

Abstract
An enantioselective synthesis of (+)-coronafacic acid has been achieved. Rhodium catalyzed cyclization of an α-diazoester provided the intermediate cyclopentanone in high enantiomeric purity. Subsequent Fe-mediated cyclocarbonylation of a derived alkenyl cyclopropane gave a bicyclic enone, that then was hydrogenated and carried on to the natural product.
Introduction
Bicyclic and polycyclic ring systems are common in structurally complex and physiologically-active natural products. Although there are many methods of preparing such ring systems, the number of approaches to carbopolycyclic scaffolds is more limited. Corey demonstrated the utility of an enzyme to convert 20,21-dehydro-2,3-oxidosqualene to a dehydroprotosterol. Hajos2 employed a catalytic amount of (S)-(−)-proline in an asymmetric aldol condensation to form optically pure bicyclic intermediates. We3 were able to demonstrate that a single stereogenic center on the bridge between the diene and dienophile could set the absolute center of an intramolecular Diels-Alder reaction leading to a carbobicyclic 6,6-system. Corey4 reported examples of catalytic enantioselective [2+2] cycloaddition catalyzed by chiral aluminum bromide complexes to form 5,4-, 6,4-, and 7,4-carbobicyclic systems. We5 also reported another approach to enantioselective polycyclic construction, utilizing Shi epoxidation followed by selective ring opening, and then intramolecular alkylation to set the stereogenic centers. We6 further showed in our synthesis of (+)-sulcatine G, that enantiospecific C-H insertion, followed by intramolecular alkylation, could also be applied to carbobicycle construction. Schaus7 demonstrated the utility of the Morita-Baylis Hillman reaction, coupled with the Hosomi-Sakauri, to construct 6,6-bicyclic systems. Gaunt8 reported a strategy using oxidative dearomatization and amine-catalyzed enantioselective desymmetrizing Michael reaction to form 6,6-carbobicycles. In the Phillips9 synthesis of (+)-cyanthiwigin U, tandem metathesis was used to enantioselectively construct the polycarbocycle. Recently, Ishihara10 utilized enantioselective Robinson annulation and cycloaddition reactions catalyzed by chiral Lewis acids for polycarbocycle construction.
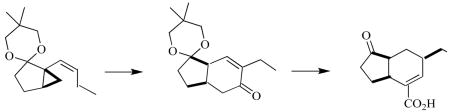
We had developed (Scheme 1) a general route to enantiomerically pure 5,3- and 6,3-carbobicyclic scaffolds by cyclization of menthyl esters such as 1 to the cyclopentanone 2.11a Nakada11l,r,s later reported an enantioselective preparation of similar carbocyclic esters and sulfones by copper catalyzed asymmetric intramolecular cyclopropanation. We have improved upon our intramolecular cyclization11a by increasing the selectivity and preparative yield of the cyclization. We anticipated that we could convert the ester to the alkenyl cyclopropane 3. We speculated12 that Fe-mediated cyclocarbonylation of the alkenyl cyclopropane could deliver the bicyclic enone 4. If this were successful, the enone 4 could then be transformed in a few steps to the natural product (+)-coronafacic acid 5.
SCHEME 1.

Background and Results
Coronatin 6 (Eq. 1) is a phytotoxin produced by Pseudomonas syringae. Its structure is composed of coronafacic acid 5, a bicyclic core with three stereogenic centers, and coronamic acid 7, a cyclopropane amino acid derived from isoleucine. Both coronatin and coronafacic acid have been reported to mimic jasmonic acid. All three compounds induce tubers, induce cell expansion, inhibit cell division, and promote senescence in plants.13 While there were 15 previous total syntheses of coronafacic acid,14 only one enantioselective route had been reported.14m
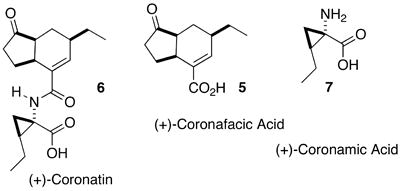 |
(1) |
Intramolecular Cyclopropanation
Dirhodium tetracarboxylate catalysts are known to cyclize α-diazo esters, such as 1. Earlier, we had cyclized 1 to a 1:1 mixture of 2 and 8 (Eq. 2), using a copper bronze catalyst.11a We separated the menthyl esters 2 and 8 by column chromatography, leading to pure 2 in 21% yield. We have investigated a diverse selection of dirhodium tetracarboxylate catalysts to improve upon those earlier results.
