

Abstract
In its native form, the chemokine CX3CL1 is a firmly adhesive molecule promoting leukocyte adhesion and migration and hence involved, along with its unique receptor CX3CR1, in various inflammatory processes. Here we investigated the role of molecular aggregation in the CX3CL1 adhesiveness. Assays of bioluminescence resonance energy transfer (BRET) and homogeneous time-resolved fluorescence (HTRF) in transfected cell lines and in primary cells showed specific signals indicative of CX3CL1 clustering. Truncation experiments showed that the transmembrane domain played a central role in this aggregation. A chimera with mutations of the 12 central transmembrane domain residues had significantly reduced BRET signals and characteristics of a non-clustering molecule. This mutant was weakly adhesive according to flow and dual pipette adhesion assays and was less glycosylated than CX3CL1, although, as we demonstrated, loss of glycosylation did not affect the CX3CL1 adhesive potency. We postulate that cell surfaces express CX3CL1 as a constitutive oligomer and that this oligomerization is essential for its adhesive potency. Inhibition of CX3CL1 self-assembly could limit the recruitment of CX3CR1-positive cells and may be a new pathway for anti-inflammatory therapies.
Migration of circulating leukocytes to injury sites is the first step of the inflammation process, which involves a sequence of coordinated interactions between leukocytes and endothelial cells (1). Central to this physiological and pathological event are chemokines, a family of low molecular weight soluble proteins, that function to attract leukocytes bearing the appropriate receptors (2). Chemokines trigger activation of leukocytes and their firm adhesion to inflamed endothelium, mainly through the mediation of integrins and their cognate ligands (3).
Among chemokines, there are two exceptions: CXCL16 and CX3CL1 are type-I membranous proteins. In addition to their chemokine domain (CD),4 they are composed of a long mucin-like stalk, a transmembrane domain, and a cytoplasmic tail (4, 5) (Fig. 1). The CX3CL1 molecule, with its unique CX3CR1 receptor (6), has been shown to be central in defenses against neurodegenerative disorders (7, 8) and against several cancers in murine models (9, 10). The CX3CL1-CX3CR1 axis is also involved in various inflammatory diseases (2), including renal inflammation (11) and atherosclerosis (12, 13). Understanding the structure of this pair of molecules is necessary for exploring pharmacological methods to regulate their activity. The structure of CX3CR1, a receptor of the G protein-coupled receptor (GPCR) family, is relatively well known, but that of CX3CL1 much less so.
FIGURE 1.

Schematic drawing of the structure of the CX3CL1 molecule. The domains of CX3CL1 are drawn according to the available structural data (6, 14).
CX3CL1-CD (76 residues), a globular protein domain 3 nm in diameter (14) and maintained by two disulfide bridges, is structurally similar to other chemokines. It is composed of a disordered N terminus up to the first cysteine (Cys-8), a long loop followed by a three-stranded antiparallel β-sheet (residues 24–51) and a C-terminal α-helix (residues 56–67) packed against the β-sheet (15). The stalk (241 residues) is 26 nm in length (14) and highly glycosylated with 17 degenerate mucin-like repeats (6, 16). This native form can be cleaved by various metalloproteinases (17, 18) to release the soluble form, which, like other chemokines, is chemoattractant. In contrast, membrane-associated CX3CL1, along with CX3CR1, mediates stable adhesion of leukocytes such as integrins (16, 19). The mechanism of this adhesive property is poorly understood. Although we know that the potent adhesiveness of CX3CR1 under flow requires that CX3CL1-CD have a high affinity for it (19), nothing is known about the quaternary structure of the ligand.
Most of the soluble chemokines bind sulfated glycoaminoglycans of the extracellular matrix and endothelial cell surfaces. This immobilization leads to chemokine clustering and is essential for establishing the chemokine gradient that leads to directed migration. Moreover, numerous chemokines tend to self-associate at high concentrations in solution, and this clustering sometimes appears to be essential to their in vivo activity (20). For CX3CL1, however, the CD itself remains monomeric in solution, even at high concentrations (15). The CD structure has been studied in crystals (21), where it includes four monomers arranged as two asymmetric dimers. However, the potential aggregation of the complete CX3CL1 molecule has not yet been investigated. We reasoned that CX3CL1 clustering could be important for its adhesive properties, as it is for integrins (22).
Among the many methods for studying the aggregation of cellular proteins, resonance energy transfer techniques have emerged as useful tools for analyzing the proximity of membrane molecules in living cells (23). We used these techniques here to study the aggregation status of CX3CL1 in its native membranous state. We report that CX3CL1 is indeed clustered and that this clustering takes place primarily through its transmembrane domain. Moreover, we show that this aggregation is associated with the mature glycosylation of the protein and, most importantly, is required for its adhesive potency in flow conditions (flow adhesion assay) and under normal pulling (dual pipette assay).
EXPERIMENTAL PROCEDURES
Chemicals, Proteins, and Monoclonals—Benzyl-2-acetamido-2-deoxy-α-d-galactopyranoside was purchased from Calbiochem. Neuraminidase (acylneuraminyl hydrolase) and O-glycosidase (endo-d-galactosyl-N-acetyl-α-galactosamine hydrolase) came from Roche Diagnostics. Monoclonal antibodies against CX3CL1-CD (clone 51637) and full-length CX3CL1 tagged with His6 were purchased from R&D Systems (Lille, France) and CX3CL1-CD from Peprotech (Levallois-Perret, France).
Plasmid Constructs—The CX3CL1 constructs in pcDNA3 (Invitrogen), pEYFP-N1 (Clontech), and pRluc-N2 (Perkin-Elmer) were made with CX3CL1-pBLAST (Invivogen Cayla, Toulouse, France) as template and primers containing a HindIII restriction site in 5′position and a BamHI restriction site in the 3′-position for PCR amplification as explained in supplemental Table S1. The HindIII/BamHI fragment was then cloned in the different plasmids. The truncated and mutated constructs were generated using the QuikChange II site-directed mutagenesis kit according to the manufacturer's instructions (Stratagene). Briefly, 10 ng of various plasmid constructions was used as a template, with mutated nucleotide primers as described in supplemental Table S1. PCR conditions were as follows: predenaturing at 95 °C for 3 min followed by 18 cycles of denaturing at 95 °C for 1 min, annealing at 60 °C for 50 s, and extension at 68 °C for 7 min. After digestion with DpnI, 2 μl of PCR product was used to transform the XL10-Gold Ultracompetent cells provided with the kit. Appropriate clones were identified by sequencing.
Cell Culture—HEK293 cells were maintained in Dulbecco's modified Eagle's medium plus GlutaMax I (Invitrogen) supplemented with 10% fetal bovine serum and 1 mm sodium pyruvate. To obtain HEK clones expressing CX3CL1 or ALA12, CX3CL1-pEYFP and ALA12-pEYFP constructs were transfected into HEK cells with the cationic polymer transfection reagent jetPEI (Polyplus transfection, Ozyme, St. Quentin-en-Yvelines, France). A stable transformant resistant to 0.5 mg/ml G418 (Invitrogen) was selected with a FACS Aria cell sorter (BD Biosciences). HUVEC were cultured in endothelial cell medium (PAA Laboratories, Les Mureaux, France), supplemented with 10% fetal bovine serum. HUVEC were incubated for 20 h with tumor necrosis factor-α at 20 ng/ml and interferon-γ at 500 units/ml (Peprotech) to induce CX3CL1 expression. Expression of CX3CL1 on HUVEC and HEK cells and clones was tested with phycoerythrin-labeled murine anti-CX3CL1 mAb (mAb clone 51637, R&D Systems) and analyzed by flow cytometry with a FACScan (BD Biosciences).
