

Abstract
Genetic and biochemical data demonstrate a pivotal role for S-nitrosothiols (SNOs) in mediating the actions of nitric oxide synthases (NOSs). SNOs serve to convey NO bioactivity and to regulate protein function. This understanding is of immediate interest to the pulmonary clinical and research communities. This article reviews the following: (1) biochemical and cellular evidence that SNOs in amino acids, peptides, and proteins elicit NOS-dependent signaling in the respiratory system and (2) studies that link SNO signaling to pulmonary medicine. SNO-mediated signaling is involved in the regulation of minute ventilation, ventilation–perfusion matching, pulmonary arterial pressure, basal airway tone, and respiratory and peripheral muscle function. Derangements in SNO signaling are implicated in many disorders relevant to pulmonary and critical care medicine, including apnea, hypoxemia, pulmonary hypertension, asthma, cystic fibrosis, pneumonia, and septic shock.
Keywords: asthma, cystic fibrosis, pulmonary hypertension, S-nitrosoglutathione, S-nitrosohemoglobin, S-nitrosylation
BACKGROUND: S-NITROSOTHIOL BIOCHEMISTRY AND BIOLOGY
S-Nitrosylation Signaling
Nitric oxide synthases (NOSs) are extensively involved in the functions of the respiratory system (Table 1). As originally conceived, NOS generates the NO radical, which diffuses into a target cell; NO either signals by activating guanylate cyclase, is inactivated by binding to heme proteins, or contributes to cytotoxicity, primarily by reacting with superoxide. However, extensive biochemical and genetic data—including both mutational analyses of cysteine (Cys) residues in over 30 proteins that are targets of NO, and creation of plants and mice deficient in S-nitrosothiol (SNO) metabolism—have led to the current understanding that most actions of NOSs are in fact conveyed by S-nitrosylation, the modification of protein Cys thiols by NO (1–5) (Figures 1 and 2). By contrast, enzymes that would selectively block the actions of NO radical have not been identified in mammals; the extent to which NOSs may act independently of SNOs is therefore unclear (Table 1). It is important to note that signaling by S-nitrosylation and guanylate activation may not be mutually exclusive (6). Our focus will be on SNO signaling of relevance to pulmonary biology, a subject that has not been covered in previous reviews.
TABLE 1.
NITRIC OXIDE SYNTHASES AND S-NITROSOTHIOLS IN RESPIRATORY BIOLOGY
| Respiratory Effect | NOS Isoforms Involved (reviewed elsewhere) | S-NO Signaling Involved | Studies of SNOs Performed in Human Disease | NO Radical May Be Involved, Independent of SNOs‡ |
|---|---|---|---|---|
| Control of breathing | Yes (25) | Yes (25) | No† | Unknown |
| Ventilation–perfusion matching | Yes (56) | Yes (28, 42, 50) | Yes (28, 42, 50) | Presumed (43, 56)§ |
| Pulmonary vascular tone and pulmonary hypertension | Yes (56)* | Yes (20, 26, 28, 33, 42) | Yes (20, 28, 33, 42) | Presumed (43, 56)§ |
| Human airway smooth muscle tone | Yes (56) | Yes (20, 44, 47, 48) | Yes (44) | Unlikely (44, 47, 48) |
| Asthma | Yes (56) | Yes (20), 44, 57, 58) | Yes (57, 58) | Unlikely, though NO is a biomarker of interest |
| Cystic fibrosis | Yes (56, 60) | Yes (38, 44, 50, 59, 61) | Yes (44, 50, 59) |
Definition of abbreviations: NOS = nitric oxide synthase; SNO = S-nitrosothiol.
Reviewed extensively in many references.
Through guanylate cyclase activation or inflammatory cytotoxicity, as reviewed elsewhere (56).
Figure 1.

Overview of S-nitrosothiol (SNO) signaling in the respiratory system. SNO signaling affects respiratory control, airway function, and pulmonary vascular tone. AE1 = anion exchange protein 1 on the erythrocyte membrane; CFTR = cystic fibrosis transmembrane regulatory protein; GSNO = S-nitrosoglutathione; Hb = hemoglobin; HbFeNO = hemoglobin iron nitrosyl; HbSO2 = oxyhemoglobin saturation; HIF = hypoxia inducible factor; HVR = hypoxic ventilatory response; PAH = pulmonary arterial hypertension; RBC = red blood cell.
Figure 2.