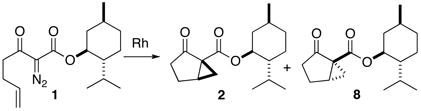 |
(2) |
We have found (Table 1) that, by using Rh (II) catalysts, we could induce some diastereoselectivity in this cyclization. We found that dirhodium pivalate at room temperature provided the best preparative yield, 63%, of the desired cyclopentanone 2.
Table 1.
Cyclization of Diazo Menthyl Ester 1
| Entry | Rhodium Ligand | Solvent | Temperature (°C) | Diastereoselectivity (2:8) | Yield (%)a |
|---|---|---|---|---|---|
| 1 | pivb | CH2Cl2 | 25 | 2.5:1 | 95 |
| 2 | pivb | PhCH3 | 25 | 1.5:1 | 91 |
| 3 | pivb | C6H12 | 25 | 1.2:1 | 80 |
| 4 | pivb | CCl4 | 25 | 1.2:1 | 70 |
| 5 | pivb | CH2Cl2 | −78 | 3.0:1 | 90 |
| 6 | tfac | CH2Cl2 | 25 | 0.9:1 | 70 |
| 7 | octd | CH2Cl2 | 25 | 1.2:1 | 89 |
| 8 | Ph3CCO2e | CH2Cl2 | 25 | 0.7:1 | 75 |
| 9 | OAcf | CH2Cl2 | 25 | 1.0:1 | 80 |
| 10 | Espg | CH2Cl2 | 25 | 1.3:1 | 88 |
| 11 | R-ptpah | CH2Cl2 | 25 | 1.7:1 | 71 |
| 12 | R-pttli | CH2Cl2 | 25 | 1.3:1 | 82 |
| 13 | S-dospj | CH2Cl2 | 25 | 1.7:1 | 91 |
| 14 | S-tbspk | CH2Cl2 | 25 | 1.5:1 | 89 |
Preparation of alkenyl cyclopropane 6
With the initial two stereocenters set, we protected (Scheme 2) the ketone as the ketal 9. Attempts to reduce the ester directly to the aldehyde with less than an equivalent of lithium aluminum hydride or with DIBAL delivered a mixture of alcohol, aldehyde, and the starting ester. We found that completely converting the ester 2 to the alcohol followed by oxidization to aldehyde 10 with Dess-Martin reagent was more effective. We were then able to perform a Wittig reaction on aldehyde 10 to give alkenyl cyclopropane 3.
SCHEME 2.

Preparation of enone 9
We12b,c,d had reported the preparation of 2,5 and 5-substituted cyclohexenones via Fe-mediated cyclocarbonylation. More recently, we found that we could couple the Wittig reaction with Fe-mediated cyclocarbonylation to convert aldehydes to 2-substituted cyclohexenones.12e Sarel12a demonstrated the photochemical expansion of a methylene spiroalkane 11 to the bicyclic enones 12 and 13 in his studies of (+)-α-thujene (Eq. 3). The cyclocarbonylation to a bicyclic enone from a 5,3- or 6,3-ring fused alkenyl cyclopropane such as 3 had not yet been reported.
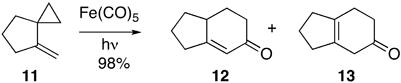 |
(3) |
We found (Eq. 4) that UV irradiation of 3 in the presence of Fe(CO)5 converted the alkenyl cyclopropane 3 to the bicyclic cyclohexenone 4 in low yield with accompanying deketalization. We found that by adding K2CO3 to the irradiation, the reaction was buffered and ketal deprotection was avoided. The use of tetrahydrofuran, rather than isopropanol,12b–e as the reaction solvent doubled the conversion of 3 to 4. We found it practical to run the cyclocarbonylation to about 40% conversion (TLC), as longer irradiation led to diminished yield. The unreacted starting material was easily separated and recycled.
The kinetic product from the cyclocarbonylation was the β,γ-unsaturated ketone. This was not purified, but was converted to the desired α,β-unsaturated ketone 4 by stirring for an hour at room temperature with DBU.
