

Abstract
Sulfate reduction is one of the earliest types of energy metabolism used by ancestral organisms to sustain life. Despite extensive studies, many questions remain about the way respiratory sulfate reduction is associated with energy conservation. A crucial enzyme in this process is the dissimilatory sulfite reductase (dSiR), which contains a unique siroheme-[4Fe4S] coupled cofactor. Here, we report the structure of desulfoviridin from Desulfovibrio vulgaris, in which the dSiR DsrAB (sulfite reductase) subunits are bound to the DsrC protein. The α2β2γ2 assembly contains two siroheme-[4Fe4S] cofactors bound by DsrB, two sirohydrochlorins and two [4Fe4S] centers bound by DsrA, and another four [4Fe4S] centers in the ferredoxin domains. A sulfite molecule, coordinating the siroheme, is found at the active site. The DsrC protein is bound in a cleft between DsrA and DsrB with its conserved C-terminal cysteine reaching the distal side of the siroheme. We propose a novel mechanism for the process of sulfite reduction involving DsrAB, DsrC, and the DsrMKJOP membrane complex (a membrane complex with putative disulfide/thiol reductase activity), in which two of the six electrons for reduction of sulfite derive from the membrane quinone pool. These results show that DsrC is involved in sulfite reduction, which changes the mechanism of sulfate respiration. This has important implications for models used to date ancient sulfur metabolism based on sulfur isotope fractionations.
The dissimilatory reduction of sulfur compounds is one of the earliest energy metabolisms detected on earth, at ∼3.5 billion years ago (1, 2). At the end of the Archean (∼2.7 billion years ago), the advent of oxygenic photosynthesis led to a gradual increase in the levels of atmospheric oxygen, which in turn caused an increasing flux of sulfate to the oceans from weathering of sulfide minerals on land (3). As a consequence of this process, reduction of sulfate became a dominant biological process in the oceans, resulting in sulfidic anoxic conditions from about 2.5 to 0.6 billion years ago (3, 4). During this extended period, sulfate-reducing prokaryotes were main players in marine habitats where most evolutionary processes were taking place. Today, these organisms are still major contributors to the biological carbon and sulfur cycles, and their activities have important environmental and economic consequences.
A key enzyme in sulfur-based energy metabolism is the dissimilatory sulfite reductase (dSiR),3 which is present in organisms that reduce sulfate, sulfite, and other sulfur compounds. This enzyme is also found in some phototrophic and chemotrophic sulfur oxidizers, where it is proposed to operate in the reverse direction (reverse sulfite reductase, rSiR). The dSiR is minimally composed of two subunits, DsrA and DsrB, in an ∼200-kDa α2β2 arrangement. The dsrA and dsrB genes are paralogous and most likely arose from a very early gene duplication event that preceded the separation of the archaea and bacteria domains (5–8), in agreement with a very early onset of biological sulfite reduction. The dSiR belongs to a family of proteins that also include the assimilatory sulfite (aSiR) and nitrite (aNiR) reductases, the monomeric low molecular mass aSiRs, and other dSiRs like asrC and Fsr (9–11). This family has in common a characteristic cofactor assembly that includes an iron tetrahydroporphyrin of the isobacteriochlorin class, termed siroheme (see supplemental Fig. S1), that is coupled through its cysteine axial ligand to a [4Fe4S] iron-sulfur cluster (12–14). The aSiR and aNiR, found in plants, fungi, and bacteria, are monomeric enzymes that display an internal two-fold symmetry of a module that is related to DsrA/DsrB, suggesting that these assimilatory proteins also resulted from a gene duplication event (9, 10, 14). Phylogenetic sequence analysis indicates that both dSiRs and aSiRs diverged from a common ancestral gene that was present in one of the earliest life forms on earth (7, 10, 14).
Despite its central role in anaerobic metabolism, many aspects of dSiRs remain poorly understood. One of these is the nature of their physiological product because in vitro, they reduce sulfite to a mixture of trithionate, thiosulfate, and sulfide in proportions that depend on the reaction conditions (15), in contrast to aSiRs, which reduce sulfite directly to sulfide. This has led to some controversy, still unresolved, over whether the biological mechanism of dissimilatory sulfite reduction involves the formation of thiosulfate and trithionate as necessary intermediates (15, 16). Another open question regarding dSiRs is the content and the actual nature of the cofactors present. It is not clear whether both DsrA and DsrB subunits contain the coupled siroheme-[4Fe4S] cofactor because its characteristic binding site in DsrB is missing the first cysteine (5). Both DsrA and DsrB contain a ferredoxin-like domain, not present in aSiRs, which should bind an extra [4Fe4S] cluster. Cofactor quantification of dSiRs is quite disparate, with studies reporting from 2 to 4 sirohemes and from 10 to 32 non-heme irons per α2β2 module (13, 17–20). The nature of the catalytic cofactor has also been disputed, with some studies proposing a cubanesiroheme arrangement similar to that found in aSiRs (13, 19) and other studies proposing the presence of higher nuclearity high spin iron-sulfur clusters (18, 20, 21). Finally, a most important and unresolved question is the nature of the physiological electron donor to dSiR.
Most studies of dSiRs have focused on desulfoviridin (Dvir), the dSiR of Desulfovibrio spp. This enzyme has a characteristic and redox-insensitive band at ∼628 nm due to the presence of sirohydrochlorin, the iron-free form of siroheme (supplemental Fig. S1). It is reported that up to 80% of its siroheme lacks iron and is in the form of sirohydrochlorin (13, 18, 22), but this has been disputed (19). Another particular feature of Dvir is that it forms a stable complex with DsrC, a small protein of 11 kDa. Initial reports described DsrC as a γ subunit of dSiRs present in a stoichiometry of α2β2γ2 (23). However, the dsrC gene is located separately from dsrAB in several sulfate-reducing organisms, like Desulfovibrio vulgaris Hildenborough, where its expression is not coordinately regulated (24). There are also several dSiRs that do not include DsrC (5, 7), but all organisms that contain the dsrAB genes include also dsrC. These points indicate that DsrC is not a subunit of dSiR, but rather, a protein with which it interacts. In several sulfur-oxidizing and sulfite-reducing bacteria, dsrC is located in the same operon as dsrAB (25–27). It is actually one of the few proteins, apart from DsrAB (sulfite reductase) and the DsrMKJOP membrane complex (a membrane complex with putative disulfide/thiol reductase activity), to be conserved in both sulfur oxidizers and sulfate/sulfite reducers (26, 27). In addition, D. vulgaris dsrC is one of the most highly expressed genes in the cell with twice the expression level of dsrAB (28), pointing to an important role in cellular metabolism. Interestingly, homologues of DsrC, like YccK, are also present in organisms that do not contain dSiRs such as Escherichia coli and Hemophillus influenzae. YccK (renamed as TusE) was recently shown to be involved in sulfur-transfer reactions as part of the biosynthesis of thio-modifications of bacterial tRNA wobble positions (29). This work provided the first functional assignment of a DsrC-like protein in sulfur metabolism, with important implications to the dissimilatory processes where the function of DsrC has not yet been elucidated. The functional part of DsrC seems to be a C-terminal flexible arm, which displays several strictly conserved residues including a cysteine that is the penultimate one (30).