Airways of GSNOR–/– mice are hyporeactive to methacholine (MCh) after allergen challenge. Total pulmonary resistances (RT) of wild-type (WT) and GSNOR–/– (knockout [KO]) mice after control (nonallergic; phosphate-buffered saline [PBS]) (A) and allergen (ovalbumin [OVA]) (B) treatment were determined in the absence or presence of various concentrations of MCh administered intravenously. RT values in PBS-treated and in OVA-treated GSNOR–/– mice were significantly lower than in WT controls (KO PBS vs. WT PBS, p < 0.001; KO OVA vs. WT OVA, p < 0.004; analysis of variance [ANOVA] and post hoc analyses at 3 to 5 MCh doses). Data represent the mean + SE of at least 7 to 10 mice per group. (C) The incremental effect of OVA (over PBS control) on WT and GSNOR–/– mice (OVA minus PBS [OVA−PBS]). Although RT of WT mice increased significantly after OVA treatment (WT PBS vs. WT OVA, p < 0.04; ANOVA), the RT of GSNOR–/– mice did not change significantly (KO PBS vs. KO OVA, p = 0.1; ANOVA). (D) Effect of the inducible nitric oxide synthase (iNOS) inhibitor 1400W on airway responsiveness in PBS- and OVA-treated WT mice (WT PBS vs. WT PBS + 1400W, p = 0.12, n = 5–9; WT OVA vs. WT OVA + 1400W, p = 0.22, n = 7–9). (E) Effect of iNOS inhibition by 1400W on airway responsiveness in GSNOR–/– mice. Administration of 1400W to OVA-treated GSNOR–/– mice resulted in a significant increase in airway resistance (KO OVA vs. KO OVA + 1400W, p < 0.02, n = 5–9; ANOVA). (F) Protein S-nitrosylation (SNO) in lung homogenates of OVA-treated mice. iNOS inhibition (1400W) reduces SNO levels in GSNOR–/– mice (*p < 0.05, n = 3). Reprinted by permission from Reference 2.
S-nitrosylation is in many ways analogous to phosphorylation. Specific amino acid motifs are targeted for S-nitrosylation; the post-translational modification leads to changes in protein activity, protein–protein interactions, or subcellular location of target proteins (5–9). The effects of S-nitrosylation are not restricted to any cell type or function, and indeed, all major classes of proteins are candidates for S-nitrosylation (5–11). S-nitrosylation is primarily regulated enzymatically: NOSs target specific Cys residues for S-nitrosylation (5, 7), and additional enzymes that may perform S-nitrosylation and denitrosylation reactions are discussed below (1–4, 12–16). Thus, cells may precisely regulate subcellular SNO-protein concentration and localization (5, 7, 9, 11, 17–21). More generally, nitrosylation and phosphorylation may provide parallel effector arms across a range of cellular functions (5). We will review this signal transduction mechanism as it relates to pulmonary physiology and disease.
Regulation of SNO Metabolism
Synthesis.
NOS activity leads directly to SNO formation; each NOS isoform is capable of producing SNOs in multiple tissues, cells, and subcellular compartments (5–11, 17, 20). A variety of chemical pathways have been invoked to explain S-nitrosylation. NO may react directly with thiyl radicals or with thiols to form SNOs or SNO radicals, respectively; SNO radicals may be stabilized through the loss of an electron (to form SNO) or through protonation (SNOH radical) (5, 10). Oxidation of NO to NO+ (or a molecule of equivalent reactivity) followed by a reaction with thiols will also produce SNOs (5, 10, 12, 16, 17). Oxidation of NO can be readily catalyzed by oxygen, aromatic residues, and various transition metal ion complexes, which are competent as electron acceptors or facilitators of oxidative chemistry (5, 12, 20–23). SNOs are often formed and stabilized in hydrophobic compartments (e.g., in membranes or discrete protein pockets) (11, 16). Acidic conditions such as those in the lysosome and the mitochondrial intermembrane space—as well as transition metals, including those found in certain metalloproteins and enzymes (e.g., myeloperoxidase or hemoglobin [Hb])—may also promote SNO formation from NO or nitrite, a ubiquitous oxidation product of NOS and dietary substance (10, 11).