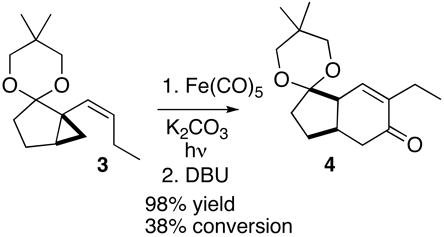 |
(4) |
Synthesis of (+)-Coronafacic Acid
Pd-mediated hydrogenation of the cyclohexenone 4 (Scheme 4) at ambient temperature and pressure provided the bicyclic ketone 11 as a single diastereomer. Carbomethoxylation followed by reduction with sodium borohydride gave the alcohol 12 as a mixture of diastereomers. Dehydration of the mixture by mesylation and subsequent addition of DBU gave the ketal-protected coronafacic methyl ester 13 as a single diastereomer. Deprotection and ester hydrolysis were then completed in one step with aqueous HCl at reflux to give the natural product 5. The physical data, including optical rotation (obs., [α]20D +104, lit.,14o [α]20D +105) were consistent with those previously reported for (+)-coronafacic acid.
Conclusion
We have completed an enantioselective synthesis of (+)-coronofacic acid. We were able to set the initial two stereogenic centers utilizing rhodium catalyzed intramolecular cyclopropanation. We were then able to expand the cyclopropane utilizing Fe-mediated cyclocarbonylation. We expect that this will be a general route to enantiomerically pure 6,5- and 6,6-carbobicyclic systems.
Experimental Section
(1S,2R,5S)-5-Methyl-2-(propan-2-yl)cyclohexyl 2-oxobicyclo[3.1.0]hexane-1-carboxylate 2 and 8
Rh2(Piv)415a (5 mg) was added to α-diazoester 111a (6.12 g, 20 mmol) in 20 mL of dry CH2Cl2 (1.0 M) at rt. The solution was stirred at rt for 1 h, and then additional Rh2(Piv)4 (5 mg) was added and the reaction was allowed to stir for an additional 2 h. The mixture was concentrated and the residue chromatographed using 94:6 hexanes/EtOAc as elution solvent to give the cyclopentanone 2 (3.61 g, 63% yield) and 8 (1.78 g, 32% yield) as an oil. Cyclopentanone 2 was then crystallized at −78°C in petroleum ether (5 mL) to give 2 (3.49 g, 62% as a white crystalline solid, mp = 34–36°C, [α]20D +47.2 (c=1.0, CH2Cl2); TLC Rf = 0.27 (9:1 hexanes/EtOAc); 1H NMR δ4.71 (dt, J = 4.9, 11.5 Hz, 1H), 2.54 (m, 1H), 2.22 (m, 3H), 2.01 (m, 4H), 1.68 (d, J = 12.1 Hz, 2H), 1.34–1.48 (m, 3H), 0.88–1.05 (m, 9H), 0.75 (d, J= 7.6 Hz, 3H); 13C NMR δ u:16 207.0, 167.6, 40.6, 37.8, 34.2, 33.5, 21.4, 21.0; d: 74.9, 46.8, 32.4, 31.3, 25.8, 23.1, 21.9, 20.8, 16.0; IR 2952, 1736, 1454, 1383, 1311 cm−1; MS m/z (%) 279 (M+H, 100), 263 (48), 141 (98), 123 (79); HRMS: Calcd for C17H27O3 (M+H) 279.1960, obsd 279.1967. For cyclopentanone 8: (1.45 g, 27% as an oil, [α]20D −47.0 (c=1.0, CH2Cl2); TLC Rf = 0.25 (9:1hexanes/EtOAc); 1H NMR δ 4.65 (dt, J = 4.9, 11.5 Hz, 1H), 2.49 (m, 1H), 2.16 (m, 3H), 1.93 (m, 3H), 1.75 (m, 1H), 1.60 (d, J = 12.1 Hz, 2H), 1.34–1.48 (m, 3H), 0.88–1.05 (m, 9H), 0.67 (d, J = 7.6 Hz, 3H); 13C NMR δu: 207.0, 167.6, 40.7, 37.7, 34.2, 33.6, 21.6, 20.7; d: 74.9, 46.8, 32.2, 31.3, 26.2, 23.4, 22.0, 20.9, 16.3; IR 2950, 1735, 1454, 1384, 1311 cm−1; MS m/z (%) 279 (M+H, 100), 263 (49), 141 (98), 123 (79); HRMS: Calcd for C17H27O3 (M+H) 279.1960, obsd 279.1967.