In this work, we report the structure of Dvir, the dSiR from D. vulgaris Hildenborough in which the DsrAB proteins are bound to DsrC. This structure resolves several long standing questions about dSiRs and suggests a function for DsrC that has important implications regarding the mechanism of sulfate reduction.
EXPERIMENTAL PROCEDURES
Protein Crystallization and X-ray Data—Dvir from D. vulgaris Hildenborough (DSM 644) was purified as described previously (32). A detailed description of the crystallization and structure solution of Dvir by multiple-wavelength anomalous dispersion based on the iron is presented in Ref. 32. In summary, small dark green crystals were obtained in 12.5% polyethylene glycol 4000, 0.1 m Tris-HCl, pH 8.5, 0.2 m MgCl2 and were cryoprotected with the crystallization solution supplemented with 20% glycerol. Crystals belong to the monoclinic space group P21 (a = 122.7, b = 119.4, and c = 146.68 Å, β = 110.0°) with two α2β2γ2 units per asymmetric unit corresponding to a solvent content of ∼51%. A three-wavelength multiple-wavelength anomalous dispersion data collection to 2.9 Å was carried out on the tunable beamline ID29 at the European Synchrotron Radiation Facility (ESRF), Grenoble, France.
Further efforts were undertaken to improve crystal x-ray diffraction, which included many crystallization set-ups with the same or similar experimental conditions to overcome low crystallization reproducibility and very poor crystal quality (low resolution, multiple and anisotropic diffraction patterns). Additional x-ray data were later measured at the ESRF ID23-2 beamline using an ADSC Q315 CCD detector. A data set with 300 images was collected at 0.933 Å, with an oscillation angle of 0.5° and an exposure time of 9 s/image. The x-ray data were integrated using the XDS program (33). Data merging and conversion to structure factor amplitudes were carried out respectively with SCALA (34) and TRUNCATE (35) from the CCP4 suite (36). Crystals diffract to 2.1 Å and belong to the same space group (P21) as the 2.9 Å data set, but with different cell parameters: a = 65.41, b = 118.91, and c = 132.25 Å, β = 104.1° with one α2β2γ2 assembly in the asymmetric unit and solvent content of ∼50%.
Structure Determination and Refinement—Because the two measured crystals are not isomorphous, the 2.1 Å data set structure was solved by molecular replacement with MOLREP (37) using the 2.9 Å model as a template (two αβγ units were found). Further refinement (using non-crystallographic symmetry restraints) was carried out with BUSTER (38) and model building using Coot (39). 5% of reflections were randomly excluded from the refinement for cross-validation (Rfree calculation). The electron density maps are generally well defined (better for chains ABC than DEF, as reflected by the higher average isotropic thermal motion parameters), except for some surface loops. The two αβγ units are very similar (r.m.s. deviation of 0.2 Å for 919 aligned residues), so chains A, B, and C will be used to describe the Dvir structure. The final model comprises 1818 amino acid residues, 2 sirohemes, 2 sirohydrochlorins, 8 [4Fe4S] clusters, 2 sulfite ions, and 1016 water molecules. The Ramachandran plot shows that 87.4% of the residues lie in most favored regions and only 0.4% fall in disallowed regions. Figs. 1, 2, 3, 4 were generated using PyMOL (40).
FIGURE 1.

Structure of the DsrAB sulfite reductase bound to DsrC. A, secondary structure representation of the α2β2γ2 assembly (DsrAB sulfite reductase bound to DsrC), with the cofactors in ball-and-stick mode. DsrA (chains A and D) is colored blue, DsrB (chains B and E) is magenta, and DsrC (chains C and F) is green. The distance between the cofactors from one αβγ unit is displayed on the right side. Color code is yellow, carbon; red, oxygen; blue, nitrogen; brown, iron; and green, sulfur. B, molecular surface of theα2β2γ2 assembly with oneαβγ unit in gray and the other colored according to A. C, superposition of DsrA and DsrB. N-term, N terminus; C-term, C terminus.
FIGURE 2.

The sirohydrochlorin group of DsrA and the siroheme-[4Fe4S] active site
of DsrB. A–C, electron density maps
(2Fo - Fc contoured at 1. 5 σ)
around the sirohydrochlorin group and cluster 1 of DsrA (A), the
siroheme and cluster 1 of DsrB (B); and the siroheme iron of DsrB,
bound 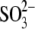 and C-terminal arm of DsrC
(in stereo) (C). The Fo - Fc
OMIT map is shown for sulfite ion and Sγ of Cys-104C (3.5 σ
contour). The amino acid residues and water molecules (red spheres)
that are hydrogen-bonded with sulfite are also displayed. The color code is
the same as in Fig. 1, except
for carbon atoms of substrate interacting residues (gray) and DsrC
C-terminal arm (orange). SRM B, siroheme B.
and C-terminal arm of DsrC
(in stereo) (C). The Fo - Fc
OMIT map is shown for sulfite ion and Sγ of Cys-104C (3.5 σ
contour). The amino acid residues and water molecules (red spheres)
that are hydrogen-bonded with sulfite are also displayed. The color code is
the same as in Fig. 1, except
for carbon atoms of substrate interacting residues (gray) and DsrC
C-terminal arm (orange). SRM B, siroheme B.
FIGURE 3.

Substrate and DsrC-binding channels. A, molecular surface
of one αβγ unit showing the substrate channel, with a zoomed
view of the channel entrance, containing a randomly placed
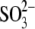 ion for scale; the distal site of
the siroheme (in yellow) is solvent-accessible. The color scheme is
as in Fig. 1. B,
surface representation of DsrAB with DsrC displaced from its binding position.
The siroheme (in yellow) can be seen in the interior of the cleft
formed between DsrAB. C, secondary structure view of one DsrABC unit
with A. fulgidus DsrC (PDB code: 1SAU) superposed. The zoomed image
shows the extended C-terminal arm of the D. vulgaris DsrC reaching
the heme and the retracted arm from A. fulgidus DsrC. The two
conserved cysteines of each DsrC are represented in stick mode, a dashed
black line showing the close contact between Cys-103 and Cys-114 in
A. fulgidus DsrC. Some water molecules at the interface are displayed
as red spheres.
ion for scale; the distal site of
the siroheme (in yellow) is solvent-accessible. The color scheme is
as in Fig. 1. B,
surface representation of DsrAB with DsrC displaced from its binding position.