Enzymes other than NOS may catalyze S-nitrosylation. The coupling of NO and glutathione in the presence of ceruloplasmin provides one example (12):
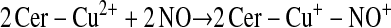 |
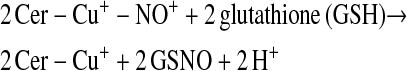 |
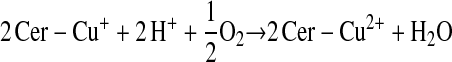 |
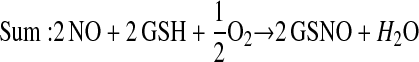 |
This scheme presents general chemical steps of NO oxidation, nitrosation of GSH (i.e., the attachment of NO+), and regeneration of the enzyme, although the precise reaction sequence is not established. Another example of an SNO synthase is Hb: in concert with auxiliary compounds that maintain redox status, it plays the enzymatic role in both autonitrosylation of a conserved Cys as well as S-nitrosylation of the red blood cell (RBC) membrane protein (anion exchange protein 1 [AE1]) and low-molecular-weight thiols (20–22, 24) (Figures 1 and 3).
Figure 3.


Erythrocyte SNO content (SNORBC) and O2 content are functionally coupled. (A) Washed red blood cells (RBCs) from normal humans suspended with (open circles) or without (solid circles) extracellular glutathione (GSH) were steadily deoxygenated under argon. The SNORBC/Hb ratio is plotted against Hb SO2 (inset). Washed RBCs without extracellular GSH were treated in the same fashion as described for A but not deoxygenated. The ratio of SNORBC to Hb was stable over time. (B) The natural logarithm of the ratio of SNORBC to Hb was modeled as a function of Hb SO2; extraerythrocytic GSH was included as a covariate, generating two lines describing the decay rate of the ratio of SNORBC to Hb with or without extraerythrocytic GSH. These rates differed (p < 0.0001). A and Breprinted by permission from Reference 28. As a functional correlate, the low-mass fraction from deoxygenated blood signals an increase in V̇e at the level of the nucleus tractus solitarius (nTS). (C) Microinjection into the nTS of conscious rats of the GSH-derived fraction from deoxygenated blood (black line; see Figure 1) stimulated a V̇e increase that was absent when the low-mass fraction from oxygenated blood was injected (gray line). (D) The differences in V̇e before and after injection of deoxygenated fractions (black bars; n = 14) were highly reproducible (*p < 0.0001), whereas oxygenated fractions (gray bars; n = 12) had no effect (p = not significant). Reprinted by permission from Reference 25.
Catabolism.
SNO breakdown is subject to precise regulation. For example, GSNO reductase (GSNOR) breaks down cytosolic GSNO, ultimately to oxidized GSH and ammonia (1–4). GSNOR, in turn, modulates the levels of some S-nitrosylated proteins by shifting the following transnitrosylation equilibrium to the right:
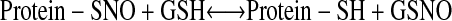 |
The physiologic relevance of GSNOR has been established in knockout mice and plants (i.e., by strict genetic criteria [1–4]); e.g., see Figure 2. Additional proteins my break down SNOs in vitro (13, 14), but none has been firmly established to play a role in physiologic context (1–4).
Transmembrane transport.
Transporters regulate cellular access of low-molecular-weight SNOs. For example, S-nitroso-l-cysteine (LCSNO) has stereoselective effects not replicated by the D-isomer in physiology; stereoselectivity results, in part, from transmembrane transport (18, 25). In addition, γ-glutamyl transpeptidase (GGT) facilitates SNO uptake by cleaving GSNO, which is not membrane permeable, to S-nitroso-cysteinyl glycine (CGSNO) dipeptide, which is readily imported by cells (15, 25). Other membrane proteins, including protein disulfide isomerase, appear also to play a role in SNO transport (19).
SNOs SIGNAL Hb DESATURATION
Background
The influence of Hb on NO bioactivity cycles in tandem with O2 loading/offloading in RBCs (20, 21, 24) (Figures 1 and 3). Thus, coupling by RBCs of the release of NO bioactivity to tissue O2 gradients may subserve the matching of blood flow to O2 demand; furthermore, disordered NO processing by RBCs may subvert normal vascular control, contributing to pulmonary pathology (26–29, 31–33, 35). There is agreement that the Cys at position 93 of the Hb β-chain can be S-nitrosylated; indeed, the crystal structure of β93 Cys SNO-Hb has been solved (22, 26, 27, 31, 32, 36). The following data suggest that the β93 SNO bond is stabilized when tetrameric Hb is in the oxygenated (relaxed [R]) conformation and destabilized when it is in the deoxygenated (tense [T]) conformation, consonant with both preferential formation of SNO in R structure and increased reactivity of SNO in T structure (Figure 1):
Oxygen binding is increased by S-nitrosylation of Hb versus the unmodified protein; thus, thermodynamic principles of Hb (“linkage”) require that SNO is stabilized by oxygenation (24).
The β93 SNO is detected by X-ray crystallography in R-state (oxy) Hb, but not in the T (deoxy) conformation (36).