Ketal 9
2, 2-Dimethyl-1,3-propanediol (2.11g, 24 mmol), p-toluenesulfonic acid monohydrate (5 mg, 0.02 mmol), and cyclopentanone 2 (3.33 g, 12 mmol) were combined in 24 mL of dry toluene. The solution was maintained at reflux for 8 h. The reaction was concentrated, and the residue was chromatographed to give the ketal 6 (3.89 g, 89% yield) as a white solid, mp = 97–99°C; [α]20D +32.1(c=1.0, CH2Cl2); TLC Rf = 0.49 (8:2 petroleum ether/MTBE); 1H NMR δ 4.69 (dt, J = 4.9, 11.5 Hz, 1H), 3.62 (d, J =11.2 Hz, 1H), 3.47 (m, 3H), 2.43 (dd, J =8.2, 13.6 Hz, 1H) 1.87–2.10(m, 4H), 1.66–1.74 (m, 3H), 1.39–1.48 (m, 5H), 1.14 (m, 2H), 1.06 (m, 3H), 0.868–1.05 (m, 7H), 0.74 (m, 6H); 13C NMR δ u: 170.6, 107.1, 73.2, 71.7, 40.7, 37.0, 34.5, 30.2, 25.2, 23.6, 23.3, 23.0, 16.8; d: 74.3, 47.0, 31.4, 26.0, 24.6, 22.1, 22.0, 20.8, 16.4; IR 2950, 1705, 1455, 1384, 1309 cm−1; MS m/z (%) 365 (M+H, 87), 279 (7), 227 (100), 209 (63), 182 (36), 141 (65), 128 (60); HRMS: Calcd for C22H36O4 364.2614, obsd 364.2623.
(1S,5R)-5′,5′-Dimethyl-1H-spiro[bicyclo[3.1.0]hexane-2,2′-[1,3]dioxane]-1-carbaldehyde 10
LiAlH4 (1.0 M Solution in THF, 12 mL, 12 mmol) was added over 10 min to ketal 9 (3.64 g, 10 mmol) in 10 mL (1.0 M) of dry THF at rt The solution was stirred at rt for 2 h. Ethyl acetate (5 mL) was added dropwise over 30 minutes followed by sodium sulfate decahydrate (3.2 g, 10 mmol). The mixture was stirred at rt for 2 h, then filtered through a pad of silica gel with ethyl acetate (20 mL) and concentrated. The residue was chromatographed to give the crude alcohol. Dess-Martin periodinane (4.24 g, 11 mmol) was added to the alcohol in 10 mL of dry dichloromethane at rt. The mixture was concentrated and the residue was chromatographed to give the aldehyde 10 (1.62 g, 77% yield from 9) as an oil, TLC Rf = 0.34 (8:2 petroleum ether/MTBE); [α]20D −59.1 (c=1.0, CH2Cl2); 1H NMR δ 10.36 (s, 1H), 3.72 (d, J = 11.2 Hz, 1H), 3.54 (d, J =11.2 Hz, 1H), 3.48 (s, 2H), 2.47 (dd, J =8.6, 14.3 Hz, 1H), 2.07 (m, 1H), 1.90 (m, 1H), 1.79 (m, 1H), 1.49 (dd, J =5.2, 13.0 Hz, 1H), 1 30 (m, 3H), 1.17 (m, 2H), 0.77 (s, 3H); 13C NMR δ u: 107.7, 73.3, 71.6, 44.8, 30.0, 25.6, 24.2, 18.6; d: 201.1, 30.5, 22.9, 22.0; IR 2953, 1700, 1467, 1335, 1116 cm−1; MS m/z (%) 211 (M+H, 58), 181 (55), 141 (67), 123 (100); HRMS: Calcd for C12H18O3 210.1256, obsd 210.1262.
(1S,5R)-1-[(1Z)-But-1-en-1-yl]-5′,5′-dimethylspiro[bicyclo[3.1.0]hexane-2,2′-[1,3]dioxane] 3
Potassium t-butoxide (1.0 M solution in THF, 20 mL, 20 mmol) was added over 20 min to a suspension of propyltriphenylphosphonium bromide (3.50 g, 9 mmol) in 20 mL of dry THF at 0°C. The external cooling was removed and the mixture was stirred for 30 min. The aldehyde 10 (1.57 g, 7.5 mmol) was added, and the mixture was stirred at rt for 1 h. The mixture was quenched with water (50 mL) and then partitioned between water and EtOAc. The combined organic extract was dried (MgSO4) and concentrated. The residue was chromatographed to give the alkenyl cyclopropane 3 (1.84 g, 78% yield) as an oil, [α]20D +20.8 (c=1.0, CH2Cl2); TLC Rf = 0.74 (8:2 petroleum ether/MTBE); 1H NMR δ 5.70 (d, J = 8.0 Hz, 1H), 5.48 (m, 1H), 3.56 (d, J =11.6 Hz, 1H), 3.43 (d, J =11.6 Hz, 1H), 3.40 (s, 2H), 2.18–2.29 (m, 3H), 1.93 (m, 1H), 1.72 (dd, J =8.0, 12.0 Hz, 1H), 1.41 (m, 1H), 1.17 (m, 4H), 0.95 (t, J =7.2 Hz, 3H), 0.85 (m, 1H), 0.71 (s, 3H), 0.65 (dd, J =4.2, 7.2 Hz, 1H); 13C NMR δ u: 110.0, 73.1, 71.4, 33.2, 30.1, 24.7, 24.6, 21.8, 14.7; d: 136.6, 125.5, 22.7, 22.5, 22.1, 14.3; IR 2950, 2859, 1211, 1153, 1117 cm−1; MS m/z (%) 237 (M+H, 18), 201 (100), 183 (54), 141 (71); HRMS: Calcd for C15H24O2 236.1776, obsd. 236.1767.
