuterosulbactam with SHV-1 in H2O at room temperature at a) 10 min., b) 60 min., and c) 90 min. The spectra at 10 and 90 min. are superimposed in panel d.
Figure 5.

Steady-state Raman difference spectra of the acyl-enzyme derived intermediates from clavulanate with SHV-1 (top) or SHV E166A in H2O at room temperature.
From the data presented in this section, we offer two hypotheses explaining why sulbactam is a less effective inhibitor of SHV-1 than tazobactam: 1) it forms absolutely less trans-enamine with the β-lactamase and 2) it forms relatively more of other the reaction intermediates, such as the cis-enamine and deacylation-prone imine. The only difference between sulbactam and tazobactam is the presence of a triazolyl moiety on the C2 β-methyl group of the later. There are two possible explanations as to how the triazolyl group might favor formation of the trans-enamine: 1) rotation of the imine intermediate about the C5-C6 bond is faster in the case of tazobactam than it is when the triazole is absent, as in sulbactam, probably due to some steric clashes in the active site and 2) once the trans-enamine of tazobactam is formed, it is stabilized via interactions between the triazole ring and the protein.19 While these explanations are not mutually exclusive, one thing is clear: removing the triazole group results in an inferior inhibitor.
Clavulanic Acid Predominantly Forms a trans-Enamine with the E166A Mutant but an Imine with SHV-1
In the previous section, we showed that the acyl-enzymes formed between E166A and sulbactam consist predominantly of trans-enamine with minor populations of cis-enamine and imine. The same species occur in the acyl-enzymes formed by wild-type SHV-1 except that the relative populations of cis-enamine and imine increase significantly (Figure 3). The findings with clavulanic acid are even more striking since the spectra for the E166A intermediates reveal an overwhelming population of trans-enamine, with, at best, minimal populations of cis-enamine and imine, whereas in the wild-type enzyme greatly increased populations of imine bring about profound spectral changes (Figure 5). Discussions in the literature emphasize that clavulanic acid and the sulfones inhibit β-lactamases by a similar mechanism. However, the Raman difference analysis emphasizes important mechanistic differences between clavulanate and the sulfones. The spectrum of the clavulanate/SHV E166A complex is shown in Figure 5 (bottom). The Raman line at 1602 cm−1 marks the presence of a predominant population of trans-enamine. This finding is in agreement with an inhibitor-bound X-ray structure of E166A, which also revealed a linear trans-enamine covalently attached to S70.18 Moreover, both the Raman data and X-ray omit density maps indicate that the trans-enamine intermediate is decarboxylated. This is evidenced in the Raman spectrum as an absence in the intensity of the –CO2−1 symmetric stretch near 1400 cm−1 and the coupled –CO2−1 antisymmetric stretch near 1630 cm−1 (see Table 2 for assignments).17, 18 A similar decarboxylated trans-enamine intermediate is made by clavulanate with wild-type SHV-1; however, its precise conformation and relative population in the SHV-1 active site differ from E166A. The frequency of the trans-enamine stretch in wild-type is increased by 10 cm−1. The O=C-C=C-NH stretching frequency is dependent on the O-C-C-C dihedral angle; thus, the conformation of the linear trans-enamine must vary between E166A and wild-type, which leads to minor frequency differences.17–19 To assess the relative amount of trans-enamine formed by either E166A or wild-type SHV-1, the 1696 cm−1 stretch of the C2 ketone can serve an internal standard (see species F in Table 4). This moiety is common to all clavulanate acyl-enzymes following opening of the oxazolidinium ring. Peak intensity ratios of the trans-enamine stretch to the carbonyl stretch are approximately 10 in E166A and 3 in wild-type. These intensities suggest that clavulanate forms a predominant population of trans-enamine with E166A but only a fraction of trans-enamine with wild-type SHV-1. The identities of the other acyl-enzymes are discussed below.
Clavulanate generates a minor population of cis-enamine with both enzymes. Like sulbactam, the 1585 cm−1 shoulder features of both difference spectra in Fig. 5 are assigned to cis O=C-C=C-NH stretch (Table 3 and Figure 5). While a crystal structure is not available for the clavulanate/SHV-1 complex, this intermediate was not detected in the clavulanate/E166A structure. Its absence is attributed to two factors: 1) The cis-enamine is a minor product in the reaction between clavulanate and E166A. Intermediates constituting less than 30% active site occupancy are difficult to observe in X-ray crystallography. 2) Crystals of β-lactamase/inhibitor complexes are flash-cooled to 120 K after ‘soak-in’ and their structures are determined at cryogenic temperatures. Compared to Raman crystallography, where the spectra are recorded at room temperature, the colder temperature used in X-ray analysis favors the more enthalpically stable trans-enamine species.