The siroheme (in yellow) can be seen in the interior of the cleft
formed between DsrAB. C, secondary structure view of one DsrABC unit
with A. fulgidus DsrC (PDB code: 1SAU) superposed. The zoomed image
shows the extended C-terminal arm of the D. vulgaris DsrC reaching
the heme and the retracted arm from A. fulgidus DsrC. The two
conserved cysteines of each DsrC are represented in stick mode, a dashed
black line showing the close contact between Cys-103 and Cys-114 in
A. fulgidus DsrC. Some water molecules at the interface are displayed
as red spheres.
FIGURE 4.

Schematic representation of the proposed sulfate reduction mechanism. Sat, sulfate adenylyltransferase; ApsAB, adenosine phosphosulfate reductase; QmoABC, membrane complex that is the probable electron donor to ApsAB. C-SH represents the thiol group of Cys-93, C-S-SH a persulfide group of Cys 104, and C-S-S-C the disulfide bond between the two Cys. SH, Src homology.
RESULTS
The Crystal Structure of Dvir—The 2.1 Å final model of Dvir was refined to an R-factor of 19% (Rfree of 21.9%). Statistics of data processing and refinement are listed in Table 1. This model contains two αβγ units (α: residues 2–437 of DsrA, chains A and D; β: residues 2–381 of DsrB, chains B and E; and γ: residues 3–105 of DsrC, chains C and F) (Fig. 1A). The α2β2γ2 assembly has overall dimensions of ∼125 × 100 × 60 Å and a total surface area of about 55,720 Å2. The interface area between the two αβ units is 6,100 Å2, which represents ∼11% of the total dimer surface area. This interface is mainly hydrophilic with an important contribution from the DsrA C-terminal tail that embraces DsrB from the other αβ unit, and to a lesser extent, the C terminus of DsrB (Fig. 1B). This demonstrates a strong interaction between the two αβ units that corroborates the α2β2 minimal composition proposed for all dSiRs.
TABLE 1.
Crystallographic data processing and refinement statistics Values in parentheses show the statistics of the highest resolution shell (2.21–2.10 Å).
| Data collection | |
| Wavelength (Å) | 0.933 |
| Space group | P21 |
| Unit cell parameters (Å, °) | a = 65.41, b = 118.90, c = 132.45, β = 104.1 |
| Resolution range (Å) | 40.29–2.10 (2.21–2.10) |
| Unique reflections | 112244 (14772) |
| I/σ (I) | 13.2 (2.0) |
| Rmerge (%)a | 4.7 (44.9) |
| Rpim (%)b | 3.8 (38.1) |
| Completeness (%) | 98.1 (88.7) |
| Multiplicity | 3.1 (2.4) |
| Wilson B (Å2) | 36.8 |
| Refinement | |
| No. amino acid residues in asymmetric unit | 1818 |
| Other moieties | |
| Siroheme (SRM) | 2 (with iron) + 2 (iron-free) |
| Fe4S4 clusters | 8 |
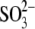
|
2 |
| Water | 1016 |
| R/R-free (%) | 19.0/21.9 |
| Average B-factor (main-chain) (Å2) | |
| Chains A and B/C | 31.9/42.9 |
| Chains D and E/F | 47.2/56.1 |
| Solvent molecules | 43.7 |
| r.m.s. bond length deviation from ideal values (Å) | 0.005 |
| r.m.s. bond angle deviation from ideal values (°) | 0.944 |
Rmerge = Σh Σi |Ii(h) – 〈I(h)〉|/Σh Σi Ii (h), where I is the observed intensity, 〈I〉 is the average intensity of multiple observations from symmetry-related reflections, and N is redundancy
Rpim = Σh [1/(N-1)]1/2 Σi |Ii(h) – 〈I(h) 〉|/Σh Σi Ii (h), where I is the observed intensity, 〈I〉 is the average intensity of multiple observations from symmetry-related reflections, and N is redundancy
DsrAB Structure and Cofactor Binding—The α-, β-, and γ-proteins correspond respectively to DsrA, DsrB, and DsrC. The structures of the DsrA and DsrB subunits are very similar. Both proteins can be divided into three main domains (A1/B1, A2/B2, A3/B3), which can be superimposed with an r.m.s. deviation of 1.96 Å for 321 equivalent Cα atoms, despite sharing only 20% sequence identity (Fig. 1C). Apart from the three similar domains, the structures of DsrA and DsrB include N- and C-terminal tails that are distinct in the two proteins.
The A1 and B1 domains (residues 19A-168A and 24B-134B, respectively) show an antiparallel four- or five-stranded β-sheet, flanked by a pair of α-helices with two additional helices in the N-terminal region of the A1 domain. The A2 and B2 domains (residues 169A-241A and 323A-402A and residues 135B-207B and 278B-365B, respectively) consist of a five-stranded β-sheet bundled with several α-helices. These domains bind a [4Fe4S] cluster (cluster 1) that is part of the siroheme-[4Fe4S] cofactor (Fig. 1, A and C). In the A2 domain, this cluster is coordinated by four cysteines (Cys-177A, -183A, -221A, -225A), which are strictly conserved among dSiRs and form the CX5CXnCX3C motif previously identified as the sequence motif for binding of the siroheme-[4Fe4S] cofactor in both aSiRs and dSiRs (5, 41). Notably, the [4Fe4S] cluster bound by DsrA is in close proximity to a sirohydrochlorin group, i.e. a siroheme that is demetallated (Fig. 2A). The sirohydrochlorin is buried in the interior of the protein and sits on the interface between DsrA and DsrB. In the B2 domain, the iron-sulfur cluster is also coordinated by four cysteines (Cys-151B, -188B, -189B, and -193B) but in a different sequence motif CXnCCX3C. The absence of the CX5CXnCX3C motif in DsrB led to the idea that this protein would not bind a siroheme-[4Fe4S] cofactor, which is now shown not to be true. The CXnCCX3C motif responsible for binding the catalytic cluster 1 in DsrB is conserved in most dSiRs (with the exception of DsrB from Pyrobaculum aerophilum, which has the same CX5CXnCX3C motif in both DsrA and DsrB) and is quite unusual in having two consecutive cysteines ligating the cluster. There are only two examples where this has been reported structurally: for cluster N2 in the Nqo6 subunit of Thermus thermophilus complex I (42) and for Pseudomonas aeruginosa adenosine phosphosulfate reductase (43). Recently, tandem cysteine coordination of a novel [4Fe4S] cluster has also been proposed for a large family of CCG proteins (which includes DsrK) by site-directed mutagenesis (44). Close to cluster 1 of DsrB is a siroheme group that is coupled to the cluster through Cys-193B, positioned at ∼2.4 Å from both (Fig. 2B). Thus, in Dvir, the DsrB protein contains the typical exchange-coupled siroheme-[4Fe4S] cofactor previously identified in aSiRs, aNiRs, and dSiRs as the catalytic site for sulfite or nitrite reduction (9, 12, 13), whereas the DsrA protein contains a demetallated siroheme that is obviously not catalytic.