(3a′R,7a′R)-6′-Ethyl-5,5-dimethyl-2′,3′,3a′,7a′-tetrahydrospiro[1,3-dioxane-2,1′-inden]-5′(4′H)-one 4
To the alkenyl cyclopropane 3 (236 mg, 1.0 mmol) and potassium carbonate (280 mg, 2.0 mmol) in 10 mL of 2-propanol and 5 mL of tetrahydrofuran (0.075 M) was added Fe(CO)5 (392 mg, 2.0 mmol). The Pyrex reaction vessel was purged with CO, a CO balloon was attached, and the mixture was irradiated for 4 h at room temperature as a thin film in a Rayonet apparatus (300 nm) set for autocooling. The reaction was halted every hour to agitate the tube inside the larger tube, after which photolysis was restarted. At the end of the irradiation, DBU (304 mg, 2.0 mmol) was added, and the mixture was stirred at room temperature for 1 h under nitrogen. The mixture was diluted with 40 mL of EtOAc and filtered through a small pad of packed silica gel. The eluate was concentrated and the residue was chromatographed to give 152 mg of unreacted 3 and 90 mg of 4 (98% yield based on 3 not recovered) as an oil, [α ]20D +102.5 (c=1.0, CH2Cl2); TLC Rf= 0.30 (8:2 petroleum ether/MTBE); 1H NMR δ 6.45 (s, 1H), 3.45 (m, 4H), 2.95 (br s, 1H), 2.64 (m, 1H), 2.41 (m, 2H), 2.17 (m, 2H), 1.70– 1.97 (m, 3H), 1.35 (m, 1H), 0.95 (m, 6H), 0.85 (m, 3H); 13C NMR δ u: 198.4, 140.7, 108.2, 71.4, 70.7, 39.9, 30.5, 29.3, 25.0, 21.7; d: 138.9, 45.0, 33.1, 21.5, 21.3, 12.1; IR 2952, 1672, 1493, 1312, 1153 cm−1; HRMS: Calcd for C16H24O3 264.1725, obsd. 264.1731.
(3a′R,6′R,7a′S)-6′-Ethyl-5,5-dimethylhexahydrospiro[1,3-dioxane-2,1′-inden]-5′(4′H)-one 14
Pd/C (5 wt%, 10 mg) was added to the bicyclic enone 4 (85 mg, 0.32 mmol) in 5 mL of methanol. The reaction vessel was purged with hydrogen, a hydrogen balloon was attached, and the mixture was stirred at rt for 4 h until the TLC indicated disappearance of the UV absorbent spot. The mixture was filtered through a plug of Celite and concentrated. The residue was chromatographed to give 11 (81 mg, 95% yield) as an oil, [α]20D +124.6 (c=1.0, CH2Cl2), TLC Rf = 0.30 (8:2 petroleum ether/MTBE); 1H NMR δ 3.48 (m, 4H), 2.62 (m, 1H), 2.51 (m, 2H), 2.25 (dd, J =5.6, 14.4 Hz, 1H), 2.15 (m, 1H), 2.00 (m, 3H), 1.82 (m, 2H), 1.46 (m, 1H), 1.24 (m, 2H), 1.04 (m, 3H), 0.91 (m, 6H); 13C NMR δ u: 214.2, 109.6, 72.4, 71.5, 43.8, 30.7, 30.0, 28.3, 26.6, 22.3; d: 49.2, 44.3, 37.1, 22.5, 11.6; HRMS: Calcd for C16H26O3 266.1882, obsd 266.1880.