Hb is preferentially S-nitrosylated at high Po2, whereas NO groups are preferentially released at low Po2 (22, 24, 26).
The half-life of infused SNO-Hb is prolonged in animals breathing high FiO2 versus room air (30).
Erythrocytic SNO content is essentially static at 100% oxygen saturation, but decays with desaturation (26, 28, 29).
The release of NO bioactivity by erythrocytes, which occurs via transnitros(yl)ation of AE1 (forming SNO-AE1) or reduced GSH (forming GSNO) is favored with decreasing Po2 (21, 25, 28, 29) (Figure 3).
SNO-Hb content of RBCs, as measured by photolysis-chemiluminescence, by chemical- and fluorescent-based probes and chemical reduction followed by chemiluminescence, is higher in oxygenated blood than in deoxygenated blood of mammals (26, 28, 30, 31). It is important to emphasize in this context that some assay techniques may radically alter Hb allostery, structure, or solubility, as well as iron nitrosyl (FeNO) or SNO stability (27, 28, 32). (Of five methods that have been used to measure SNO-Hb, four are in general agreement, the exception being a technique called triodide-chemiluminescence that, in our hands, cannot accurately measure either SNO-Hb or FeNO [22, 24, 26–28, 32]).
Vasodilation by RBCs is proportional to the extent of hypoxemia (21, 28, 33). Thus, although the rate of decay of SNO-Hb has varied in different hands—reflecting different experimental conditions—in intact erythrocytes (28) and in vivo (30), the experimental data leave no doubt as to the role of deoxygenation in facilitating NO group release (in keeping with theoretical predictions); there is also agreement that NO radical itself cannot account for the vasodilatory actions of SNO-Hb (37), consistent with data that erythrocyte deoxygenation produces SNOs (21, 25, 28, 29). Thus, SNO-based signaling cascades are specifically coupled to RBC oxyHb/deoxyHb cycling, as discussed below.
Relevance to Pulmonary and Critical Care Medicine
SNOs signal hypoxic ventilatory drive.
The Cys thiols of certain proteins and peptides, including those of AE1 and GSH, undergo S-nitrosylation during erythrocyte deoxygenation (21, 25, 28, 29) (Figures 1 and 3). These SNOs, in turn, may signal tissue responses to hypoxia (21, 25, 28, 38) (Figure 3). Thus, SNOs enable hypoxic signaling by RBCs. Hypoxia-sensing cells in the carotid body stimulate neurons that project to NOS1-rich neurons in the nucleus tractus solitarius (nTS); NOS1 activation in the nTS is critical to hypoxic ventilatory response. NOS1 activation in the brain forms SNOs (20, 39); GSNO, a major bioactive product of NOS1 (6), is processed via GGT to L-CSNO. L-CSNO (but not D-CSNO) injected into the nTS of conscious rats dramatically increases minute ventilation (V̇e), with a time course that mimics the physiologic effect of hypoxia (25). Similarly, GSNO formed during erythrocyte deoxygenation is precisely hypoxia-mimetic when directly injected into the rat nTS (24) (Figure 3). Strikingly, GSNO-stimulated increases in V̇e are prevented by GGT inhibitors, and mice deficient in GGT have abnormal hypoxic responses, including a paradoxical apneic recovery response reminiscent of the preterm human newborn response to hypoxia (25). Collectively, these observations suggest that GSNO may serve as a signal through which erythrocytes can regulate the drive to breathe, and that the GSNO signal is elicited through deoxygenation of Hb.
Hildebrandt and coworkers have shown that systemic N-acetylcysteine (NAC) therapy has hypoxia-mimetic effects in humans, including augmentation of hypoxic ventilatory drive (40). NAC recapitulates the effects of endogenous GSH: deoxygenation of human blood in the presence of added NAC yields SNO-NAC (SNO-AC), depleting SNO-Hb in the process (29). High doses of NAC will deplete SNO-Hb even under normoxia (by direct transnitrosation). Like GSNO, SNO-AC can signal to increase V̇e. This finding has led to interest in short-term use of NAC to augment hypoxic ventilatory drive in patients with impairments, such as preterm newborns and subjects with chronic obstructive pulmonary disease (COPD).
Deoxyhemoglobin-derived SNOs signal hypoxic gene regulation.