The A1A2B1B2 domains of Dvir form a unit that is comparable with the structures of siroheme aSiR and aNiR. There are two structures of aSiRs described, the truncated sulfite reductase hemoprotein from E. coli (14) and the monomeric NirA protein from Mycobacterium tuberculosis (45), and one structure of an aNiR from spinach (46). Despite the low degree of sequence identity (below 25%), these three structures show an overall similarity in the folding arrangement, and all contain one siroheme-[4Fe4S] cofactor. A structural comparison of Dvir with the aSiRs and aNiR shows that the domains A1A2 of DsrA can be superimposed with half of the parachute domain (domain 1 in aSiRs) and the siroheme binding domain (domain 2 in aSiRs), whereas the domains B1B2 of DsrB can be superimposed with the other half of the parachute domain and the [4Fe4S] coordinating domain (domain 3 in aSiRs). In this orientation, the catalytic siroheme-[4Fe4S] cofactor of Dvir is in a similar position to the aSiR cofactors. Superposition of these Dvir domains with the aSiR and aNiR structures yields r.m.s. values in the range 2.4–2.8 Å for the aligned Cα atoms. This similarity supports the proposals of a common ancestor for dSiRs, aSiRs, and aNiRs (7, 10, 14).
The A2/B2 domains of DsrA/DsrB are interrupted by insertion of the A3/B3 domains (residues 242A-322A and 208B-277B) that display a typical ferredoxin fold. These domains fold into two two-stranded β-sheets surrounded by three α-helices and bind one [4Fe4S]cluster (cluster 2), coordinated by four cysteines (Cys-284A, -288A, -306A, and -309A in DsrA and Cys-231B, -258B, -261B, and -264B in DsrB) (Fig. 1, A and C). Cluster 2 of the DsrB ferredoxin domain is 13 Å from the siroheme-coupled cluster 1 and 6.5 Å from the protein surface. This arrangement suggests that cluster 2 is positioned to transfer electrons from a yet unidentified external electron donor to cluster 1 of the catalytic cofactor. The way the ferredoxin domain interrupts the A2/B2 domain agrees with the proposal that a ferredoxin gene was inserted into an ancestral gene for a siroheme-[4Fe4S] binding reductase (5). In this respect, it is quite interesting to note that when the structure of a DsrAB dimer is aligned with the structure of the spinach aNiR (for which ferredoxin is the electron donor), with the two sirohemes superimposed, the ferredoxin domain of DsrB is in the actual position where spinach ferredoxin has been modeled to bind to the aNiR (46). This indicates that the ancestral siroheme-[4Fe4S] binding reductase was probably made more efficient by incorporating its electron donor as another domain in its sequence.
The recently reported structure of the Archaeoglobus fulgidus dSiR (31) includes only the α2β2 unit of DsrAB. Overall, this structure is quite similar to the structure of the DsrAB proteins from D. vulgaris, apart from some small differences in the N and C termini of both subunits and two longer loops in the ferredoxin domains (see supplemental Fig. S2). The most noteworthy difference lies in the fact that dSiR from A. fulgidus has four sirohemes and no sirohydrochlorin.
The Catalytic Site—The siroheme and sirohydrochlorin groups are located in the interior of the molecule, in the interface between DsrA and DsrB, with the closest distance to the protein surface of ∼14 Å. The catalytic siroheme is surrounded on the proximal side by residues that belong to DsrB. In contrast, the distal side of the heme, where the substrate will bind, is surrounded by basic residues that belong to DsrA, namely Arg-83, Arg-101, Arg-172, Lys-213, Lys-215, Lys-217, Arg-231, Arg-376, and Arg-378. These residues are strictly conserved in dSiRs and create a positive pocket in the active site that favors the binding of the negatively charged sulfite and may also be involved in providing some of the protons necessary for sulfite reduction. Moreover, several of these residues are establishing H-bonds or salt bridges with the siroheme carboxylate groups and are important for counterbalancing the negative charge of these groups and thus for protein stabilization. Residues Arg-71, His-150, and His-152 of DsrB are also involved in this role. These features of the active site of Dvir are also present in the aSiRs and aNiR (14, 32, 33), and a structure-based sequence alignment shows conservation of the residues involved in forming the active site and stabilizing the siroheme carboxylates.
At the distal side of the catalytic siroheme, residual density is observed, indicating the presence of an axial ligand bound to the iron, which was assigned as a sulfite ion due to its trigonal pyramidal shape combined with the presence of positively charged residues oriented toward the negatively charged oxygen atoms of the sulfite ion. The electron density was well fitted with sulfite as there were no significant residual negative or positive electron density peaks. It is quite clear that an atom lighter than sulfur (e.g. carbon, nitrogen, oxygen) cannot be fitted at the central position. Sulfite is coordinating the siroheme iron through its sulfur atom (∼2.4 Å), with its oxygen atoms establishing H-bonds with Arg-101A, Arg-172A, Lys-213A, and Lys-215A and also with two water molecules (Fig. 2C). The four basic residues that interact directly with the substrate are strictly conserved in dSiRs and aSiRs, whereas in spinach aNir, Lys-215 is replaced by Asn-226, which has been associated with a switch in substrate preference from sulfite to nitrite (45, 46). At the distal side of the sirohydrochlorin group in Dvir, the crucial residues Arg-172A and Lys-213A of the active site are replaced by Ser-140B and Pro-181B (Ser-130B and Met-170B for the structural heme in A. fulgidus dSiR).
We have also identified a possible substrate channel (Fig. 3A). The molecular surface of Dvir shows that the distal face of the catalytic siroheme B is solvent-accessible through a channel (∼9 Å wide, 6 Å high, and 20 Å deep), with one side formed by residue Tyr-212A and loops 334A-336A and 374A-381A and the other side formed by residues on helices 225B-230B and 263B-267B. The positive electrostatic potential at the entrance of this channel, which extends into the active site, facilitates the entrance of the negatively charged substrate molecule. This substrate channel is not present in the case of the DsrA sirohydrochlorin group due to the presence of bulkier residues, such as Arg-283A and Tyr-334B, which block the access to the demetallated siroheme in DsrA. In the A. fulgidus dSiR, the substrate binding site of the DsrA siroheme is also blocked by a tryptophan residue that is sitting right above the iron. In addition, there is an extended loop that blocks access to this heme, which was proposed to have a structural role (31). This loop is absent from Dvir, but in its place, there is an extended loop from the DsrA ferredoxin domain (Gly-257A to Asp-275A) that also shields access to the sirohydrochlorin.