Methyl (3a′S,5′S,6′R,7a′S)-6′-ethyl-5′-hydroxy-5,5-dimethyloctahydrospiro[1,3-dioxane-2,1′-indene]-4′-carboxylate 15
Sodium hydride (60%, 24 mg, 0.6 mmol) and dimethyl carbonate (3 mL, 35 mmol) were heated to reflux for 20 min. A drop of methanol was added via an addition funnel over a condenser and stirred at reflux for 5 min. Bicyclic ketone 14 (80 mg, 0.3 mmol) in 1 mL dry toluene was added via an addition funnel over a condenser, and the mixture was stirred at reflux for 3 h. The mixture was cooled to rt and quenched with saturated aqueous NH4Cl and then partitioned between water and EtOAc. The combined organic extract was dried (MgSO4) and concentrated. The residue was dissolved in methanol (5 mL) and NaBH4 (19 mg, 0.5 mmol) was added. The mixture was stirred at rt for 2 h. The mixture was concentrated and the residue was chromatographed to give alcohol 15 (44 mg, 45% yield) as an oil, [α]20D+10.4 (c=1.0, CH2Cl2); TLC Rf = 0.19 (8:2 petroleum ether/MTBE); 1H NMR δ 4.17 (s, 1H), 3.72 (s, 3H), 3.50 (s, 2H), 3.42 (s, 2H), 2.64 (m 1H), 2.56 (m, 1H), 2.21 (m, 2H), 2.01 (m, 1H), 1.92 (m, 1H), 1.56 (m, 3H), 1.38 (m, 4H), 0.95 (m, 9H); 13C NMR δ u: 176.1, 109.5, 72.7, 71.0, 31.6, 30.1, 30.0, 25.3, 22.3; d: 67.6, 51.7, 46.8, 44.5, 42.0, 36.5, 22.5, 11.7; IR 3488, 2913, 1713, 1289, 1152 cm−1; MS m/z (%) 327 (M+H, 6), 141 (100); HRMS: Calcd for C18H30O5 326.2093, obsd 326.2095.
Methyl (3a′S,6′R,7a′S)-6′-ethyl-5,5-dimethyl-2′,3′,3a′,6′,7′,7a′-hexahydrospiro[1,3-dioxane-2,1′-indene]-4′-carboxylate 16
Methanesulfonyl chloride (60 mg, 0.36 mmol) was added slowly to alcohol 12 (40 mg, 0.12 mmol) and triethylamine (1 mL, 7.2 mol) in 1 mL of dichloromethane at 0°C. The solution was stirred to rt over 2 h. DBU (2 mL, 2.04g, 14 mmol) was added and the solution was stirred at rt for 18 h. The solution was filtered through a pad of silica gel with EtOAc and concentrated. The residue was chromatographed to give ketal ester 15 (21 mg, 56% yield) as an oil, [α]20D +25.5 (c=1.0, CH2Cl2); TLC Rf = 0.44 (8:2 petroleum ether/MTBE); 1H NMR δ 6.84 (s, 1H), 3.72 (s, 3H), 3.52 (s, 2H), 3.45 (d, J =4.4 Hz, 2H), 2.95 (m 1H), 2.34 (m, 1H), 2.12 (m, 3H), 1.84 (m, 2H), 1.47 (m, 4H), 0.98 (m, 9H); 13C NMR δ u: 185.1, 133.3, 109.5, 72.9, 71.2, 31.2, 30.1, 28.2, 27.7, 25.9; d: 143.2, 51.8, 42.8, 38.1, 36.0, 22.5, 11.3; IR 2958, 1714, 1440, 1249 cm−1; MS m/z (%) 309 (M+H, 8), 279 (7), 237 (12), 222 (94), 209 (37), 165 (55), 141 (100), 119 (99); HRMS: Calcd for C18H29O4 (M+H) 309.2066, obsd. 309.2066.
(+)-Coronafacic Acid 5
Hydrochloric acid (20% aqueous, 2 mL) was added to ketal ester 16 (11 mg, 0.07 mmol). The mixture was heated to reflux for 4 hours. The solution was allowed to cool to rt then partitioned between water and EtOAc. The combined organic extract was dried (MgSO4) and concentrated. The residue was chromatographed to give 5 (7 mg, 55% yield) as an oil, TLC Rf = 0.25 (7:3 petroleum ether/EtOAc).; [α]20D +104 (c=0.1, MeOH), lit.,14o [α]20D +105. The spectroscopic data of the natural product 5 were compared with those of earlier syntheses14o and found to be identical.
Supplementary Material
General experimental procedures, details of the photochemical apparatus, preparation of 1 and spectroscopic data. This material is available free of charge via the Internet at http://pubs.acs.org.
SCHEME 3.