GSNO increases hypoxia inducible factor 1 (HIF1)–DNA binding through stabilization of the α subunit of the HIF1 heterodimer (34). This finding suggests a mechanism by which Hb desaturation can alter expression of genes under hypoxemic conditions. Further, GSNO produced on deoxygenation of RBCs has been found to regulate the expression and DNA binding of specificity protein 1 (Sp1) and Sp3 (38). GSNO transcriptional effects in cells are GGT dependent (34, 38). Again, SNO-AC reproduces the effect of GSNO. Thus, treatment of mice with NAC, which forms circulating SNO-AC, produces a tonic signal in pulmonary vascular endothelial cells that mimics hypoxia in up-regulating HIF1- and Sp-dependent effects in the murine lung (29). Systemic NAC therapy is hypoxia mimetic in humans, increasing erythropoietin production (40). These data raise the concern that up-regulation of hypoxia-associated genes may be augmented by NAC treatment for conditions such as idiopathic pulmonary fibrosis (41).
The Role of RBC SNO in Ventilation–Perfusion Matching and Pulmonary Hypertension
SNO-enriched RBCs improve oxygenation (33). Thus, RBCs may play a previously unappreciated role in matching ventilation to perfusion. Deployment of RBC-derived SNO bioactivity is increased in human sepsis (28), an effect that can blunt hypoxic pulmonary vasoconstriction (HPV). Unregulated vasodilation by RBCs may provide mechanistic insight into disrupted ventilation–perfusion matching that occurs early in the course of acute lung injury (28).
Sustained hypoxia both impairs the synthesis of SNO-Hb and increases the rate at which SNO is released from human RBCs, thereby leading to depletion of SNO-Hb (33). Sustained hypoxemia thus impairs hypoxia-induced vasodilation by RBCs. Indeed, patients with pulmonary hypertension and mild hypoxemia exhibit SNO RBC depletion and their pulmonary arterial pressures are inversely related to the amount of NO bound to Hb (33). Chemical repletion of RBC SNO restores hypoxia-induced vasodilation by RBCs and reverses hypoxia-associated human pulmonary hypertension (33, 42). Likewise, humans with sickle cell disease show depleted levels of SNO-Hb and, in addition, fail to process NO normally within RBCs (abnormal intramolecular heme-thiol transfer within Hb as well as abnormal transfer of NO between Hb and the RBC membrane protein AE1), with a net reduction in the hypoxia-responsive export of NO bioactivity from RBCs (35). This NO-processing defect in sickle cell RBCs is most pronounced in patients with more severe illness (predisposing to acute chest syndrome); the defect is not noted in patients without frequent occlusive episodes (35). Conversely, RBCs obtained from humans with septic shock have excessive amounts of SNO-Hb (28). Notably, although inhaled NO therapy is used to treat acute chest syndrome (43), it does not readily reverse the SNO deficit (35). Restoration of physiologic SNO-Hb ratios, recovery of vasodilation by RBCs, reduction of pulmonary vascular resistance (PVR), and improved oxygenation is, how- ever, achieved in humans by inhalation of an SNO-generating gas (O-nitrosoethanol) (42).
SNOs IN AIRWAY DISEASES
Background
General effects of SNOs in the airways.
GSNO and SNO proteins are normally present in the human airways at concentrations of approximately 0.5 to 1 μM (44). GSNO increases ciliary beat frequency and dilates human airways with potency of approximately two log orders greater than theophylline (44, 45). The human bronchodilatory effects of GSNO are largely cGMP independent (44, 47) and likely result from S-nitrosylation of ion channels, receptor systems, and other myocyte proteins (47–49, 51). SNOs also have pulmonary vascular smooth muscle relaxant effects (28, 42, 51), and in humans, SNOs can augment perfusion to well-ventilated lung units, improving oxygenation and decreasing dead space (42, 50).
Ion channel and receptor effects of SNOs.
SNOs affect the expression and/or activities of a broad range of ion channels that are of potential importance to lung physiology, including sodium, chloride (cystic fibrosis transmembrane conductance regulator [CFTR]), potassium, and calcium channels of different types (5, 49). Phosphorylation by cGMP-dependent kinases and direct S-nitrosylation are well-established mechanisms by which NOSs can regulate ion channel activity. A principle that emerges in the regulation of calcium channels by NO—including the l-type, N-methyl-D-aspartate, and ryanodine receptors—is that NOSs associate with either the ion channels themselves or with scaffolding proteins that serve to place the NOS in close proximity to the channel. NOSs can thereby selectively S-nitrosylate–critical thiols to influence channel activity (5, 49).
SNOs can also regulate signaling through both G-protein–coupled receptors (e.g., seratonergic and adrenergic) and receptor tyrosine kinases (9, 51). One target of S-nitrosylation by NOS is the master G-protein dynamin, which regulates the surface expression levels of receptors. S-nitrosylation of dynamin leads to its assembly (protein–protein interaction), redistribution from cytosol to membrane, and increased enzymatic activity (9). Stimulation of the β2-adrenergic receptor initiates dynamin S-nitrosylation.