BRET—Cells were seeded at a density of 100,000 cells/well in 12-well dishes 24 h before transfection. Transient transfections were performed with the cationic polymer transfection reagent, JetPEI (Polyplus transfection, Ozyme) in 150 mm NaCl. In most experiments, 0.2 μg of various CX3CL1-pRluc constructs was transfected alone or with increasing quantities of various CX3CL1-pEYFP constructs. The total amount of DNA transfected in each well was completed to 2.2 μg with empty pcDNA3 vector. After overnight incubation, the transfected cells were detached with phosphate-buffered saline (PBS) and washed with HBSS buffer supplemented with 10 mm HEPES, 1 mm CaCl2, and 0.5 mm MgCl2. The cells were then seeded in 96-well black plates (PerkinElmer) in 100 μl of supplemented HBSS. Coelenterazine H (Interchim, Montluçon, France) was added to reach a final concentration of 5 μm. Readings were collected with a microplate analyzer (Fusion; PerkinElmer) that allowed the sequential integration of signals detected in the 485 ± 20-nm window for luciferase and the 540 ± 20-nm window for yellow fluorescence protein (YFP) light emissions. The BRET signal was determined by calculating the ratio of the light intensity emitted by the CX3CL1-YFP over the light intensity emitted by the CX3CL1-luciferase (Luc). The values were corrected by subtracting the background BRET signal detected when the CX3CL1-Luc construct was expressed alone. The values are the mean over 15 measurements. To relate the BRET measurements to the actual CX3CL1 concentration, we measured the luminescence or fluorescence of HEK transfected either with CX3CL1-Luc or CX3CL1-YFP constructs and quantified their CX3CL1 content in Western blot experiments with CX3CL1-His6 tag (R&D Systems) as standard (Fig. 2B).
FIGURE 2.

Characteristics of BRET of CX3CL1 in HEK cells. A, HEK293 cells were transfected with 0.05 μg of CX3CL1-Luc and 0.5 μg of various YFP constructs: empty pEYFP (left), CXCL16-YFP (middle), CX3CL1-YFP (right). The values are the mean ± S.D. of 15 measurements. Insert, fluorescence microscopy imaging of HEK cells transfected with CX3CL1-YFP (bar = 50 μm). B, HEK cells were transfected with 1, 2, or 3 μg of CX3CLI-Luc or CX3CL1-YFP constructs. The luminescence of fluorescence of 50,000, 100,000, and 200,000 cells was then measured. In addition, the CX3CL1 content of each transfectants was analyzed by SDS-PAGE and Western blot and evaluated with CX3CL1-His6 as standard. C, BRET variation using HEK cells transfected with a constant amount of donor (CX3CL1-Luc) and increasing amount of acceptor (CX3CL1-YFP) (▪). CX3CL1-Luc and CX3CL1-YFP contents in each sample were quantified with the calibration curve in B. Here the CX3CL1-Luc was kept constant at 1.5 fmol/mg total protein. The same measurements were taken with CXCL16-YFP as acceptor (□). The data were fitted with GraphPad Prism 4 with one-site binding hyperbola. The resulting BRETmax was 0.230 for CX3CL1-Luc/CX3CL1-YFP and 0.027 for CX3CL1-Luc/CXCL16-YFP. D, BRET variation using HEK cells co-transfected with CX3CL1-Luc and CX3CL1-YFP at a constant ratio ([YFP]/[Luc] = 5). The linear regression is given and indicates that BRET is constant in this range. E, BRET variation versus CX3CL1-YFP concentration using HEK cells transfected with various amounts of CX3CL1-Luc and CX3CL1-YFP. F, HEK293 cells were transfected with 0.1 μg of CX3CL1-Luc and 0.2 μg of CX3CL1-YFP supplemented with 2 μg of empty pcDNA3 (left) or CX3CL1-pcDNA3 (right). The values are the mean ± S.E. of 15 measurements. The difference between the CX3CL1 BRET ratio with or without native CX3CL1 was significant (***, p < 0.0001). The CX3CL1 content of each sample was analyzed by Western blot to check that the untagged CX3CL1 expression did not change expression of CX3CL1-Luc and CX3CL1-YFP (data not shown).
Homogeneous Time-resolved Fluorescence (HTRF)—Monoclonal anti-CX3CR1 (clone 51637) was labeled with the different fluorescent donors or acceptors as described previously (24, 25). The donor was a pyridine-bipyridine (PBP) europium cryptate (europium cryptate-PBP). The number of europium cryptate-PBP per antibody (molar ratio) was determined spectrophotometrically by measuring their absorbance at 280 and 317 nm and inserting these values into the equation
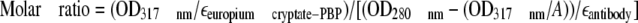 |
(Eq. 1) |
where the molar extinction coefficient of the europium cryptate-PBP (εeuropium cryptate-PBP) was taken as 19,800 m-1 cm-1 and that of the antibodies as 210,000 m-1 cm-1. Factor A expressed the ratio (OD317 nm/OD280 nm) for europium cryptate-PBP and was determined to be 2. The final molar ratio was determined to be 1.1.
The anti-CX3CR1 monoclonal was labeled with the N-hydroxysuccinimide ester derivative D2 acceptor as described previously (26). The final number of dyes per antibody was determined spectrophotometrically. By using the maximum absorption for D2 at 650 nm and its molar extinction coefficient value (240,000 m-1 cm-1), the final ratio was determined to be 1.3. HEK clone cells and HUVEC (activated or not) were harvested and resuspended in KREBS buffer (Tris/HCL 20 mm, pH 7.4, NaCl 118 mm, glucose 5.6 mm, KH2PO4, MgSO4 1.2 mm, KCl 4.7 mm, CaCl2 1.8 mm, and bovine serum albumin 0.1%). Fifty thousand cells in 100 μl were distributed in 96-well black plates (PerkinElmer) where 4 nm europium cryptate-labeled donor anti-CX3CL1 and 4 nm D2-labeled acceptor antibody (anti-CX3CL1 or anti-FLAG as control) were already present, diluted in the same buffer. After incubation at 37 °C for 4 h, fluorescence emissions were measured, at both 620 and 665 nm, on a multidetection microplate reader (PHERAstar, BMG Labtech).
The HTRF ratio was calculated as r = (fluorescence at 665 nm/fluorescence at 620 nm) × 104. The specific signal over background, noted as ΔF, was calculated with the following formula,
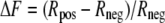 |
(Eq. 2) |
where Rpos corresponds to the ratio with anti-CX3CL1 mAb as the acceptor and Rneg to the ratio with anti-FLAG mAb as the acceptor.
Static Adhesion Assay—Two different assays were performed. In the cell-cell model, performed as described previously (27), CHO-CX3CR1 and parental CHO were grown in supplemented Dulbecco's modified Eagle's medium in 96-well plate (5.104 cells/well) and washed in HBSS buffer. Then 105 HEK cells expressing variants of CX3CL1 were added for 45 min at room temperature. In the cell-to-protein adhesion model, a monoclonal anti-polyhistidine antibody (mAb 050, R&D Systems) (250 ng in 25 mm Tris, pH 8, 150 mm NaCl) was adsorbed for 2 h at room temperature in flat-bottom 96-well microtiter plates (Nunc A/S, Roskilde, Denmark). Then CX3CL1-His6, diluted in the same buffer, was incubated overnight at 4 °C at a concentration of 1.8 μg/ml (50 μl/well in triplicate). Adhesion of HEK-CX3CR1 cells was performed as described previously (27) with an incubation time of 45 min at room temperature in HBSS buffer with or without enzymatic treatment. In both cases, before the adhesion assay the cells were labeled with 1 μm 5(6)carboxyfluorescein diacetate, succinimidyl ester (Interchim) and the microplate was read at 535 nm with a microplate analyzer (Fusion; PerkinElmer).
Deglycosylation Treatment—The adsorbed protein was digested with 4 milliunits of neuraminidase in 40 mm Tris, pH 8, and 4 mm CaCl2 for 2 h at 37 °C before incubation with 1 milliunit of O-glycosidase in PBS for 4 h at 37 °C. Western blotting using an anti-CX3CL1 antibody analyzed the digested protein.