Acknowledgments
We thank John Dykins for high-resolution mass spectrometry under the financial support of NSF 054177. We thank the NIH (GM060287) for financial support of this work.
References
- 1.Corey EJ, Lin K, Yamamoto H. J Am Chem Soc. 1969;91:2132. doi: 10.1021/ja01036a059. [DOI] [PubMed] [Google Scholar]
- 2.Hajos ZG, Parrish DR. J Org Chem. 1974;39:1615. [Google Scholar]
- 3.Taber DF, Gunn BP. J Am Chem Soc. 1979;101:3992. [Google Scholar]
- 4.Corey EJ, Canales E. J Am Chem Soc. 2007;129:12686. doi: 10.1021/ja0765262. [DOI] [PubMed] [Google Scholar]
- 5.Taber DF, He T, Xu M. J Am Chem Soc. 2004;126:13900. doi: 10.1021/ja045849k. [DOI] [PubMed] [Google Scholar]
- 6.Taber DF, Frankowski KJ. J Org Chem. 2005;70:6417. doi: 10.1021/jo0508752. [DOI] [PubMed] [Google Scholar]
- 7.Pfeiffer MWB, Phillips AJ. J Am Chem Soc. 2005;127:5334. doi: 10.1021/ja0509836. [DOI] [PubMed] [Google Scholar]
- 8.Rodgen SA, Schaus SE. Angew Chem, Int Ed. 2006;45:4929. doi: 10.1002/anie.200601076. [DOI] [PubMed] [Google Scholar]
- 9.Vo NT, Pace RDM, O’Hara F, Gaunt MI. J Am Chem Soc. 2008;130:404. doi: 10.1021/ja077457u. [DOI] [PubMed] [Google Scholar]
- 10.Ishihara K, Fushimi M. J Am Chem Soc. 2008;130:7532. doi: 10.1021/ja8015318. [DOI] [PubMed] [Google Scholar]
- 11.For previous preparations of cyclohexanones and cyclopentanones via intramolecular cyclopropanation, see Taber DF, Saleh SA, Korsmeyer RW. J Org Chem. 1980;45:4699.Taber DF, Malcolm SC. J Org Chem. 1998;63:3717. doi: 10.1021/jo001519g.Dauben WG, Hendricks RT, Luzzio MJ, Ng HP. Tetrahedron Lett. 1990;31:6969.Pique C, Fahndrich B, Pfaltz A. Synlett. 1995;(Special):491.Park SW, Son JH, Kim SG, Ahn KH. Tetrahedron: Asymmetry. 1999;10:1903.Kim SG, Cho CW, Ahn KH. Tetrahedron. 1999;55:10079.Barberis M, Estevan F, Lahuerta P, Perez-Prieto J, Sanau M. Inorg Chem. 2001;40:4226. doi: 10.1021/ic010257d.Barberis M, Prieto J, Stiriba SE, Lahuerta P. Org Lett. 2001;3:3317. doi: 10.1021/ol010170w.Saha B, Uchida T, Katsuki T. Chem Lett. 2002;8:846.Barberis M, Perez-Prieto J, Herbst K, Lahuerta P. Organometallics. 2002;21:1667.Saha B, Uchida T, Katsuki T. Tetrahedron: Asymmetry. 2003;14:823.Honma M, Sawada T, Fujisawa Y, Utsugi M, Watanabe H, Umino A, Matsumura T, Hagihara T, Takano M, Nakada M. J Am Chem Soc. 2003;125:2860. doi: 10.1021/ja029534l.Honma M, Nakada M. Tetrahedron Lett. 2003;44:9007.Wong A, Welch CJ, Kuethe JT, Vazquez E, Shaimi M, Henderson D, Davies IW, Hughes DL. Org Biomol Chem. 2004;2:168. doi: 10.1039/b312180c.Estevan F, Lahuerta P, Lloret J, Perez-Prieto J, Werner H. Organometallics. 2004;23:1369.Estevan F, Lahuerta P, Lloret J, Penno D, Sanau M, Ubeda MA. J Organomet Chem. 2005;690:4424.Uchida T, Katsuki T. Synthesis. 2006:1715.Ida R, Nakada M. Tetrahedron Lett. 2007;48:4856.Takeda H, Honma M, Ida R, Sawada T, Nakada M. Synlett. 2007;4:579.