Antimicrobial effects of SNOs.
Antiviral effects of SNOs can involve inhibition of viral Cys proteases (52). Antibacterial and antimycobacterial effects are more complex, involving inhibition of key prokaryotic proteins and inactivation of cellular defenses that protect against nitrosative stress (1, 3, 5, 52, 53). Not all actions of NOS are microbicidal. NO produced by NOS can be exploited by certain microbes to enhance their own transmembrane import (9).
SNO signaling and cell survival.
NOSs have both pro- and antiapoptotic actions that are mediated predominantly by S-nitrosylation (8, 11, 46, 54, 55). Two general mechanisms by which NO regulates cell death have been described. First, NO inhibits apoptosis through S-nitrosylation of caspases; some caspases are constitutively S-nitrosylated and thereby maintained in an inactive state. In human lymphocytes, denitrosylation of caspase 3 and caspase 9 is coupled to ligand (Fas)-induced release of caspases from the mitochondrial intermembrane space (11). Second, the principal proapoptotic effect of NO appears to be mediated through the S-nitrosylation of GAPDH, promoting its interaction with the E3 ligase Siah1, leading to nuclear translocation and ubiquitin-mediated degradation of nuclear target proteins (nitrosative stress) (54). Other mechanisms (e.g., the inhibition of nuclear factor [NF]-κB or Jun NH2-terminal kinase [JNK]) may subserve context- and cell-specific regulation of apoptosis by NO (8, 55). Indeed, inhibition of NF-κB by NO may contribute to apoptosis of airway epithelial cells and infiltrating inflammatory cells, as seen in patients with asthma. This balance of pro- and antiapoptotic effects can thus be best understood in terms of the specific Cys-containing proteins that are targets of NO in the context of cell type and stimulae.
SNO signaling and the cellular inflammatory response.
S-nitrosylation signaling has an important role in endotoxin-induced injury (3). Thus, altered amounts or spatiotemporal production of SNOs may contribute to inflammation. Although the proinflammatory effects of NO have been held to result from widespread injury, the picture that emerges from the recent work of Snyder and colleagues is different (7, 54). SNOs stimulate the injurious response through precise effects on key enzymes, such as cyclooxygenase 2 (7) and GAPDH (54).
Specific Lung Diseases
SNOs in asthma.
Low pH and inducible NOS (iNOS) up-regulation in the asthmatic airway should favor formation of SNOs (20, 56). However, airway SNO levels are substantially lower in children intubated for asthmatic respiratory failure than in children intubated for elective surgery (57). Furthermore, in adults with asthma undergoing segmental allergen challenge, baseline SNO levels are decreased, and the post-challenge increase in SNO levels does not match the post-challenge increase in other nitrogen oxides (58). These findings are consistent with animal data showing that GSNO breakdown is up-regulated after ovalbumin challenge, and that genetic deficiency of GSNOR protects mice from antigen sensitization–induced methacholine hyperresponsiveness (2) (Figure 2). These data suggest that GSNO turnover in general, and GSNOR activity in particular, are increased in the asthmatic airway, particularly in severe asthma.
SNOs in cystic fibrosis.
As in asthma, levels of SNOs are lower than normal in the cystic fibrosis (CF) airway (59). In CF, however, the deficiency may result from decreased synthesis, reflecting decreased GSH transport into the airway and decreased airway epithelial iNOS expression (60). SNO deficiency may result in a complex defect, including impairments in airway smooth muscle relaxation (44, 47, 48), mucociliary clearance (45), neutrophil apoptosis (46), and host defense (53, 60). Repletion of GSNO increases expression, maturation, and function of the common CF mutant ΔF508 (38, 61). Importantly, the effects of GSNO are concentration dependent. At high concentrations, GSNO may promote CFTR degradation (62), may inhibit its transcription (38), and may inhibit the activity of the wild-type protein (63). Clinically, replacement therapy with GSNO modestly improves oxygenation in CF, and the beneficial effect is not related to the amount of free NO liberated by GSNO (50).
Other lung and respiratory muscle disorders.