Parallel Plate Laminar Flow Chamber Adhesion Assay—The technique we used has been thoroughly described previously (28). Briefly, the coverslips were cultured with adherent clone HEK cells expressing either CX3CL1 or ALA12 mutant. The coverslip was mounted in a chamber set on the stage of an inverted microscope (TE300, Nikon) equipped with a phase contrast 10× objective (Nikon, n.a. 0.25) and a cooled charge-coupled device camera (Sensicam, PCO, Kelheim, Germany). The entire apparatus was kept at 37 °C by a thermostatic chamber (Life Imaging Services, Reinach, Switzerland). HEK-CX3CR1 clone cells were suspended in PBS, incubated for 30 min at 37 °C with 5 μm carboxy-SNARF-1-AM (seminaphtorhodafluor-1-acetoxymethylester, Molecular Probes-Invitrogen) for labeling, and resuspended in flow buffer (HBSS supplemented with 2.5 mm EDTA, 2.5 mm EGTA, 10 mm HEPES, and 2 mg/ml bovine serum albumin) at 106 cells/ml. A syringe pump (PHD 2000; Harvard Apparatus, Les Ulis, France) drove 0.5 ml of cell suspension through the chamber at a wall shear stress of 1.5 dynes.cm-2. After a 10-min wash at 1.5 dynes.cm-2, fluorescent images of three separate 0.5 mm2 fields were recorded to count the adherent cells (excitation 450–500 nm, emission 510–560 nm, dichroic filter Q505lp, Chroma, Brattleboro, VT). The number of nonspecific adhering cells was obtained in the same manner, except the HEK-CX3CR1 clone cells were preincubated for 2 min with 100 nm soluble CX3CL1-CD (chemokine domain alone, R&D Systems).
Dual Pipette Aspiration Technique—The dual pipette adhesion assay was performed as described previously (28). Briefly, two cells, collected by gentle aspiration onto the tip of each pipette (cell 1 in pipette A, cell 2 in pipette B), were brought into contact with the micromanipulators and allowed to remain in contact for 4 min. To separate the cells, aspiration in pipette B was maintained at a level sufficiently high to hold cell 2 tightly, and the aspiration in pipette A was increased in steps measured with a pressure sensor (Validyne, model DP103-38; ranging from 0 to 50,000 pascals). After each step, the pipettes were moved apart in an effort to detach the adherent cells from one another. A pair pulled intact from pipette A was moved back to the pipette orifice, the aspiration in the pipette was increased, and another attempt was made to detach the cells from each other. The cycle was repeated until the level of aspiration in pipette A was sufficient to pull one cell apart from the other. The aspiration employed in each cycle was monitored continuously. The values recorded for each of the last two cycles in the series (Pn - 1 and Pn) were used to calculate the separation force, F, for the pair tested, with the equation,
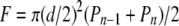 |
(Eq. 3) |
with d the internal diameter of pipette A. The results were expressed as the mean ± S.E. for 14 measurements.
RESULTS
BRET of CX3CL1 in the HEK Cell Line—To investigate by BRET the aggregation state of the chemokine CX3CL1 in its native membranous form, we developed various constructs of CX3CL1 chimera that expressed Luc or YFP on the cytoplasmic C-terminal side. As expected, the tagged CX3CL1 was expressed at the plasma membrane in transiently transfected HEK cells (Fig. 2A, insert). The membrane-targeted CX3CL1-YFP or CX3CL1-Luc accounted for about 50% of the total cellular CX3CL1, as already shown for the CX3CL1-GFP chimera (29). Finally, our CX3CL1 chimera also functioned as native CX3CL1 according to adhesion tests (data not shown), as the GFP (green fluorescent protein) chimera did (29).
Using such chimera with a ratio of 1:10 YFP (acceptor) over Luc (donor), we measured a high BRET ratio (Fig. 2A, right), higher than that obtained when the CX3CL1-YFP plasmid was replaced by the empty pEYFP construct (Fig. 2A, left), by CXCL16-YFP (Fig. 2A, middle), or by ICAM-4-YFP (not shown). These data suggest the specificity of the homotypic CX3CL1 BRET and show for the first time the probable self-assembly of CX3CL1 proteins in a cellular context. After transfecting HEK cells with various amounts of CX3CL1-YFP and a constant amount of CX3CL1-Luc (yielding 1.5 fmol of CX3CL1-Luc/mg total protein; see Fig. 2B), we plotted the BRET data versus the CX3CL1-YFP/CX3CL1-Luc ratio and observed a saturation curve (Fig. 2C, filled squares) as expected for specific clustering (30, 31). In contrast, the same curve plotted with CXCL16-YFP replacing CX3CL1-YFP remained at low amplitude (Fig. 2C, empty squares). Working with a constant CX3CL1-YFP/CX3CL1-Luc ratio, here equal to 5 (Fig. 2D), we verified that the BRET level was constant when plotted against the total amount of tagged CX3CL1 (donor plus acceptor) (Fig. 2D) and against the CX3CL1-YFP concentration (acceptor) (Fig. 2E). Indeed, when BRET is specific, its amplitude should not depend on the total amount of interacting proteins (30–34) or on the concentration of the donor moiety (30). Finally, the significant BRET decrease in the presence of an excess of an untagged unit (Fig. 2F) showed that the BRET was specific and not due to random collision. We concluded that CX3CL1 appears to aggregate specifically, at least in the HEK cell line.
HTRF Measurements with Cellular CX3CL1—To test whether native CX3CL1 aggregates similarly in primary cells, we used the method called HTRF (35), which has been applied to detect biomolecular interactions (36) including ligand-enzyme relations (37) and protein clustering (24). It consisted here of measuring the resonance energy transfer between labeled antibodies specific against CX3CL1. Using both stably CX3CL1-expressing HEK cells (data not shown) and cytokineactivated HUVEC, also expressing CX3CL1 (16, 38) (Fig. 3A), we observed a marked level of energy transfer (Fig. 3B, right, solid bar), substantially higher than the level observed with irrelevant monoclonals (Fig. 3B, right, empty bar). This result was confirmed by plotting HTRF signals as the difference between specific and nonspecific data (ΔF; see “Experimental Procedures”) (Fig. 3B, insert). In contrast, measurements of resting HUVEC, which do not express CX3CL1 (Fig. 3A), showed no specific HTRF (Fig. 3B, left, and insert). Accordingly, CX3CL1 appears natively aggregated when it is expressed at the external membrane of primary cells.
FIGURE 3.

HTRF measurements of CX3CL1 in HUVEC. A, resting HUVEC (dotted line) and activated HUVEC (continuous line) were stained with phycoerythrin murine mAb anti-CX3CL1 and analyzed by flow cytometry. B, resting HUVEC (left) and activated HUVEC (right) were stained with anti-CX3CL1 mAb labeled with cryptate (donor) and with D2-labeled anti-CX3CL1 mAb (acceptor) (filled bars) as described under “Experimental Procedures.” Control measurements were made with anti-FLAG D2-labeled mAb as acceptor (empty bars). The FRET signals were plotted as HTRF ratio R (r = (fluorescence at 665 nm/fluorescence at 620 nm) × 104). The HTRF difference between HUVEC and activated HUVEC was significant (**, p < 0.0018). Insert, the specific signal over background called ΔF was calculated with the following formula: ΔF = (Rpos - Rneg)/Rneg where Rpos corresponds to the HTRF ratio with anti-CX3CL1 mAb as acceptor and Rneg to the ratio with anti-FLAG mAb as acceptor.
CX3CL1 Clustering Is Mainly Due to Its Transmembrane Domain—We next sought to identify the domain of CX3CL1 involved in clustering, by progressively deleting the external (first the CD and then the mucin stalk) and intracellular domains of the molecule (Fig. 4, top). We first checked that all these constructs when transfected in HEK produced proteins that effectively targeted the membrane (Fig. 4, A–C) and were expressed at levels similar to that of whole CX3CL1 (Fig. 2A, insert). After transfection with these constructs, the HEK still exhibited BRET. Each construct produced BRET with saturation curves (BRET versus [YFP]/[Luc] at a constant Luc concentration) (Fig. 4D). Moreover, the BRET of these truncated CX3CL1 constructs met the criteria for specificity described above (Fig. 2D), even for the TM domain alone (Fig. 4E). Yet, the Muc and TM constructs gave a BRETmax lower than that of CX3CL1 (Fig. 2C), whereas the TM&cyto construct gave a higher level (Fig. 4, D and E). One should recall here that the BRET was not only dependent on the proximity of the donor and acceptor moieties but also on their relative orientation (39). Hence, a small conformational change could dramatically affect the BRET level. Nevertheless, all of the constructs we tested gave specific BRET signals (Fig. 4, D and E). So our data strongly suggest that the TM domain alone still aggregates and that this domain is the primary site involved in CX3CL1 clustering.