Structure of DsrC Bound to DsrAB—The evidence so far available for DsrC indicates that it is likely to have an important role in sulfite metabolism, and there have been several proposals for its involvement in the sulfite reduction/sulfide oxidation pathway (25, 26, 30, 47). DsrC contains a highly conserved C-terminal sequence that includes two cysteines. The penultimate residue, Cys-104C in D. vulgaris, is strictly conserved in all family members (including YccK/TusE), whereas the previous one (Cys-93C) is conserved only in DsrC proteins that are involved in dissimilatory sulfur metabolism. This suggests the possible involvement of a disulfide bridge between these two cysteines as a redox-active center in the sulfite reduction pathway, and one of the proposals is that DsrC could act as an electron donor for DsrAB (30, 47).
The structure of DsrC bound to DsrAB comprises 105 residues (Glu-3 to Val-105) and has a mainly helical fold (six α-helices and one 3/10 helix) with a two-stranded-β-sheet at the N terminus (Fig. 1C). DsrC is enclosed in a cleft formed by the DsrA and DsrB subunits and establishes several hydrogen bonds with both (Fig. 3, B and C). The C-terminal arm of DsrC extends into the interface between DsrA and DsrB, where it reaches the siroheme (Figs. 2C and 3C). DsrC and DsrAB are associated through a large interface area of 3,090 Å2 that corresponds to ∼24% of the total area of DsrC, 4% of DsrA, and only 2.4% of DsrB, which become inaccessible to solvent due to the complex formation. The DsrC-DsrAB interface has a pronounced polar character comprising several hydrogen bonds (10 H-bonds between DsrC and DsrA and 3 H-bonds with DsrB), many water molecules, and only a few hydrophobic contacts.
The D. vulgaris DsrC structure is quite similar to the other known DsrC structures from P. aerophilum (30), A. fulgidus (47), and Allochromatium vinosum (Protein Data Bank code: 1YX3). The only major structural difference among these structures is in the C-terminal segment. In the NMR structures of P. aerophilum and A. vinosum DsrC, the C-terminal arm is very disordered and in an extended configuration, whereas in the A. fulgidus crystal structure, the C-terminal arm is retracted and in contact with the rest of the protein, such that the two conserved cysteines come in van der Waals contact (Fig. 3C). Treatment with an oxidizing agent leads to the formation of a disulfide bond between them (47). In the D. vulgaris model, the extended C-terminal arm of DsrC has its Cys-104C positioned right next to the substrate binding side (Figs. 2C and 3C). This configuration strongly suggests an involvement of Cys-104C in the reduction of sulfite. Cys-93C is 18 Å away from Cys-104C, so a disulfide bond between the two is not possible in this configuration.
Remarkably, the Sγ of Cys-104C is only 1.9 Å from the 20′-meso carbon of the porphyrin ring of siroheme B, indicating that there is a cross-link from Cys-104C to the heme (Fig. 2C). This observation is unexpected as it fixes the interaction between DsrC and DsrAB and conflicts with the observation that for most dSiRs, the DsrC protein is not found associated with DsrAB. Therefore, the interaction between Cys-104C and siroheme B should be transient in nature. There are several possible mechanisms that could generate this cross-link, like formation of a π-cation radical in the siroheme, which could be quenched by reaction with the nearby Cys-104C or generation of a sulfenic acid on Cys-104 followed by attack and displacement by the ring. Whatever its nature, this side reaction is most probably caused by contact with oxygen during aerobic purification of the enzyme because it is highly unlikely that the link could be formed during normal turnover conditions as it would be highly inhibitory. The observed DsrAB-DsrC cross-linked complex can explain the very low levels of activity of aerobically isolated Dvir when compared with the activity of whole cells. This cross-link also explains why only sirohydrochlorin and not the siroheme can be extracted from Dvir (22).
DISCUSSION
The structure of D. vulgaris Dvir shows that it contains two sirohemes, two sirohydrochlorins and 34 irons per α2β2 unit, finally settling the debate on the cofactor content of this protein. The nature of the catalytic site was confirmed to be a coupled siroheme-[4Fe4S] cofactor. The presence of the catalytic cofactor in DsrB rather than DsrA was unexpected, as well as the presence of fully demetallated siroheme in DsrA. To the best of our knowledge, this is the first time that a metal-free heme has been reported in a protein structure. However, it has long been reported that Dvir contained sirohydrochlorin in uncertain amounts (22), which is responsible for its characteristic absorption peak at ∼628 nm. This peak is detected even in whole bacterial cells (48), revealing that the presence of this chromophore is not the result of iron loss during protein purification. Several factors, such as the absence of a substrate channel and the lack of two of the four crucial positive residues conserved at the active site of the several siroheme-containing reductases, indicate that the cofactor in DsrA would not be catalytic even if it had iron. Most dSiRs are reported to contain two sirohemes per α2β2 unit (for example, see Refs. 5, 7, 13 and 21). However, apart from Dvir, no dSiR has the characteristic absorption at 628 nm, indicating that they do not contain sirohydrochlorin. Because only two sirohemes are catalytically active in Dvir, it is possible that only the two catalytic sirohemes are present in other dSiRs. The presence of four sirohemes in the structure of A. fulgidus dSiR was unforeseen because siroheme quantification of this protein had yielded two hemes per α2β2 unit (5). This suggests that the heme quantification method is not reliable, so the actual number of sirohemes in other dSiRs is yet to be firmly established. Nevertheless, only two of the four A. fulgidus dSiR sirohemes are proposed to be catalytic because in the other two, the substrate binding site is blocked, and some crucial positive residues are also missing. Therefore, for both dSiRs, only two catalitically active sirohemes are present per α2β2 unit, whereas in aSiRs, the second catalytic heme that should be present was lost during evolution (9). These observations suggest that in both families (aSiRs and dSiRs), the process of gene duplication was associated with loss of function from one of the catalytic sites. In aSiRs, this center is no longer present, whereas in dSiRs, it may still be present, but without a catalytic role. In the particular case of Dvir from Desulfovibrio spp., this center has no iron bound to the porphyrin. It is quite intriguing why this iron is not present as this is an absolutely unprecedented situation. By comparing the structures of A. fulgidus and D. vulgaris dSiRs, we could find no obvious difference surrounding the prosthetic group of DsrA that could explain why one retains the iron and not the other. Nothing is really known about how siroheme (or sirohydrochlorin) is inserted into proteins, so it is difficult to speculate as to in which step iron is lost (or not inserted) into the DsrA heme of Dvir.