In the context of nonasthmatic airway inflammation (i.e., pneumonia and after lung transplantation), SNO levels may be elevated, perhaps augmenting antimicrobial host defense (44, 60). In addition, NOSs play an important role in the process of excitation–contraction coupling in skeletal muscles, and the altered respiratory muscle performance in myopathies of sepsis and COPD may relate to a deficiency of NOS bioactivity. In particular, S-nitrosylation of a critical Cys within the RyR1 controls the release of calcium, which governs the contractile performance of skeletal muscles, such as diaphragm. S-nitrosylation by NO is impaired by pathologic Po2 and oxidative stress (64). Myopathies are frequently associated with altered expression, distribution, or activity of skeletal NOSs and/or impairment of oxygenation or blood flow. In many cases, chronic deprivation of NO is associated with oxidative stress. Thus, NO/redox imbalance may provide a novel molecular basis for respiratory muscle weakness: conditions such as sepsis and COPD, previously subsumed under the rubric of oxidative and nitrosative stress, may be viewed as symptomatic of a disruption of nitrosylation/redox-based signaling.
CONCLUSIONS
For over a decade, the attention of the pulmonary research community has been focused on NO diffusion and free-radical reactions to explain signals elicited by NOS. Exhaled NO has been used as a marker of NOS activity, and inhaled NO has emerged as an important therapy. However, NO in exhaled breath is a limited measure of the panoply of NO functions in solution, and inhaled NO can reconstitute only a small part of NOS bioactivity. In addition, exhaled NO is generated, at least partly, by metabolism of alternative nitrogen oxides, including SNOs, which provide the majority of NO-related bioactivity in the lungs as determined by stringent genetic criteria. Inhaled NO is a relatively ineffective means of raising SNO levels or targeting critical cysteines. Against this background, endogenous NO is converted efficiently and selectively into SNOs. SNOs play central roles in the control of breathing, ventilation–perfusion matching, vascular tone, diaphragmatic performance, and airway resistance; furthermore, altered SNO signaling is linked to pulmonary hypertension, septic shock, asthma, and CF. The biology of SNO signaling, which is of primary relevance to pulmonary physiology in health and disease, remains largely unexplored and ripe for discovery.
This work was supported by National Institutes of Health (NIH) 2RO1 HL59337 (B.G.), NIH RO1 Severe Asthma Research Program (SARP) HL 69170 (B.G.), NIH K08 GM 069977 (A.D.), and NSF MCBS 00981228 (D.S.).
Originally Published in Press as DOI: 10.1164/rccm.200510-1584PP on March 9, 2006
Conflict of Interest Statement: B.G. is a consultant for and owns “B Unit” equity in Nitrox, LLC. He also owns intellectual property related to the treatment of lung disease with S-nitrosothiols. D.S. does not have a financial relationship with a commercial entity that has an interest in the subject of this manuscript. A.D. has served as a consultant to INO Therapeutics and Nitrox, LLC. He has received research support ($95,000) from Nitrox, LLC, and INO Therapeutics ($15,000), and he has a patent related to the topic of the manuscript. J.S.S. has a financial interest in Nitrox, LLC.
References
- 1.Liu L, Hausladen A, Zeng M, Que L, Heitman J, Stamler JS. A metabolic enzyme for S-nitrosothiol conserved from bacteria to humans. Nature 2001;410:490–494. [DOI] [PubMed] [Google Scholar]
- 2.Que LG, Liu L, Yan Y, Whitehead G, Gavett SH, Schwartz DA, Stamler JS. Protection from experimental asthma by an endogenous bronchodilator. Science 2005;308:1618–1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liu L, Yan Y, Zeng M, Zhang J, Hanes MA, Ahearn G, McMahon TJ, Dickfeld T, Marshall HE, Que LG, et al. Essential roles of S-nitrosothiols in vascular homeostasis and endotoxic shock. Cell 2004;116:617–628. [DOI] [PubMed] [Google Scholar]
- 4.Feechan A, Kwon E, Yun B, Wang Y, Wang Y, Pallas JA, Loake GJ. A central role for S-nitrosothiols in plant disease resistance. Proc Natl Acad Sci USA 2005;102:8054–8059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hess DT, Matsumoto A, Kim SO, Marshall HE, Stamler JS. Protein S-nitrosylation: purview and parameters. Nat Rev Mol Cell Biol 2005;6:150–166. [DOI] [PubMed] [Google Scholar]