FIGURE 4.

BRET of CX3CL1 deletion mutants. A–C, HEK cells were co-transfected with YFP-tagged constructs containing the various truncated CX3CL1-YFP constructs and observed with fluorescence microscopy (bar = 50 μm). D, BRET variation using HEK cells co-transfected with a constant amount of Luc-tagged truncated CX3CL1 constructs (4, 4, and 7 fmol/mg total protein for Muc, TM&cyto, and TM mutants, respectively) and increasing amount of YFP-tagged constructs as indicated. The data were calibrated (Fig. 2B) and fitted using GraphPad Prism 4 with one-site binding hyperbola. E, BRET variation using HEK cells co-transfected with Rluc- and YFP-tagged constructs containing truncated CX3CL1 at a constant [YFP]/[Luc] ratio. Circles, Muc ([YFP]/[Luc] = 20); triangles, TM&cyto ([YFP]/[Luc] = 40); diamonds, TM ([YFP]/[Luc] = 12).
The ALA12 CX3CL1 Mutant Is Not Fully Glycosylated and Does Not Cluster—For additional evidence that CX3CL1 clusters through its TM domain, we looked for the TM residues with mutations that could suppress the specific BRET observed with the entire protein. We mutated in alanine the residues thought to be important for self-association of transmembrane helices (Fig. 5, upper panel), such as glycine (40), leucine (41) and phenylalanine (42), and observed no significant decrease of BRET (Fig. 5, lower panel).
FIGURE 5.

BRET of the CX3CL1 mutants with modifications in the TM domain. HEK cells were co-transfected with a constant amount of CX3CL1-Luc-tagged constructs and increasing amounts of CX3CL1-YFP constructs with various mutations in the TM domain, as indicated at the top of this figure. In each case, BRETmax ± S.E. was analyzed as in Fig. 2C.
We therefore decided to proceed to larger modifications. Residues 321–325 and 326–332 were replaced by their alanine counterparts for the ALA5 and ALA7 mutants (Fig. 6, top panel). We also mutated residues 321–332 together for an ALA12 mutant (Fig. 6, top panel). Protein expression of ALA5 and ALA7 at the membrane, assessed by fluorescence imaging (Fig. 6, A and B) and flow cytometry (data not shown), was substantial, similar to that of the native CX3CL1 (Fig. 2A, insert). The ALA12 mutant, however, was mainly intracellular (Fig. 6C). Flow cytometry assays revealed that it was expressed overall at the same level as the other mutants (data not shown) but that the expression of the ALA12 protein at the membrane level was actually half that of that of CX3CL1 and of the ALA5 and ALA7 constructs.
FIGURE 6.

BRET of the CX3CL1 mutants with modifications in the TM. A–C, the various CX3CL1-YFP constructs were transiently expressed in the HEK cell line and observed with fluorescence microscopy (bar = 50 μm). D, lysates of HEK transiently transfected with the various CX3CL1-YFP constructs were analyzed by SDS-PAGE and Western blot with a goat anti-CX3CL1 antibody. E, HEK cells were co-transfected with a constant amount of Luc-tagged constructs (giving 1.5 fmol/mg total protein) and increasing amount of YFP-tagged constructs as indicated. In each case, data were calibrated (see Fig. 2B) and fitted with GraphPad Prism 4 with one-site binding hyperbola. F, BRET variation using HEK cells co-transfected with various amounts of Luc- and YFP-tagged constructs at a constant ratio. Triangles, ALA7 ([YFP]/[Luc] = 5); diamonds, ALA5 ([YFP]/[Luc] = 9). G, BRET variation using HEK cells co-transfected with various amounts of ALA12 Luc-tagged constructs and ALA12 YFP-tagged constructs at a constant ratio ([YFP]/[Luc] = 9).
Moreover, Western blot analysis revealed that each construct gave rise to two CX3CL1-YFP species (Fig. 6D), one mature and complete, with a molecular weight of 120 kDa (90 kDa for CX3CL1 plus 30 kDa for YFP), and then a smaller one (60 kDa plus 30 kDa for YFP). The latter did not seem to be a truncated fragment of the mature form, as it contained both the CD and YFP moieties as assayed by Western blot (data not shown). It most probably corresponds to the 60-kDa form indicated in an earlier study (17) as an immature, intracellular form of CX3CL1. Interestingly, this shorter form, which accounted for only a small proportion of the expression of the ALA5 and ALA7 mutants, was the major form in the ALA12 mutant (Fig. 6D). This is consistent with its intracellular location (Fig. 6C).
The ALA5 and ALA7 mutants exhibited BRET saturation curves (Fig. 6E) similar to those of CX3CL1 (Fig. 2C). Fig. 6F shows that the BRET data for the ALA5 and ALA7 mutants was characteristic of specific BRET (constant versus acceptor plus donor). The ALA12 mutant, in contrast, had a very low BRET amplitude (Fig. 6E), which was not constant when plotted against the total amount of tagged protein (Fig. 6G); the BRET data were clearly linear, going to almost zero, i.e. they corresponded to a nonspecific aggregation (30, 33). The BRET curve versus the acceptor concentration behaved similarly (data not shown) and thus confirmed that the BRET of ALA12 was mainly unspecific. Mutation of the 12 central residues of the TM domain of CX3CL1 led to less protein maturation (Fig. 6D) and, according to the BRET assays, to a reduced ability to aggregate (Fig. 6, E and G).
Role of Clustering in the Adhesive Potency of CX3CL1—To test the non-clustering CX3CL1 mutant functionally, we derived HEK clones that stably expressed either the CX3CL1-YFP or the ALA12-YFP chimera. We obtained two clones with similar YFP fluorescence and expressing the same level of CX3CL1 at the cell membrane according to flow cytometry (Fig. 7A) and confocal microscopy (Fig. 7B). Both clones expressed the two forms of CX3CL1 (90 and 120 kDa), both of which were full-length peptides, as they were recognized by both anti-CX3CL1-CD (Fig. 7C, left) and anti-YFP (Fig. 7C, middle). The complete, mature form is the major form in the CX3CL1-YFP clone, either in the whole cell (Fig. 7C, left) or at the external membrane level, as shown by immunoprecipitation of the CX3CL1 protein accessible from outside (Fig. 7C, right). Conversely, in confirmation of the data obtained with transient transfectants (Fig. 6D), the smaller form is the main component of the ALA12-YFP clone (Fig. 7C, left) and is dominantly expressed at the surface membrane of the clone (Fig. 7C, right). Finally, both clones released a similar amount of the 80-kDa soluble form of CX3CL1 (data not shown), probably after cleavage by ADAM10 and ADAM17 (17, 43–46).
FIGURE 7.