Peaks at 1662 and 1647 cm−1 are assigned to a pair of imine acyl-enzymes (Figure 5, top). As demonstrated by the spectra, these intermediates are only made in appreciable amounts with the wild-type enzyme. More specifically, the 1662 cm−1 band in the SHV-1/clavulanate complex is assigned to a C5=N4 imine (species 7, Scheme III) that results from cleavage of the C-O bond and concomitant opening of the oxazolidinium ring. Structurally, this imine is analogous to those formed by sulbactam and tazobactam. Tautomerization of imine 7 to imine 8 in Scheme III is driven by resonance with the adjacent ketone at C2. The N4=C3 imine, which is marked by the 1647 cm−1 band in the Raman spectrum, is unique to clavulanate and was first detected by mass spectrometry.37 QM calculations on compound F and G, which are models for the C5=N4 and N4=C3 imines, predict strong C=NH+ stretching modes near 1663 cm−1 and 1648 cm−1, respectively (Table 4). Lastly, the frequency of both the 1662 and 1647 cm−1 species shifts down by approximately 20 cm−1 in D2O due to NH/ND exchange, which further confirms their assignments (data not shown).
Scheme 3.

Revised reaction scheme for clavulanate in SHV-1. The N4=C5 imine is unique to clavulanate and a direct result of decarboxylation.
Until now, assignment of the imine features were based on their frequency in the Raman spectra, response to NH/ND exchange, and QM calculations. Here, we provide additional support for these assignments by chemical modification of the imine moiety using sodium cyanoborohydride, a mild reducing agent. In this experiment, a crystal of SHV-1 is soaked in a 5 mM bath of clavulanate for 20 minutes to allow for the maximum amount of acyl-enzyme to accumulate. Then, the crystal complex is treated with a 2-fold excess of sodium cyanoborohydride. This reducing agent is less reactive than its parent compound, sodium borohydride, and is considered to react only with protonated imines and not with their unprotonated counterparts.38 The relevant portion of the difference spectrum of the clavulanic acid complex following treatment with sodium cyanoborohydride is shown in Figure 6. Although the O=C-C=C-NH- stretch of clavulanate’s trans-enamine species is unaffected, the intensities of the 1662 and 1649 cm−1 bands are markedly reduced, providing evidence they are associated with protonated imines.
Figure 6.

Partial Raman difference spectrum of the acyl-enzyme derived intermediates from clavulanate with SHV-1 following reduction by sodium cyanoborohydride, a mild-reducing agent that is specific for protonated imines.
Conclusion
In order to design an effective inhibitor, it is necessary, as a starting point, to identify large populations of stable intermediates that are formed by mechanism-based inhibitors. Various pathways have been proposed for the inhibition of β-lactamases by mechanism-based inhibitors; however, all share the first two steps. 1) The active site serine residue reacts with the carbonyl carbon atom to form a tetrahedral intermediate at the carbonyl carbon atom. 2) The C-N bond in the β-lactam ring is cleaved and an acyl enzyme is formed. Analysis of the resulting acyl-enzymes is complicated by the number of potential intermediates and branched reaction pathways. Extensive early studies via kinetics, X-ray, and mass spectrometry provide a fragmented picture in which there are no integrated data showing which chemical species are present and providing information on their relative populations. There are only three crystallographic studies in the literature of complexes formed between wild-type β-lactamases and the clinical inhibitors. These studies underline the fact that a wide variety of intermediates, differing from enzyme to enzyme, can be detected by X-ray. Consequently, there is no consistent message as to whether the cis- or trans-enamine, imine, or the S130 adduct is responsible for inhibition on a relevant time-scale. Moreover, studies which provide high-resolution X-ray data on stoichiometric complexes of enzyme and inhibitor often use a catalytically impaired form of the enzyme, i.e. E166A. Here, we show that it may be hazardous to employ structure-based design based on the latter results as comparative studies on the wild-type enzyme reveal a much higher level of complexity. For instance, several intermediates are formed in the inactivation of SHV-1 by clavulanate. In distinction, only a single adduct, the trans-enamine, is found when E166A β-lactamase is inactivated by clavulanate under the same conditions. One notable exception is tazobactam, which forms a predominant population of trans-enamine with both wild-type and E166A. The ability of tazobactam to form a stable population of trans-enamine, even in the wild-type background, explains its superior performance in both enzyme- and cell-based assays. This ability is directly associated with the triazole group of tazobactam, which provides for hydrogen-bonding and hydrophobic interactions with the enzyme. The trans-enamine forms of sulbactam and clavulanate do not exploit any protein/ligand interactions in the ‘tail’ of the molecule. Therefore, one approach to design more effective mechanism-based compounds is to increase the complexity and diversity of C2 substituents of penam sulfone and clavam scaffolds. These compounds must then be evaluated on their ability to form stable trans-enamine populations.