Regarding the positively charged active site of Dvir, it is quite striking that so many of its characteristics are shared with those of aSiRs, indicating that this is not the main factor affecting the difference in products observed between both types of enzymes. However, there are a few noteworthy differences in the active site residues of Dvir in comparison with the aSiRs. In particular, two positive residues of the aSiRs that are making salt bridges to the carboxylate groups of acetate 12′ (Arg-117 of E. coli aSiR) and acetate 18′ of the siroheme (Arg-214 of E. coli aSiR) are replaced by Thr-136A and Tyr-212A, respectively, two residues that are conserved in dSiRs (except in P. aerophilum). Also, a strictly conserved glutamine in aSiRs and aNiRs (Gln-121 of E. coli aSiR), which is H-bonded with the carboxylate of propionate 3′, is absent in dSiRs. A strictly conserved arginine of dSiRs, Arg-83A in Dvir, is also making a salt bridge with the same carboxylate and is absent in aSiRs. These localized differences may affect the electronic density at the siroheme in such a way that may have an effect on the sulfite reduction mechanism of dSiRs relative to aSiRs. It is also interesting to compare the active site residues of dSiRs with the rSiRs, which in theory catalyze the reverse reaction, i.e. oxidation of sulfide to sulfite. It is striking that all of the residues highlighted above for Dvir are also conserved in rSiRs, with the exception of Arg-376 of DsrA and His-152 of DsrB, which are establishing H-bonds with acetates 18′ and 2′ of the siroheme. The acetate 2′ has been proposed to contain an amide instead of a carboxylic group (49). However, at the current resolution, it is not possible to differentiate between oxygen and nitrogen atoms at this position.
The elucidation of the interaction between DsrAB and DsrC is a major step in our understanding of the role of this small protein in sulfite reduction. DsrC has a highly conserved C terminus that is very disordered in solution, indicating that it is a site of interaction with other proteins. In contrast, in the crystal structure of A. fulgidus DsrC, the C terminus is found in a different conformation, close to the rest of the protein, such that the two strictly conserved cysteines come within bonding distance of each other. Our structure shows that when DsrC interacts with DsrAB, its C terminus is extended and inserts into a cleft between both proteins, in a way that brings its penultimate cysteine in close contact to the siroheme. The position of DsrC Cys-104 near the siroheme active site is extremely relevant and has important implications for the mechanism of sulfite reduction. It is important to note that aSiRs, which do not interact with DsrC-like proteins, reduce sulfite directly to sulfide, whereas in vitro, dSiRs display a very low activity (suggesting the need for partner proteins) and form a mixture of products that is probably not physiologic. The presence of Cys-104C so close to the siroheme strongly suggests its involvement in binding either the substrate or the product or in the catalytic reaction. Given the difference in products between aSiRs and dSiRs and the fact that a sulfite ion is found at the active site, it seems more likely that DsrC is involved in the catalytic reaction and/or binding the product. In turnover conditions, the Sγ atom of Cys-104C may be positioned right next to the substrate molecule because a rotamer of Cys-104C can be easily oriented toward the siroheme distal site so that its Sγ atom is only 2.4 Å away from one of the oxygen atoms of the sulfite ion. We propose that in dSiRs, the reduction of sulfite involves not a six-electron but a four-electron reduction to form an S0 intermediate, which is then transferred to Cys-104C of DsrC to form a persulfide (Fig. 4). Once DsrC dissociates from DsrAB, Cys-93C can reduce this persulfide, releasing H2S and forming a Cys-93–Cys-104 disulfide in DsrC. This oxidized form of DrsC may then be reduced by the membrane-bound DsrMKJOP complex, which contains a cytoplasmic catalytic subunit (DsrK) similar to heterodisulfide reductases and has an unusual catalytic iron-sulfur center for putative reduction of disulfide bonds (26). The reduced DsrC can then enter another catalytic cycle and bind to DsrAB. Thus, two of the electrons for sulfite reduction would derive from the quinone pool (via DsrMKJOP and DsrC), and the other four would derive from the unknown electron donor to Dvir. The involvement of the DsrMKJOP complex provides a link between membrane quinol oxidation and dSiR, which can explain the fact that proton translocation is observed upon reduction of sulfite (50). Thus, a persulfide of DsrC would be a crucial intermediate in the reduction of sulfite. The involvement of persulfides as a form of “activated sulfur” is well known in several biological pathways such as the biosynthesis of FeS clusters and other cofactors (51). In the DsrC homologue YccK/TusE, a persulfide of the cysteine corresponding to Cys-104C is also involved in the sulfur transfer reactions performed by this protein (29). The SoxYZ protein of sulfur oxidizers is another example of a protein that carries sulfur intermediates on an external mobile arm containing a conserved cysteine (52).
The observed cross-link between DsrC and the siroheme explains why in Dvir the association between this protein and DsrAB is stable, whereas in other dSiRs it must be transient. This unexpected feature may result from one of several possible mechanisms, most likely as a result of aerobic purification of the enzyme. One possibility is the formation of a π-cation radical in the siroheme. One of the characteristics of isobacteriochlorins (porphyrins in which two adjacent pyrrole rings are reduced, as sirohydrochlorin) relative to porphyrins and chlorins is the greater ease with which these macrocycles can be oxidized to generate a π-cation radical species (53). In model compounds, ring oxidation in isobacteriochlorins occurs before oxidation of the metal. The possibility of forming a π-cation radical has been invoked as one of the reasons for which sirohemes may have been selected for the reduction of sulfite and nitrite as it permits an extra electron to be generated at the active site (54). In the structure of the dSiR from M. tuberculosis, an unusual covalent bond is also found between the side chains of Tyr-69 and Cys-161, in a position relative to the siroheme very similar to that of Cys-104C (45). We believe that this covalent link may also be the result of oxidation by a π-cation radical species. There are other reports of the presence of cross-linked amino acids close to the active sites of redox metalloenzymes such as galactose oxidase, cytochrome c oxidase, catechol oxidase, and others (Ref. 55) and references therein), for which the significance is mostly unknown. For example the Tyr-Cys cross-link present in galactose oxidase forms spontaneously upon exposure to Cu(I) and oxygen through a free radical mechanism (55). Autocatalytic reactions are common in heme modifications like in heme oxygenase, mammalian peroxidases, and cytochrome P450 (56). However, covalent cross-links between porphyrin meso carbons and proteins are very unusual but have been reported in hydroxylamine oxidoreductase and cytochrome P460 from Nitrosomonas europea (57). To the best of our knowledge, this is the first time a covalent link is observed involving a heme meso carbon and a cysteine residue. The A. fulgidus dSiR, which was purified anaerobically, does not include DsrC (31). We recently obtained crystals from anaerobically purified Dvir, which still showed the presence of the cross-link. However, it is not possible to discard the possibility of transient contact with oxygen during protein or crystal manipulations. Further work will be necessary to elucidate the conditions that lead to this unusual link between the siroheme and DsrC.
The mechanism proposed herein can explain several pending questions regarding the process of sulfite reduction, namely the role of DsrC and its two conserved cysteines, the role of the DsrMKJOP complex, and why in vitro the dSirs do not form sulfide but a mixture of products that depend on the reaction conditions (and are probably the result of sulfite reacting with semireduced species at the siroheme). It is interesting that in whole cell extracts of D. vulgaris, the product detected for reduction of sulfite was only sulfide, whereas after removal of the membrane fraction, the mixture of products was observed (58). It has also been reported that a form of Dvir purified from the membranes (possibly containing small amounts of the DsrMKJOP complex) forms sulfide as the major product, in contrast to the same protein isolated from the soluble fraction, which formed a mixture of products (59). In the phototrophic sulfur oxidizer A. vinosum, the DsrKJO proteins were co-purified with DsrAB and DsrC, supporting an association between these proteins also in the reverse pathway (25).