Adhesive properties of HEK clone cells expressing CX3CL1-YFP or ALA12-YFP. A, parental HEK cells (continuous line), stable HEK clone expressing CX3CL1-YFP (fine dotted line), and ALA 12-YFP (heavier dotted line) were stained with phycoerythrin (PE)-labeled murine anti-CX3CL1 mAb and analyzed by flow cytometry. B, the two clones were observed with a Leica SP2-AOBS confocal microscope (bar = 50 μm) C, the two clones were analyzed by SDS-PAGE and Western blot (IB) using antibodies against CX3CL1 (left) or YFP (middle). For immunoprecipitation of antigen located at the cell surface (right), 2 × 106 cells of both clones were incubated in suspension for 1 h at 4 °C with 5 μg of murine mAb anti-human CX3CL1 in PBS plus 0.2% bovine serum albumin. After two washings in cold PBS, the cells were lysed in lysis buffer (150 mm NaCl, 50 mm Tris-HCl, pH 8, 1% Nonidet P-40 (Sigma), Complete protease inhibitor mixture (Roche Diagnostics). The immune complex was separated on protein-G-agarose (Sigma) and analyzed by Western blotting, with a goat anti-human CX3CL1. D, HEK-CX3CL1 or HEK-ALA12 clone cells (105 cells/well) labeled with carboxyfluorescein succinimidyl ester (1 μm, 30 min, 37 °C) were deposited onto confluent CX3CR1-CHO cell or parental CHO cell monolayers. At the end of incubation period and after washings, the plate was read at 535 nm as described under “Experimental Procedures.” The adherent cells are expressed as percentage of total cells minus the mean background corresponding to the number of HEK-CX3CL1 or ALA12 cells adhering on parental CHO cells. E, HEK clone cells expressing CX3CR1 were suspended and assayed for adhesion in a parallel plate laminar flow chamber with coverslips coated with adherent HEK-CX3CL1 or HEK-ALA12 clones as described under “Experimental Procedures.” Adhering cells were counted after 10 min over four fields (mean ± S.E.). The nonspecifically adhering cell numbers were obtained in the same conditions after the addition of 100 nm CX3CL1-CD in HEK-CX3CR1 cells before injection into the flow chamber. The difference between the number of cells specifically adhering either on CX3CL1 or on ALA12 was significant (***, p = 0.002). This result was characteristic of three independent experiments. F, HEK-CX3CL1 or HEK-ALA12 clones were assayed for adhesion by the dual pipette aspiration technique with HEK-CX3CR1 clone cells as described under “Experimental Procedures.” The dissociation force was evaluated after 4 min of adhesion (mean ± S.E., n = 14). The difference between adhesion of the CX3CL1/CX3CR1 and ALA12/CX3CR1 pairs was significant (***, p = 0.008). nN, nanonewton.
Both clones were assayed for their adhesiveness to a CX3CR1-expressing HEK or CHO clone. When assayed under static conditions, both clones specifically adhered to CX3CR1-positive CHO cells (Fig. 7D). Despite a similar CX3CL1 surface expression (Fig. 7A), the number of adhering ALA12-YFP cells was significantly lower than the number of adhering CX3CL1-YFP clone cells. In addition, in flow conditions at 1.5 dyne.cm-2, the number of CX3CR1-positive HEK clone cells adhering to ALA12 cells was less than 25% of the number adhering to standard CX3CL1 cells (Fig. 7E). In the dual pipette assay, quantifying the strength required to dissociate an adhesive cell pair (28), a dissociation force of ∼1 nanonewton (Fig. 7F) was sufficient to tear apart the ALA12-CX3CR1 paired cells. This level was also obtained with parental HEK (28) and was considered as nonspecific adhesion. In contrast, a dissociation force of 4 nanonewtons was required for CX3CL1-CX3CR1 pairs. The latter value is similar to that already found for adhesion of HEK cells expressing CX3CR1 and CX3CL1 without the YFP extension (28). Taken together, our data indicate that the primary TM domain of CX3CL1 is required for the shear-resistant adhesiveness of the CX3CL1-CX3CR1 pair.
Role of Glycosylation in CX3CL1 Adhesive Potency—The preceding data show that the ALA12 mutant, containing mainly a non-clustering form of CX3CL1, was scarcely adhesive. Moreover, this form was lighter, probably because it was not fully glycosylated (Fig. 7B). To determine whether either clustering or glycosylation was required for adhesion, our HEK clone that expressed CX3CL1-YFP was treated with benzyl-2-acetamido-2-deoxy-α-d-galactopyranoside, which inhibits glycosylation of the mucin family proteins (47). Alternatively, it was incubated with neuraminidase (acylneuraminyl hydrolase) plus O-glycosidase, which deglycosylates cellular CXCL16 (45). Unfortunately, the apparent molecular weight of CX3CL1 was not altered by the application of any of these treatments to our CX3CL1-YFP clone (either on intact cells or on cell lysate). We therefore decided to work with the commercially available external moiety of the CX3CL1 molecule, containing the CD domain and most of the mucin stalk fused to a His6 tag (48). This purified protein had an apparent molecular mass of 90 kDa (Fig. 8A, lane a) and showed adhesive properties under both static (Fig. 8B, lane a) and flow conditions (Fig. 8C, lane a). After treatment with neuraminidase, the molecular mass of CX3CL1-His6 was reduced to 65–70 kDa (Fig. 8A, lane b). If O-glycosidase was included in the treatment, the CX3CL1-His6 was found to be around 60 kDa (Fig. 8A, lane c), equivalent to that of the lighter form of CX3CL1 detected in our CX3CL1 and ALA12 HEK clones (Fig. 7C). However, these lighter forms of CX3CL1 displayed the same adhesion features as the mature form (Fig. 8, B and C, lanes b and c). The failure of the deglycosylation treatment to affect either static adhesion (Fig. 8B) or adhesion under flow (Fig. 8C) suggests that mature glycosylation is not involved in the adhesive potency of CX3CR1.
FIGURE 8.

Functional analysis of the deglycosylated CX3CL1. A, 90 ng of CX3CL1-His6 was treated or not (a) with neuraminidase alone (b) or with neuraminidase and O-glycosidase (c). Then the proteins were analyzed by SDS-PAGE and Western blot. B, 90 ng of CX3CL1-His6 was absorbed onto wells of flat-bottom 96-well microtiter plates and treated or not (a) with neuraminidase alone (b) or with neuraminidase and O-glycosidase (c). Then 105 HEK-CX3CR1 cells were added and incubated for 45 min before washing. The results are expressed as percentage of total cells (mean ± S.D. of triplicates). The control corresponds to a well without CX3CL1-His6. C, HEK clone cells expressing CX3CR1 were suspended and assayed for adhesion in the presence (filled bars) or absence (empty bars) of 100 nm CX3CL1 in a parallel plate laminar flow chamber with coverslips coated with 180 ng of CX3CL1-His6 treated or not (a) with neuraminidase alone (b) or neuraminidase and with O-glycosidase (c). The results are expressed as the mean ± S.E. of triplicate experiments. The data are representative of three experiments.
DISCUSSION
The CX3CL1-CX3CR1 axis is a highly adhesive pair involved in the firm adhesion of monocytes or lymphocytes on activated endothelium (4). CX3CL1 adhesion potency is completely independent of that of selectin or integrin (16) but is similar in strength to that of integrins (16, 19). The adhesive potency of integrins relies on molecular activation and aggregation, due in part to the fact that the molecule is a flexible heterodimer forming what have been called “compliant legs” (22). In contrast, the relation between the structural characteristics of CX3CL1 and its adhesive features remains largely unknown. Earlier studies showed that the mucin stalk contributes to the ability of the ligand to act as an adhesion molecule in flow conditions (48) and that the nature of the stalk is unimportant, as it can be replaced by the rodlike segment of E-selectin (14). Nonetheless, the potential self-aggregation of CX3CL1 has not yet been investigated.
The BRET method is now widely used as a molecular proximity assay to study the clustering of membrane proteins including circadian clock proteins (49), integrins (50), growth factor receptors (51), and G protein-coupled receptors (52, 53). Working with CX3CL1, we found a specific and high BRET level in the HEK cell line (Fig. 2A) and the CHO cell line (data not shown). Several criteria are used to assess BRET and ensure that its results are not due merely to random protein (bystander) interactions but to constitutive clustering (30–33, 53). The BRET assay of CX3CL1 in the HEK cell line met all of them. (i) Replacing one of the monomers with an irrelevant protein produced no transfer (Fig. 2A). (ii) Expression of an excess of a non-tagged unit along with the Luc and YFP chimeras reduced BRET (Fig. 2F). (iii) BRET signals for a given acceptor/donor ratio (at a constant amount of donor) reached saturation; at a sufficient acceptor concentration, each donor molecule was engaged in a cluster, and the BRET signal reached a maximum (Fig. 2C). If BRET were due to random collision, BRET amplitude would increase continuously with the acceptor/donor ratio. (iv) BRET versus the total amount of tagged protein (donor plus acceptor) at a given acceptor/donor ratio should be constant or vary only slightly; this curve should not go to zero at low protein density, as recently pointed out (32, 33), and it clearly did not do so for CX3CL1 (Fig. 2D). The same criterion can be checked by directly plotting the BRET against the acceptor concentrations (30) (Fig. 2E). These data, obtained with transfected HEK cells, were supplemented with data from HTRF of primary cells (Fig. 3B). All of the data sets provided convergent evidence that CX3CL1 behaves as an aggregating protein, both as a transfected protein in cell lines and as a native protein expressed in HUVEC cells.