Supplementary Material
Supporting figures of Raman difference spectra of the tazobactam/SHV E166A complex in H2O at room temperature and Raman difference spectra of the tazobactam/SHV S130G complex in H2O at room temperature
Acknowledgement
We are grateful to Dr. V. E. Anderson for his suggestion that sodium cyanoborohydride is a reducing agent specific for protonated imines.
The National Institutes of Health Grant RO1 GM54072 (P. R. C.), Case Western Reserve University MSTP Program (M. K.), and Robert A. Welch Foundation Grant N-0871 (J. D. B) supported this study.
Abbreviations
- TEM
class A β-lactamase of E. coli first described in a Greek patient, with the name being derived from the patient's name
- SHV
class A β-lactamase of K. pneumoniae initially thought to be a "sulfhydryl variant" of the TEM enzyme
- HEPES
N-(2-hydroxyethyl)piperazine-N'-2-ethanesulfonic acid
- HPLC
high-performance liquid chromatography
- DFT
density functional theory
- B3LYP
Becke, three-parameter, Lee-Yang-Parr
- HF
Hartree-Fock
REFERENCES
- 1.Bradford PA. Extended-spectrum beta-lactamases in the 21st century: characterization, epidemiology, and detection of this important resistance threat. Clin Microbiol Rev. 2001;14(4):933–951. doi: 10.1128/CMR.14.4.933-951.2001. table of contents. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Helfand MS, Bonomo RA. Beta-lactamases: a survey of protein diversity. Curr Drug Targets Infect Disord. 2003;3(1):9–23. doi: 10.2174/1568005033342181. [DOI] [PubMed] [Google Scholar]
- 3.Kuzin AP, Nukaga M, Nukaga Y, Hujer AM, Bonomo RA, Knox JR. Structure of the SHV-1 beta-lactamase. Biochemistry. 1999;38(18):5720–5727. doi: 10.1021/bi990136d. [DOI] [PubMed] [Google Scholar]
- 4.Ambler RP. The structure of beta-lactamases. Philos Trans R Soc Lond B Biol Sci. 1980;289(1036):321–331. doi: 10.1098/rstb.1980.0049. [DOI] [PubMed] [Google Scholar]
- 5.Hall BG, Barlow M. Structure-based phylogenies of the serine beta-lactamases. J Mol Evol. 2003;57(3):255–260. doi: 10.1007/s00239-003-2473-y. [DOI] [PubMed] [Google Scholar]
- 6.Hall BG, Barlow M. Evolution of the serine beta-lactamases: past, present and future. Drug Resist Updat. 2004;7(2):111–123. doi: 10.1016/j.drup.2004.02.003. [DOI] [PubMed] [Google Scholar]
- 7.Hall BG, Barlow M. Revised Ambler classification of {beta}-lactamases. J Antimicrob Chemother. 2005;55(6):1050–1051. doi: 10.1093/jac/dki130. [DOI] [PubMed] [Google Scholar]
- 8.Page MG. b-Lactamase inhibitors. Drug Resist Updat. 2000;3(2):109–125. doi: 10.1054/drup.2000.0137. [DOI] [PubMed] [Google Scholar]
- 9.Lietz EJ, Truher H, Kahn D, Hokenson MJ, Fink AL. Lysine-73 is involved in the acylation and deacylation of beta-lactamase. Biochemistry. 2000;39(17):4971–4981. doi: 10.1021/bi992681k. [DOI] [PubMed] [Google Scholar]
- 10.Bush K, Macalintal C, Rasmussen BA, Lee VJ, Yang Y. Kinetic interactions of tazobactam with beta-lactamases from all major structural classes. Antimicrob Agents Chemother. 1993;37(4):851–858. doi: 10.1128/aac.37.4.851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brenner DG, Knowles JR. Penicillanic acid sulfone: an unexpected isotope effect in the interaction of 6 alpha- and 6 beta-monodeuterio and of 6,6-dideuterio derivatives with RTEM beta-lactamase from Escherichia coli. Biochemistry. 1981;20(13):3680–3687. doi: 10.1021/bi00516a003. [DOI] [PubMed] [Google Scholar]
- 12.Brown RP, Aplin RT, Schofield CJ. Inhibition of TEM-2 beta-lactamase from Escherichia coli by clavulanic acid: observation of intermediates by electrospray ionization mass spectrometry. Biochemistry. 1996;35(38):12421–12432. doi: 10.1021/bi961044g. [DOI] [PubMed] [Google Scholar]