- 12.For the development of Fe-mediated cyclocarbonylation, see: Victor R, Ben-Shoshan R, Sarel S. J Org Chem. 1978;43:4971.Taber DF, Kanai K, Jiang Q, Bui G. J Am Chem Soc. 2000;122:6807.Taber DF, Bui G, Chen B. J Org Chem. 2001;66:3423. doi: 10.1021/jo001737+.Taber DF, Joshi PV, Kanai K. J Org Chem. 2004;69:2268. doi: 10.1021/jo0302760.Taber DF, Sheth RB. J Org Chem. 2008;73:8030. doi: 10.1021/jo801767n.
- 13.For a recent review of chemistry and biological properties of coronatin and coronafacic acid, see Mitchell RE. Chem N Z. 2004;68:24.
- 14.For previous coronafacic acid syntheses, see Ichihara A, Kimura R, Moryasu K, Sakamura S. Tetrahedron Lett. 1977;18:4331.Jung ME, Hudspeth JP. J Am Chem Soc. 1980;102:2463.Ichihara A, Kimura R, Moryasu K, Sakamura S. J Am Chem Soc. 1980;102:6353.Tsuji J. Pure Appl Chem. 1981;53:2371.Jung ME, Halweg KM. Tetrahedron Lett. 1981;22:2735.Nakayama M, Ohira S, Okamura Y, Soga S. Chem Lett. 1981:731.Nakayama M, Ohira S. Agric Biol Chem. 1983;47:1689.Liu HJ, Brunet ML. Can J Chem. 1984;62:1747.Bhamare NK, Granger T, Macas TS, Yates P. J Chem Soc, Chem Commun. 1990:739.Mehta G, Praveen M. J Chem Soc, Chem Commun. 1993:1573.Hoelder S, Blechert S. Synlett. 1996:505.Nara S, Toshima H, Ichihara A. Tetrahedron Lett. 1996;37:6745.Nara S, Toshima H, Ichihara A. Tetrahedron. 1997;53:9509.Sono M, Hashimoto A, Nakashima K, Tori M. Tetrahedron Lett. 2000;41:5115.Mehta G, Reddy DS. J Chem Soc, Perkin Trans I. 2001;10:1153.Moreau B, Ginisty M, Alberico D, Charette AB. J Org Chem. 2007;72:1235. doi: 10.1021/jo062099j.
- 15.For rhodium (II) complexes, see (a) piv = pivalate, see Handa M, Takata A, Nakao T, Kasuga K, Mikuriya M, Kotera T. Chem Lett. 1992;10:2085. tfa = trifluoroacetate, see Duddeck H, Ferguson G, Kaitner B, Kennedy M, McKervey MA, Maguire AR. J Chem Soc, Perkin Trans I. 1990;4:1055. oct = octanoate, see Giroud-Godquin AM, Marchon JC, Guillon D, Skoullos A. J Phys Chem. 1986;90:5502.Ph3CO2 = triphenylacetate, see Hashimoto S, Watanabe N, Ikegami S. Tetrahedron Lett. 1992;33:2709.OAc = aceto, see Rempel GA, Legzdins P, Smith H, Wilkinson G. Inorg Synth. 1971;13:90.Esp = bis [rhodium (α, α, α′, α′-tetramethyl-1,3-benzenedipropionate)], see Espino CG, Flori KW, Kim M, Du Bois JD. J Am Chem Soc. 2004;126:15378. doi: 10.1021/ja0446294.R-ptpa = dirhodium tetrakis [N-phthaloyl-(R)-phenylalaninate], see Okada Y, Minami Y, Miyamoto M, Otaguro T, Sawasaki S, Ichikawa J. Heteroatom Chem. 1995;6:195.R-pttl = dirhodium tetrakis [(2R)-3,3-dimethyl-2-(phthalimido)butanoateo], see Tsutsui H, Yamaguchi Y, Kitagaki S, Nakamora S, Anada M, Hashimoto S. Tetrahedron: Asymmetry. 2003;14:817.S-dosp = dirhodium tetrakis [(S)-(−)-N-p-dodecylphenylsulfonyl)prolinato], see Davies HML, Bruzinkski P, Hutcheson DK, Kong N, Fall MJ. J Am Chem Soc. 1996;118:6897. (j) S-tbsp = dirhodium tetrakis [1-[(4-tert-butylphenyl)sulfonyl]-(2S)-pyrrolidinecarboxylate], see ref. 14i.
- 16.13C multiplicities were determined with the aid of a JVERT pulse sequence, differentiating the signals for methyl and methine carbons as “d” and for methylene and quaternary carbons as “u”.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
General experimental procedures, details of the photochemical apparatus, preparation of 1 and spectroscopic data. This material is available free of charge via the Internet at http://pubs.acs.org.