Moreover, flow cytometry and fluorescence imaging of our transfected HEK showed that approximately half of the CX3CL1 was located at the membrane (see Fig. 2A), consistent with recent findings (29). The HTRF experiment (Fig. 3B) showed that the membrane functional CX3CL1 molecules were definitely clustered. Because the BRET assay did not discriminate between intracellular and membranous proteins, we cannot assert the aggregation status of the intracellular CX3CL1 pool.
We found that the major content of the ALA12 mutant was a 60-kDa protein (90 kDa with the YFP tag), whereas the ALA5 and ALA7 mutants were, like the native CX3CL1 (90 kDa, 120 kDa with the YFP tag; Figs. 6D and 7C) composed of the mature molecule. Our data indicate that the 60-kDa species cannot be a shorter fragment of CX3CL1 and is thus likely to represent the immature, not fully glycosylated, species. First, we found that this small form exists in cells expressing the native CX3CL1 (Figs. 6D, left, and 7C, left), as observed previously (17, 45). It also has the same terminals as the native CX3CL1 (Fig. 7C, left and center). Moreover, it is preferentially located intracellularly (Fig. 6C) as is the immature form of CX3CL1 (17). Finally, enzymatic deglycosylation of the purified CX3CL1 gives rise to a 60-kDa form (Fig. 8A). Hence, our data suggest that the same conformational characteristic of the TM domain is essential for CX3CL1 clustering, mature glycosylation, and targeting of the mature form to the external membrane. Nonetheless, we isolated a HEK clone that expressed an immature, non-aggregating CX3CL1 molecule that was well inserted in the external membrane (ALA12, Fig. 7, A, B (right), and C).
To identify the domain primarily involved in the self-association, we conducted numerous mutation experiments (Figs. 4, 5, 6), which demonstrated the essential role of the TM domain. This was confirmed by our observation that the BRET of CX3CL1 was completely abolished after treatment with a concentration of Triton X-100 as low as 0.1% (data not shown). We failed, however, to pinpoint the precise sequence motif involved in the TM helix association. Self-association of transmembrane helices may involve a glycine motif, as in glycophorin A (40), a leucine zipper (41), or a phenylalanine in a “ball and socket” dimer interface (42). Our work shows that it is not the glycine, leucine, or phenylalanine residues alone that are involved (Fig. 5). In addition, replacement of residues 321–325 (ALA5) left the specific BRET unchanged and replacement of residues 326–332 (ALA7) increased it (Fig. 6). The higher BRET amplitude for some mutants, e.g. TM&cyto (Fig. 4D) and ALA7 (Fig. 6E), compared with the whole CX3CL1 molecule, is probably because of slight modifications of relative orientations of the dipole moments of the acceptor and donor, which can lead to more efficient BRET (39). Indeed, relatively small conformational changes could dramatically affect the BRET level (54–57), including conformational changes due to genetic variations (58).
Finally, the simultaneous replacement of 12 TM residues (ALA12) dramatically decreased BRET and made it nonspecific (Fig. 6, E and G). Thus, a long stretch of the TM domain appears to be involved in aggregation, but no special motif has a particular role; other examples of this type of structure have already been observed (59). One might think that CX3CL1 is clustered in some membrane microdomains, such as “lipid rafts,” although recent work has shown that CX3CL1 in renal tubular epithelial cells is found outside of lipid rafts (60). The CX3CL1 aggregation may also be due to external constraints imposed by companion molecules. However, the observation of CX3CL1 aggregation in very different cellular contexts, such as HUVEC, HEK, and CHO cell lines, suggests that it is most likely due to intrinsic properties of the CX3CL1-TM domain. Because this aggregation is functional and takes place in various cellular environments, we propose that it corresponds to oligomerization. Definitive proof of this oligomerization of CX3CL1, however, must await reconstitution in membrane models, and many questions must be answered before we can understand how it takes place. How can such small (0.5 nm in diameter) TM domains interact despite the large diameter of mucin-like glycosylated moieties (14)? How many monomers are involved in this aggregation/oligomerization?
The unique adhesive features of CX3CL1 under flow depend on its CD. They do not change after replacement of the mucin stalk by the rod-like segment of E-selectin (14). On the other hand, the chimeras that replaced the CD of CX3CL1 with those of CCL2, CCL4, CCL5, CCL21, or CXCL8 did not act as adhesion molecules with the cognate receptors (48). The ability of CX3CL1 to mediate adhesion may therefore depend on its unique slow receptor off-rate. Our data here (Fig. 7) complement these findings in showing that self-assembly is also required for the adhesive potency of CX3CL1. The ALA12 molecule behaves similarly to the CX3CL1 chimera presenting CCL2 or CXCL8 as a CD (48); it adheres to CX3CR1 in a mild static assay (Fig. 7D) but is not resistant to shear stress (Fig. 7, E and F).
Our results with the purified CX3CL1-His6 external domain (Fig. 8) show that glycosylation is not involved in adhesive potency. The immobilized protein, however, is not representative of the complete protein embedded in a cell membrane. Unfortunately, we could not test that point directly; all of our trials to deglycosylate cellular CX3CL1 were unsuccessful (see “Results”). Nonetheless, the molecular weight loss of the CX3CL1-His6 molecule induced by our deglycosylation treatment was equivalent to the difference in molecular weight between the mature CX3CL1 form and the immature form (Figs. 6 and 7), and there is evidence that the major form of the ALA12 mutant is the deglycosylated and immature form of CX3CL1. These results suggest that although glycosylation has no direct role in the adhesive CX3CL1-CX3CR1 interaction, it facilitates the emergence of the CX3CL1-CD moiety from the glycocalyx layer and therefore enables the ligand to target the receptor.
We conclude that the adhesiveness of CX3CL1 requires at least three simultaneous conditions: (i) the unique features of the CX3CL1-CD, probably related to its slow receptor off-rate (48); (ii) presentation of the CX3CL1-CD on top of a glycosylated stalk (48), the nature of which is unimportant (14); and (iii) a TM domain capable of aggregation (this work). Another chemokine, CXCL16, has a similar primary structure (5) but seems to be less adhesive in flow conditions than CX3CL1 (61).5 It is therefore essential to investigate its structure-function relations and to compare its function to that of CX3CL1.
The fact that clustering is crucial for the adhesive capacity of CX3CL1 expands its similarity with others adhesion molecules. Integrin clustering increases its avidity (62) and is required for efficient leukocyte adhesion (63). Similarly, the dimerization of P-selectin glycoprotein ligand-1 (PSGL-1) is required for optimal recognition of P-selectin (64). Moreover, as shown here for CX3CL1, mutational changes in the TM domain of PSGL-1 affect its self-assembly (65).
Our data open up the possibility of inhibiting CX3CL1 self-assembly by using competing peptides analogous to the TM domain, as already done for integrins (66). This could lead to the specific inhibition of the adhesive function of CX3CL1 and thus help to delineate the different roles of the two forms of this chemokine. More importantly, it also points to a new way of antagonizing the CX3CL1 function specifically, without interfering with the CX3CL1-CX3CR1 interaction. This type of pharmacology may be helpful in treating the ever growing number of diseases in which CX3CL1 is involved (3), especially atherogenesis (13), inflammation diseases (2), and degenerative disorders (7, 8).
Supplementary Material
Acknowledgments
We thank Monique Agrapart for providing HUVEC, Thomas Bader for help with HTRF experiments, and Stefano Marullo for the gift of pRLuc and pEYFP plasmids. We also thank Marie-Camille Delsuc and James Vigneron for preliminary experiments and Stefano Marullo, Catherine Labbé-Jullien, Ralf Jockers, and Claudine Mayer for helpful discussions. We acknowledge the local Post-genomic Platform of Pitié-Salpêtrière (P3S) for providing access to the Fusion Packard reader and the local Cellular Imaging Platform of Pitié-Salpêtrière (PICPS) for providing access to the confocal microscope.
This work was supported by grants from the Association de Recherche contre le Cancer and the Institut National de la Santé et de la Recherche Médicale (Programme National De Recherche Sur Les Maladies Cardiovasculaires) and by European FP6 “INNOCHEM” Contract LSHB-CT-2005-518167 (to C. C.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Table S1.