The elucidation of the way DsrAB interacts with DsrC has important implications because it shows that this protein has a role in sulfite reduction. The involvement of other proteins besides the DsrAB may require a reassessment of the models used to date ancient sulfur metabolism on geological samples based on sulfur isotope fractionations. Because such fractionations depend on which steps limit the sulfate reduction process, the proposal of a new mechanism involving new proteins may demand a re-evaluation of such models (60). This will in turn require a more detailed understanding of the steps involved in sulfate and sulfite reduction.
Addendum—While we were in the final stages of preparing this manuscript, the structure of the α2β2 dSiR from A. fulgidus was also published (31), which includes only the DsrAB proteins.
Supplementary Material
Acknowledgments
We thank the ESRF for financial and technical support for data collections. C. V. thanks K. Cowtan for making an early version of BUCCANEER available.
The atomic coordinates and structure factors (code 2V4J) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
This work was supported by the Research Grants PTDC/QUI/68368/06 and PPCDT/BIA-PRO/55621/2004 funded by Fundação para a Ciência e Tecnologia (Ministério da Ciência, Tecnologia e Ensino Superior, Portugal) and Fundo Europeu de Desenvolvimento Regional program and by Fundação para a Ciência e Tecnologia-Programa Operacional Ciência, Tecnologia, Inovação fellowships SFRH/BD/29519/2006 (to T. F. O.) and SFRH/BD/30648/2006 (to S. S. V.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains two supplemental figures.
Footnotes
The abbreviations used are: dSiR, dissimilatory sulfite reductase; rSiR, reverse sulfite reductase; aSiR, assimilatory sulfite reductase; aNiR, assimilatory nitrite reductase; Dvir, desulfoviridin; r.m.s., root mean square.
References
- 1.Shen, Y., Buick, R., and Canfield, D. E. (2001) Nature 410 77-81 [DOI] [PubMed] [Google Scholar]
- 2.Philippot, P., Van Zuilen, M., Lepot, K., Thomazo, C., Farquhar, J., and Van Kranendonk, M. J. (2007) Science 317 1534-1537 [DOI] [PubMed] [Google Scholar]
- 3.Canfield, D. E., Habicht, K. S., and Thamdrup, B. (2000) Science 288 658-661 [DOI] [PubMed] [Google Scholar]
- 4.Anbar, A. D., and Knoll, A. H. (2002) Science 297 1137-1142 [DOI] [PubMed] [Google Scholar]
- 5.Dahl, C., Kredich, N. M., Deutzmann, R., and Truper, H. G. (1993) J. Gen. Microbiol. 139 1817-1828 [DOI] [PubMed] [Google Scholar]
- 6.Hipp, W. M., Pott, A. S., Thum-Schmitz, N., Faath, I., Dahl, C., and Truper, H. G. (1997) Microbiology (Read.) 143 2891-2902 [DOI] [PubMed] [Google Scholar]
- 7.Molitor, M., Dahl, C., Molitor, I., Schafer, U., Speich, N., Huber, R., Deutzmann, R., and Truper, H. G. (1998) Microbiology (Read.) 144 529-541 [DOI] [PubMed] [Google Scholar]
- 8.Wagner, M., Roger, A. J., Flax, J. L., Brusseau, G. A., and Stahl, D. A. (1998) J. Bacteriol. 180 2975-2982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Crane, B. R., and Getzoff, E. D. (1996) Curr. Opin. Struct. Biol. 6 744-756 [DOI] [PubMed] [Google Scholar]
- 10.Dhillon, A., Goswami, S., Riley, M., Teske, A., and Sogin, M. (2005) Astrobiology 5 18-29 [DOI] [PubMed] [Google Scholar]
- 11.Loy, A., Duller, S., and Wagner, M. (2007) in Microbial Sulfur Metabolism (Dahl, C., and Friedrich, C., eds) pp. pp. 46-59, Springer, Heidelberg
- 12.Janick, P. A., and Siegel, L. M. (1982) Biochemistry 21 3538-3547 [DOI] [PubMed] [Google Scholar]
- 13.Moura, I., Legall, J., Lino, A. R., Peck, H. D., Fauque, G., Xavier, A. V., Dervartanian, D. V., Moura, J. J. G., and Huynh, B. H. (1988) J. Am. Chem. Soc. 110 1075-1082 [Google Scholar]
- 14.Crane, B. R., Siegel, L. M., and Getzoff, E. D. (1995) Science 270 59-67 [DOI] [PubMed] [Google Scholar]
- 15.Peck, H. D., Legall, J., and Vanbeeumen, J. (1982) Philos. Trans. R. Soc. Lond. B Biol. Sci. 298 443-466 [DOI] [PubMed] [Google Scholar]
- 16.Akagi, J. M. (1995) in Sulfate-Reducing Bacteria (Barton, L. L., ed) pp. 89-112, Plenum Press, New York
- 17.Fauque, G., Lino, A. R., Czechowski, M., Kang, L., DerVartanian, D. V., Moura, J. J., LeGall, J., and Moura, I. (1990) Biochim. Biophys. Acta 1040 112-118 [DOI] [PubMed] [Google Scholar]
- 18.Pierik, A. J., and Hagen, W. R. (1991) Eur. J. Biochem. 195 505-516 [DOI] [PubMed] [Google Scholar]
- 19.Wolfe, B. M., Lui, S. M., and Cowan, J. A. (1994) Eur. J. Biochem. 223 79-89 [DOI] [PubMed] [Google Scholar]
- 20.Marritt, S. J., and Hagen, W. F. (1996) Eur. J. Biochem. 238 724-727 [DOI] [PubMed] [Google Scholar]
- 21.Arendsen, A. F., Verhagen, M. F., Wolbert, R. B., Pierik, A. J., Stams, A. J., Jetten, M. S., and Hagen, W. R. (1993) Biochemistry 32 10323-10330 [DOI] [PubMed] [Google Scholar]
- 22.Murphy, M. J., and Siegel, L. M. (1973) J. Biol. Chem. 248 6911-6919 [PubMed] [Google Scholar]
- 23.Pierik, A. J., Duyvis, M. G., van Helvoort, J. M., Wolbert, R. B., and Hagen, W. R. (1992) Eur. J. Biochem. 205 111-115 [DOI] [PubMed] [Google Scholar]
- 24.Karkhoff-Schweizer, R. R., Bruschi, M., and Voordouw, G. (1993) Eur. J. Biochem. 211 501-507 [DOI] [PubMed] [Google Scholar]