- 13.Yang Y, Janota K, Tabei K, Huang N, Siegel MM, Lin YI, Rasmussen BA, Shlaes DM. Mechanism of inhibition of the class A beta -lactamases PC1 and TEM-1 by tazobactam. Observation of reaction products by electrospray ionization mass spectrometry. J Biol Chem. 2000;275(35):26674–26682. doi: 10.1074/jbc.M002369200. [DOI] [PubMed] [Google Scholar]
- 14.Kuzin AP, Nukaga M, Nukaga Y, Hujer A, Bonomo RA, Knox JR. Inhibition of the SHV-1 beta-lactamase by sulfones: crystallographic observation of two reaction intermediates with tazobactam. Biochemistry. 2001;40(6):1861–1866. doi: 10.1021/bi0022745. [DOI] [PubMed] [Google Scholar]
- 15.Totir MA, Helfand MS, Carey MP, Sheri A, Buynak JD, Bonomo RA, Carey PR. Sulbactam forms only minimal amounts of irreversible acrylate-enzyme with SHV-1 beta-lactamase. Biochemistry. 2007;46(31):8980–8987. doi: 10.1021/bi7006146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brenner DG, Knowles JR. Penicillanic acid sulfone: nature of irreversible inactivation of RTEM beta-lactamase from Escherichia coli. Biochemistry. 1984;23(24):5833–5839. doi: 10.1021/bi00319a024. [DOI] [PubMed] [Google Scholar]
- 17.Helfand MS, Totir MA, Carey MP, Hujer AM, Bonomo RA, Carey PR. Following the reactions of mechanism-based inhibitors with beta-lactamase by Raman crystallography. Biochemistry. 2003;42(46):13386–13392. doi: 10.1021/bi035716w. [DOI] [PubMed] [Google Scholar]
- 18.Padayatti PS, Helfand MS, Totir MA, Carey MP, Carey PR, Bonomo RA, van den Akker F. High resolution crystal structures of the trans-enamine intermediates formed by sulbactam and clavulanic acid and E166A SHV-1 {beta}-lactamase. J Biol Chem. 2005;280(41):34900–34907. doi: 10.1074/jbc.M505333200. [DOI] [PubMed] [Google Scholar]
- 19.Padayatti PS, Helfand MS, Totir MA, Carey MP, Hujer AM, Carey PR, Bonomo RA, van den Akker F. Tazobactam forms a stoichiometric trans-enamine intermediate in the E166A variant of SHV-1 beta-lactamase: 1.63 A crystal structure. Biochemistry. 2004;43(4):843–848. doi: 10.1021/bi035985m. [DOI] [PubMed] [Google Scholar]
- 20.Hermann JC, Ridder L, Holtje HD, Mulholland AJ. Molecular mechanisms of antibiotic resistance: QM/MM modelling of deacylation in a class A beta-lactamase. Org Biomol Chem. 2006;4(2):206–210. doi: 10.1039/b512969a. [DOI] [PubMed] [Google Scholar]
- 21.Meroueh SO, Fisher JF, Schlegel HB, Mobashery S. Ab initio QM/MM study of class A beta-lactamase acylation: dual participation of Glu166 and Lys73 in a concerted base promotion of Ser70. J Am Chem Soc. 2005;127(44):15397–15407. doi: 10.1021/ja051592u. [DOI] [PubMed] [Google Scholar]
- 22.Escobar WA, Tan AK, Fink AL. Site-directed mutagenesis of beta-lactamase leading to accumulation of a catalytic intermediate. Biochemistry. 1991;30(44):10783–10787. doi: 10.1021/bi00108a025. [DOI] [PubMed] [Google Scholar]
- 23.Gibson RM, Christensen H, Waley SG. Site-directed mutagenesis of beta-lactamase I. Single and double mutants of Glu-166 and Lys-73. Biochem J. 1990;272(3):613–619. doi: 10.1042/bj2720613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Knox JR, Moews PC, Escobar WA, Fink AL. A catalytically-impaired class A beta-lactamase: 2 A crystal structure and kinetics of the Bacillus licheniformis E166A mutant. Protein Eng. 1993;6(1):11–18. doi: 10.1093/protein/6.1.11. [DOI] [PubMed] [Google Scholar]
- 25.Helfand MS, Taracila MA, Totir MA, Bonomo RA, Buynak JD, van den Akker F, Carey PR. Raman crystallographic studies of the intermediates formed by Ser130Gly SHV, a beta-lactamase that confers resistance to clinical inhibitors. Biochemistry. 2007;46(29):8689–8699. doi: 10.1021/bi700581q. [DOI] [PubMed] [Google Scholar]