Footnotes
The abbreviations used are: CD, chemokine domain; BRET, bioluminescence resonance energy transfer; HTRF, homogeneous time-resolved fluorescence; HUVEC, human umbilical vein endothelial cells; TM, transmembrane; Muc, mucin-like stalk (CX3CL1 without the chemokine domain); TM&cyto, transmembrane domain and cytoplasmic tail (CX3CL1 without the chemokine domain and mucin stalk); ALA5, CX3CL1 mutant with alanines replacing residues 321–325; ALA7, CX3CL1 mutant with alanines replacing residues 326–332; ALA12, CX3CL1 mutant with alanines replacing residues 321–332; O-glycosidase, endo-d-galactosyl-N-acetyl-α-galactosamine hydrolase; CHO, Chinese hamster ovary; HEK, human embryonic kidney; mAb, monoclonal antibody; PBS, phosphate-buffered saline; HBSS, Hanks balanced salt solution; YFP, yellow fluorescent protein; EYFP, enhanced YFP; Luc, luciferase; PBP, pyridine-bipyridine.
P. Hermand and P. Deterre, unpublished data.
References
- 1.Luster, A. D., Alon, R., and von Andrian, U. H. (2005) Nat. Immunol. 6 1182-1190 [DOI] [PubMed] [Google Scholar]
- 2.Charo, I. F., and Ransohoff, R. M. (2006) N. Engl. J. Med. 354 610-621 [DOI] [PubMed] [Google Scholar]
- 3.Combadière, B., Combadière, C., and Deterre, P. (2007) Med. Sci. (Paris) 23 173-179 [DOI] [PubMed] [Google Scholar]
- 4.Bazan, J. F., Bacon, K. B., Hardiman, G., Wang, W., Soo, K., Rossi, D., Greaves, D. R., Zlotnik, A., and Schall, T. J. (1997) Nature 385 640-644 [DOI] [PubMed] [Google Scholar]
- 5.Matloubian, M., David, A., Engel, S., Ryan, J. E., and Cyster, J. G. (2000) Nat. Immunol. 1 298-304 [DOI] [PubMed] [Google Scholar]
- 6.Imai, T., Hieshima, K., Haskell, C., Baba, M., Nagira, M., Nishimura, M., Kakizaki, M., Takagi, S., Nomiyama, H., Schall, T. J., and Yoshie, O. (1997) Cell 91 521-530 [DOI] [PubMed] [Google Scholar]
- 7.Cardona, A. E., Pioro, E. P., Sasse, M. E., Kostenko, V., Cardona, S. M., Dijkstra, I. M., Huang, D., Kidd, G., Dombrowski, S., Dutta, R., Lee, J.-C., Cook, D. N., Jung, S., Lira, S. A., Littman, D. R., and Ransohoff, R. M. (2006) Nat. Neurosci. 9 917-924 [DOI] [PubMed] [Google Scholar]
- 8.Combadiere, C., Feumi, C., Raoul, W., Keller, N., Rodero, M., Pezard, A., Lavalette, S., Houssier, M., Jonet, L., Picard, E., Debre, P., Sirinyan, M., Deterre, P., Ferroukhi, T., Cohen, S. Y., Chauvaud, D., Jeanny, J. C., Chemtob, S., Behar-Cohen, F., and Sennlaub, F. (2007) J. Clin. Investig. 117 2920-2928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lavergne, E., Combadiere, B., Bonduelle, O., Iga, M., Gao, J. L., Maho, M., Boissonnas, A., Murphy, P. M., Debre, P., and Combadiere, C. (2003) Cancer Res. 63 7468-7474 [PubMed] [Google Scholar]
- 10.Yu, Y. R., Fong, A. M., Combadiere, C., Gao, J. L., Murphy, P. M., and Patel, D. D. (2007) Int. J. Cancer 121 316-322 [DOI] [PubMed] [Google Scholar]
- 11.Feng, L., Chen, S., Garcia, G. E., Xia, Y., Siani, M. A., Botti, P., Wilson, C. B., Harrison, J. K., and Bacon, K. B. (1999) Kidney Int. 56 612-620 [DOI] [PubMed] [Google Scholar]
- 12.Moatti, D., Faure, S., Fumeron, F., Amara, M., Seknadji, P., McDermott, D. H., Debre, P., Aumont, M. C., Murphy, P. M., de Prost, D., and Combadiere, C. (2001) Blood 97 1925-1928 [DOI] [PubMed] [Google Scholar]
- 13.McDermott, D. H., Fong, A. M., Yang, Q., Sechler, J. M., Cupples, L. A., Merrell, M. N., Wilson, P. W., D'Agostino, R. B., O'Donnell, C. J., Patel, D. D., and Murphy, P. M. (2003) J. Clin. Investig. 111 1241-1250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fong, A. M., Erickson, H. P., Zachariah, J. P., Poon, S., Schamberg, N. J., Imai, T., and Patel, D. D. (2000) J. Biol. Chem. 275 3781-3786 [DOI] [PubMed] [Google Scholar]
- 15.Mizoue, L. S., Bazan, J. F., Johnson, E. C., and Handel, T. M. (1999) Biochemistry 38 1402-1414 [DOI] [PubMed] [Google Scholar]
- 16.Fong, A. M., Robinson, L. A., Steeber, D. A., Tedder, T. F., Yoshie, O., Imai, T., and Patel, D. D. (1998) J. Exp. Med. 188 1413-1419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Garton, K. J., Gough, P. J., Blobel, C. P., Murphy, G., Greaves, D. R., Dempsey, P. J., and Raines, E. W. (2001) J. Biol. Chem. 276 37993-38001 [DOI] [PubMed] [Google Scholar]
- 18.Ludwig, A., Berkhout, T., Moores, K., Groot, P., and Chapman, G. (2002) J. Immunol. 168 604-612 [DOI] [PubMed] [Google Scholar]
- 19.Haskell, C. A., Cleary, M. D., and Charo, I. F. (1999) J. Biol. Chem. 274 10053-10058 [DOI] [PubMed] [Google Scholar]
- 20.Proudfoot, A. E., Handel, T. M., Johnson, Z., Lau, E. K., LiWang, P., Clark-Lewis, I., Borlat, F., Wells, T. N., and Kosco-Vilbois, M. H. (2003) Proc. Natl. Acad. Sci. U. S. A. 100 1885-1890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hoover, D. M., Mizoue, L. S., Handel, T. M., and Lubkowski, J. (2000) J. Biol. Chem. 275 23187-23193 [DOI] [PubMed] [Google Scholar]
- 22.Luo, B. H., Carman, C. V., and Springer, T. A. (2007) Annu. Rev. Immunol. 25 619-647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Veatch, W., and Stryer, L. (1977) J. Mol. Biol. 113 89-102 [DOI] [PubMed] [Google Scholar]
- 24.Maurel, D., Kniazeff, J., Mathis, G., Trinquet, E., Pin, J.-P., and Ansanay, H. (2004) Anal. Biochem. 329 253-262 [DOI] [PubMed] [Google Scholar]
- 25.Trinquet, E., Maurin, F., Preaudat, M., and Mathis, G. (2001) Anal. Biochem. 296 232-244 [DOI] [PubMed] [Google Scholar]
- 26.Trinquet, E., Fink, M., Bazin, H., Grillet, F., Maurin, F., Bourrier, E., Ansanay, H., Leroy, C., Michaud, A., Durroux, T., Maurel, D., Malhaire, F., Goudet, C., Pin, J. P., Naval, M., Hernout, O., Chretien, F., Chapleur, Y., and Mathis, G. (2006) Anal. Biochem. 358 126-135 [DOI] [PubMed] [Google Scholar]
- 27.Hermand, P., Huet, M., Callebaut, I., Gane, P., Ihanus, E., Gahmberg, C. G., Cartron, J. P., and Bailly, P. (2000) J. Biol. Chem. 275 26002-26010 [DOI] [PubMed] [Google Scholar]
- 28.Daoudi, M., Lavergne, E., Garin, A., Tarantino, N., Debre, P., Pincet, F., Combadiere, C., and Deterre, P. (2004) J. Biol. Chem. 279 19649-19657 [DOI] [PubMed] [Google Scholar]