- 25.Dahl, C., Engels, S., Pott-Sperling, A. S., Schulte, A., Sander, J., Lubbe, Y., Deuster, O., and Brune, D. C. (2005) J. Bacteriol. 187 1392-1404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pires, R. H., Venceslau, S. S., Morais, F., Teixeira, M., Xavier, A. V., and Pereira, I. A. C. (2006) Biochemistry 45 249-262 [DOI] [PubMed] [Google Scholar]
- 27.Sander, J., Engels-Schwarzlose, S., and Dahl, C. (2006) Arch. Microbiol. 186 357-366 [DOI] [PubMed] [Google Scholar]
- 28.Haveman, S. A., Brunelle, V., Voordouw, J. K., Voordouw, G., Heidelberg, J. F., and Rabus, R. (2003) J. Bacteriol. 185 4345-4353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ikeuchi, Y., Shigi, N., Kato, J., Nishimura, A., and Suzuki, T. (2006) Mol. Cell 21 97-108 [DOI] [PubMed] [Google Scholar]
- 30.Cort, J. R., Mariappan, S. V. S., Kim, C. Y., Park, M. S., Peat, T. S., Waldo, G. S., Terwilliger, T. C., and Kennedy, M. A. (2001) Eur. J. Biochem. 268 5842-5850 [DOI] [PubMed] [Google Scholar]
- 31.Schiffer, A., Parey, K., Warkentin, E., Diederichs, K., Huber, H., Stetter, K. O., Kroneck, P. M. H., and Ermler, U. (2008) J. Mol. Biol. 379 1063-1074 [DOI] [PubMed] [Google Scholar]
- 32.Oliveira, T. F., Clemens Vonrhein Matias, P., Venceslau, S. S., Pereira, I. A. C., and Archer, M. (2008) J. Struct. Biol. 164 236-239 [DOI] [PubMed] [Google Scholar]
- 33.Kabsch, W. (1993) J. Appl. Crystallogr. 26 795-800 [Google Scholar]
- 34.Evans, P. R. (1997) Joint CCP4 and ESF-EACBM Newsletter on Protein Crystallography, 33 22-24, Daresbury Laboratory, Warrington, UK [Google Scholar]
- 35.French, G. S., and Wilson, K. S. (1978) Acta Crystallogr. Sect. A 34 517-525 [Google Scholar]
- 36.Collaborative Computational Project, N. (1994) Acta Crystallogr. Sect. D Biol. Crystallogr. 50 760-76315299374 [Google Scholar]
- 37.Vagin, A., and Teplyakov, A. (1997) J. Appl. Crystallogr. 30 1022-1025 [Google Scholar]
- 38.Blanc, E., Roversi, P., Vonrhein, C., Flensburg, C., Lea, S. M., and Bricogne, G. (2004) Acta Crystallogr. Sect. D Biol. Crystallogr. 60 2210-2221 [DOI] [PubMed] [Google Scholar]
- 39.Emsley, P., and Cowtan, K. (2004) Acta Crystallogr. Sect. D Biol. Crystallogr. 60 2126-2132 [DOI] [PubMed] [Google Scholar]
- 40.Delano, W. L. (2002) The PyMOL Molecular Graphics System, Delano Scientific, Palo Alto, CA
- 41.Ostrowski, J., Wu, J. Y., Rueger, D. C., Miller, B. E., Siegel, L. M., and Kredich, N. M. (1989) J. Biol. Chem. 264 15726-15737 [PubMed] [Google Scholar]
- 42.Sazanov, L. A., and Hinchliffe, P. (2006) Science 311 1430-1436 [DOI] [PubMed] [Google Scholar]
- 43.Chartron, J., Carroll, K. S., Shiau, C., Gao, H., Leary, J. A., Bertozzi, C. R., and Stout, C. D. (2006) J. Mol. Biol. 364 152-169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hamann, N., Mander, G. J., Shokes, J. E., Scott, R. A., Bennati, M., and Hedderich, R. (2007) Biochemistry 46 12875-12885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schnell, R., Sandalova, T., Hellman, U., Lindqvist, Y., and Schneider, G. (2005) J. Biol. Chem. 280 27319-27328 [DOI] [PubMed] [Google Scholar]
- 46.Swamy, U., Wang, M., Tripathy, J. N., Kim, S. K., Hirasawa, M., Knaff, D. B., and Allen, J. P. (2005) Biochemistry 44 16054-16063 [DOI] [PubMed] [Google Scholar]
- 47.Mander, G. J., Weiss, M. S., Hedderich, R., Kahnt, J., Ermler, U., and Warkentin, E. (2005) FEBS Lett. 579 4600-4604 [DOI] [PubMed] [Google Scholar]
- 48.Postgate, J. (1959) Nature 183 481-482 [DOI] [PubMed] [Google Scholar]
- 49.Matthews, J. C., Timkovich, R., Liu, M. Y., and Le Gall, J. (1995) Biochemistry 34 5248-5251 [DOI] [PubMed] [Google Scholar]
- 50.Kobayashi, K., Hasegawa, H., Takagi, M., and Ishimoto, M. (1982) FEBS Lett. 142 235-237 [Google Scholar]
- 51.Kessler, D. (2006) FEMS Microbiol. Rev. 30 825-840 [DOI] [PubMed] [Google Scholar]
- 52.Sauve, V., Bruno, S., Berks, B. C., and Hemmings, A. M. (2007) J. Biol. Chem. 282 23194-23204 [DOI] [PubMed] [Google Scholar]
- 53.Chang, C. K., Hanson, L. K., Richardson, P. F., Young, R., and Fajer, J. (1981) Proc. Nat. Acad. Sci. 78 2652-2656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Young, L. J., and Siegel, L. M. (1988) Biochemistry 27 5984-5990 [DOI] [PubMed] [Google Scholar]
- 55.Whittaker, M. M., and Whittaker, J. W. (2003) J. Biol. Chem. 278 22090-22101 [DOI] [PubMed] [Google Scholar]
- 56.Colas, C., and de Montellano, P. R. O. (2003) Chem. Rev. 103 2305-2332 [DOI] [PubMed] [Google Scholar]
- 57.Pearson, A. R., Elmore, B. O., Yang, C., Ferrara, J. D., Hooper, A. B., and Wilmot, C. M. (2007) Biochemistry 46 8340-8349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Drake, H. L., and Akagi, J. M. (1978) J. Bacteriol. 136 916-923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Steuber, J., Cypionka, H., and Kroneck, P. M. H. (1994) Arch. Microbiol. 162 255-260 [Google Scholar]
- 60.Hoek, J., and Canfield, D. E. (2007) in Microbial Sulfur Metabolism (Dahl, C., and Friedrich, C., eds) pp. 273-284, Spinger-Verlag, Heidelberg
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.