- 26.Totir MA, Padayatti PS, Helfand MS, Carey MP, Bonomo RA, Carey PR, van den Akker F. Effect of the inhibitor-resistant M69V substitution on the structures and populations of trans-enamine beta-lactamase intermediates. Biochemistry. 2006;45(39):11895–11904. doi: 10.1021/bi060990m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hujer AM, Hujer KM, Bonomo RA. Mutagenesis of amino acid residues in the SHV-1 beta-lactamase: the premier role of Gly238Ser in penicillin and cephalosporin resistance. Biochim Biophys Acta. 2001;1547(1):37–50. doi: 10.1016/s0167-4838(01)00164-9. [DOI] [PubMed] [Google Scholar]
- 28.Padayatti PS, Sheri A, Totir MA, Helfand MS, Carey MP, Anderson VE, Carey PR, Bethel CR, Bonomo RA, Buynak JD, van den Akker F. Rational design of a beta-lactamase inhibitor achieved via stabilization of the trans-enamine intermediate: 1.28 A crystal structure of wt SHV-1 complex with a penam sulfone. J Am Chem Soc. 2006;128(40):13235–13242. doi: 10.1021/ja063715w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Carey PR. Raman crystallography and other biochemical applications of Raman microscopy. Annu Rev Phys Chem. 2006;57:527–554. doi: 10.1146/annurev.physchem.57.032905.104521. [DOI] [PubMed] [Google Scholar]
- 30.Carey PR. Spectroscopic characterization of distortion in enzyme complexes. Chem Rev. 2006;106(8):3043–3054. doi: 10.1021/cr0502854. [DOI] [PubMed] [Google Scholar]
- 31.Krezel A, Bal W. A formula for correlating pK(a) values determined in D2O and H2O. Journal of Inorganic Biochemistry. 2004;98(1):161–166. doi: 10.1016/j.jinorgbio.2003.10.001. [DOI] [PubMed] [Google Scholar]
- 32.Frisch MJTGW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery JA, Jr, Vreven T, Kudin KN, Burant JC, Millam JM, Iyengar SS, Tomasi J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Petersson GA, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Klene M, Li X, Knox JE, Hratchian HP, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Ayala PY, Morokuma K, Voth GA, Salvador P, Dannenberg JJ, Zakrzewski VG, Dapprich S, Daniels AD, Strain MC, Farkas O, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Gonzalez C, Pople J. A Gaussian 03, Revision C.02. Wallingford CT: Gaussian, Inc; 2004. [Google Scholar]
- 33.Johnson RD., III NIST Computational Chemistry Comparison and Benchmark Database. NIST Standard Reference Database Number 101 In National Institute of Standards and Technology. 2006 [Google Scholar]
- 34.Zheng R, Zheng X, Dong J, Carey PR. Proteins can convert to beta-sheet in single crystals. Protein Sci. 2004;13(5):1288–1294. doi: 10.1110/ps.03550404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sun T, Bethel CR, Bonomo RA, Knox JR. Inhibitor-resistant class A beta-lactamases: consequences of the Ser130-to-Gly mutation seen in Apo and tazobactam structures of the SHV-1 variant. Biochemistry. 2004;43(44):14111–14117. doi: 10.1021/bi0487903. [DOI] [PubMed] [Google Scholar]
- 36.Daimay L-VC, Norman B, Fateley William G, Grasselli Jeanette G. The Handbook of Infrared and Raman Characteristic Frequencies of Organic Molecules. Boston: Academic Press, Inc.; 1991. p. 501. [Google Scholar]
- 37.Sulton D, Pagan-Rodriguez D, Zhou X, Liu Y, Hujer AM, Bethel CR, Helfand MS, Thomson JM, Anderson VE, Buynak JD, Ng LM, Bonomo RA. Clavulanic acid inactivation of SHV-1 and the inhibitor-resistant S130G SHV-1 beta-lactamase. Insights into the mechanism of inhibition. J Biol Chem. 2005;280(42):35528–35536. doi: 10.1074/jbc.M501251200. [DOI] [PubMed] [Google Scholar]
- 38.Borch RF, Bernstein MD, Durst HD. Cyanohydridoborate anion as a selective reducing agent. J Am Chem Soc. 1971;93(12):2897–2904. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting figures of Raman difference spectra of the tazobactam/SHV E166A complex in H2O at room temperature and Raman difference spectra of the tazobactam/SHV S130G complex in H2O at room temperature