- 29.Liu, G. Y., Kulasingam, V., Alexander, R. T., Touret, N., Fong, A. M., Patel, D. D., and Robinson, L. A. (2005) J. Biol. Chem. 280 19858-19866 [DOI] [PubMed] [Google Scholar]
- 30.Kenworthy, A. K., and Edidin, M. (1998) J. Cell Biol. 142 69-84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mercier, J.-F., Salahpour, A., Angers, S., Breit, A., and Bouvier, M. (2002) J. Biol. Chem. 277 44925-44931 [DOI] [PubMed] [Google Scholar]
- 32.James, J. R., Oliveira, M. I., Carmo, A. M., Iaboni, A., and Davis, S. J. (2006) Nat. Methods 3 1001-1006 [DOI] [PubMed] [Google Scholar]
- 33.Meyer, B. H., Segura, J. M., Martinez, K. L., Hovius, R., George, N., Johnsson, K., and Vogel, H. (2006) Proc. Natl. Acad. Sci. U. S. A. 103 2138-2143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Marullo, S., and Bouvier, M. (2007) Trends Pharmacol. Sci. 28 362-365 [DOI] [PubMed] [Google Scholar]
- 35.Mathis, G. (1999) J. Biomol. Screen. 4 309-314 [DOI] [PubMed] [Google Scholar]
- 36.Bazin, H., Trinquet, E., and Mathis, G. (2002) J. Biotechnol. 82 233-250 [DOI] [PubMed] [Google Scholar]
- 37.Jia, Y., Quinn, C. M., Gagnon, A. I., and Talanian, R. (2006) Anal. Biochem. 356 273-281 [DOI] [PubMed] [Google Scholar]
- 38.Imaizumi, T., Matsumiya, T., Fujimoto, K., Okamoto, K., Cui, X., Ohtaki, U., Hidemi Yoshida, and Satoh, K. (2000) Tohoku J. Exp. Med. 192 127-139 [DOI] [PubMed] [Google Scholar]
- 39.Stryer, L., and Haugland, R. P. (1967) Proc. Natl. Acad. Sci. U. S. A. 58 719-726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lemmon, M. A., Treutlein, H. R., Adams, P. D., Brunger, A. T., and Engelman, D. M. (1994) Nat. Struct. Biol. 1 157-163 [DOI] [PubMed] [Google Scholar]
- 41.Gurezka, R., Laage, R., Brosig, B., and Langosch, D. (1999) J. Biol. Chem. 274 9265-9270 [DOI] [PubMed] [Google Scholar]
- 42.LeFevre, K. R., and Cordes, M. H. (2003) Proc. Natl. Acad. Sci. U. S. A. 100 2345-2350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Abel, S., Hundhausen, C., Mentlein, R., Schulte, A., Berkhout, T. A., Broadway, N., Hartmann, D., Sedlacek, R., Dietrich, S., Muetze, B., Schuster, B., Kallen, K. J., Saftig, P., Rose-John, S., and Ludwig, A. (2004) J. Immunol. 172 6362-6372 [DOI] [PubMed] [Google Scholar]
- 44.Hundhausen, C., Misztela, D., Berkhout, T. A., Broadway, N., Saftig, P., Reiss, K., Hartmann, D., Fahrenholz, F., Postina, R., Matthews, V., Kallen, K. J., Rose-John, S., and Ludwig, A. (2003) Blood 102 1186-1195 [DOI] [PubMed] [Google Scholar]
- 45.Schulte, A., Schulz, B., Andrzejewski, M. G., Hundhausen, C., Mletzko, S., Achilles, J., Reiss, K., Paliga, K., Weber, C., John, S. R., and Ludwig, A. (2007) Biochem. Biophys. Res. Commun. 358 233-240 [DOI] [PubMed] [Google Scholar]
- 46.Gough, P. J., Garton, K. J., Wille, P. T., Rychlewski, M., Dempsey, P. J., and Raines, E. W. (2004) J. Immunol. 172 3678-3685 [DOI] [PubMed] [Google Scholar]
- 47.Kuan, S. F., Byrd, J. C., Basbaum, C., and Kim, Y. S. (1989) J. Biol. Chem. 264 19271-19277 [PubMed] [Google Scholar]
- 48.Haskell, C. A., Cleary, M. D., and Charo, I. F. (2000) J. Biol. Chem. 275 34183-34189 [DOI] [PubMed] [Google Scholar]
- 49.Xu, Y., Piston, D. W., and Johnson, C. H. (1999) Proc. Natl. Acad. Sci. U. S. A. 96 151-156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Buensuceso, C., de Virgilio, M., and Shattil, S. J. (2003) J. Biol. Chem. 278 15217-15224 [DOI] [PubMed] [Google Scholar]
- 51.Nouaille, S., Blanquart, C., Zilberfarb, V., Boute, N., Perdereau, D., Burnol, A. F., and Issad, T. (2006) Biochem. Pharmacol. 72 1355-1366 [DOI] [PubMed] [Google Scholar]
- 52.Bouvier, M. (2001) Nat. Rev. Neurosci. 2 274-286 [DOI] [PubMed] [Google Scholar]
- 53.Angers, S., Salahpour, A., Joly, E., Hilairet, S., Chelsky, D., Dennis, M., and Bouvier, M. (2000) Proc. Natl. Acad. Sci. U. S. A. 97 3684-3689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Boute, N., Boubekeur, S., Lacasa, D., and Issad, T. (2003) EMBO Rep. 4 313-319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gales, C., Van Durm, J. J., Schaak, S., Pontier, S., Percherancier, Y., Audet, M., Paris, H., and Bouvier, M. (2006) Nat. Struct. Mol. Biol. 13 778-786 [DOI] [PubMed] [Google Scholar]
- 56.Zhou, J. Y., Toth, P. T., and Miller, R. J. (2003) J. Pharmacol. Exp. Ther. 305 460-466 [DOI] [PubMed] [Google Scholar]
- 57.Tateyama, M., Abe, H., Nakata, H., Saito, O., and Kubo, Y. (2004) 11 637-642 [DOI] [PubMed]
- 58.Ayoub, M. A., Levoye, A., Delagrange, P., and Jockers, R. (2004) Mol. Pharmacol. 66 312-321 [DOI] [PubMed] [Google Scholar]
- 59.Prince, S. M., Howard, T. D., Myles, D. A., Wilkinson, C., Papiz, M. Z., Freer, A. A., Cogdell, R. J., and Isaacs, N. W. (2003) J. Mol. Biol. 326 307-315 [DOI] [PubMed] [Google Scholar]
- 60.Durkan, A. M., Alexander, R. T., Liu, G.-Y., Rui, M., Femia, G., and Robinson, L. A. (2007) J. Am Soc. Nephrol. 18 74-83 [DOI] [PubMed] [Google Scholar]
- 61.Shimaoka, T., Nakayama, T., Fukumoto, N., Kume, N., Takahashi, S., Yamaguchi, J., Minami, M., Hayashida, K., Kita, T., Ohsumi, J., Yoshie, O., and Yonehara, S. (2004) J. Leukocyte Biol. 75 267-274 [DOI] [PubMed] [Google Scholar]
- 62.Stewart, M., and Hogg, N. (1996) J. Cell. Biochem. 61 554-561 [DOI] [PubMed] [Google Scholar]
- 63.Wehrle-Haller, B., and Imhof, B. A. (2003) J. Pathol. 200 481-487 [DOI] [PubMed] [Google Scholar]
- 64.Snapp, K. R., Craig, R., Herron, M., Nelson, R. D., Stoolman, L. M., and Kansas, G. S. (1998) J. Cell Biol. 142 263-270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Epperson, T. K., Patel, K. D., McEver, R. P., and Cummings, R. D. (2000) J. Biol. Chem. 275 7839-7853 [DOI] [PubMed] [Google Scholar]
- 66.Li, R., Mitra, N., Gratkowski, H., Vilaire, G., Litvinov, R., Nagasami, C., Weisel, J. W., Lear, J. D., DeGrado, W. F., and Bennett, J. S. (2003) Science 300 795-798 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.