- 6.Mayer B, Pfeiffer S, Schrammel A, Koesling D, Schmidt K, Brunner F. A new pathway of nitric oxide/cyclic GMP signaling involving S-nitrosoglutathione. J Biol Chem 1998;273:3264–3270. [DOI] [PubMed] [Google Scholar]
- 7.Kim SF, Huri DA, Snyder SH. Inducible nitric oxide synthase binds, S-nitrosylates and activates cyclooxygenase-2. Science 2005;310:1966–1970. [DOI] [PubMed] [Google Scholar]
- 8.Haendeler J, Hoffmann J, Tischler V, Berk BC, Zeiher AM, Dimmeler S. Redox regulatory and anti-apoptotic functions of thioredoxin depend on S-nitrosylation at cysteine 69. Nat Cell Biol 2002;4:743–749. [DOI] [PubMed] [Google Scholar]
- 9.Wang G, Moniri NH, Osawa K, Stamler JS, Daaka Y. Nitric oxide regulates endocytosis by S-nitrosylation of dynamin. Proc Natl Acad Sci USA 2006;103:1295–1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
-
10.Zhang H, Xu Y, Joseph J, Kalyanaraman B. Intramolecular electron transfer between tyrosyl radical and cysteine residue inhibits tyrosine nitration and induces thiyl radical formation in model peptides treated with myeloperoxidase, H2O2, and
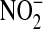 . J Biol Chem 2005;280:40684–40698. [DOI] [PubMed] [Google Scholar]
. J Biol Chem 2005;280:40684–40698. [DOI] [PubMed] [Google Scholar] - 11.Mannick J, Schonhoff C, Papeta N, Ghafourifar P, Szibor M, Fang K, Gaston B. S-nitrosylation of mitochondrial caspases. J Cell Biol 2001;154:1111–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Inoue K, Akaike T, Iyamoto Y, Okamoto T, Sawa T, Otagiri M, Suzuki S, Yoshimura T, Maeda H. Nitrosothiol formation catalyzed by ceruloplasmin: implication for cytoprottective mechanism in vivo. J Biol Chem 1999;274:27069–27075. [DOI] [PubMed] [Google Scholar]
- 13.Trujillo M, Alvarez M, Peluffo G, Freeman B, Radi R. Xanthine oxidase-mediated decomposition of S-nitrosothiols. J Biol Chem 1998;273:7929–7934. [DOI] [PubMed] [Google Scholar]
- 14.Nikitovic D, Holmgren A. S-nitrosoglutathione is cleaved by the thioredoxin system with liberation of glutathione and redox regulating nitric oxide. J Biol Chem 1996;271:19180–19185. [DOI] [PubMed] [Google Scholar]
- 15.Hogg N, Singh R, Konorev E, Joseph J, Kalyanaraman B. S-nitrosoglutathione as a substrate for γ-glutamyl transpeptidase. Biochem J 1997;323:477–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rafikova O, Rafikov R, Nudler E. Catalysis of S-nitrosothiols formation by serum albumin: the mechanism and implication in vascular control. Proc Natl Acad Sci USA 2002;99:5913–5918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ckless K, Reynaert NL, Taatjes DJ, Lounsbury KM, van der Vliet A, Janssen-Heininger Y. In situ detection and visualization of S-nitrosylated proteins following chemical derivatization: identification of Ran GTPase as a target for S-nitrosylation. Nitric Oxide 2004;11:216–227. [DOI] [PubMed] [Google Scholar]
- 18.Lewis SJ, Hoque A, Bates JN. Differentiation of L- and D-S-nitrosothiol recognition sites in vivo. J Cardiovasc Pharmacol 2005;46:660–671. [DOI] [PubMed] [Google Scholar]
- 19.Ramachandran N, Root P, Jiang XM, Hogg PJ, Mutus B. Mechanism of transfer of NO from extracellular S-nitrosothiols into the cytosol by cell-surface protein disulfide isomerase. Proc Natl Acad Sci USA 2001;98:9539–9544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gow AJ, Chen Q, Hess DT, Day BJ, Ischiropoulos H, Stamler JS. Basal and stimulated protein S-nitrosylation in multiple cell types and tissues. J Biol Chem 2002;277:9637–9640. [DOI] [PubMed] [Google Scholar]
- 21.Pawloski JR, Hess DT, Stamler JS. Export by red blood cells of nitric oxide bioactivity. Nature 2001;409:622–626. [DOI] [PubMed] [Google Scholar]
- 22.Jia L, Bonaventura C, Bonaventura J, Stamler JS. S-nitrosohaemoglobin: a dynamic activity of blood involved in vascular control. Nature 1996;380:221–226. [DOI] [PubMed] [Google Scholar]
- 23.Zhao YL, Bartberger MD, Goto K, Shimada K, Kawashima T, Houk KN. Theoretical evidence for enhanced NO dimerization in aromatic hosts: implications for the role of the electrophile (NO) (2) in nitric oxide chemistry. J Am Chem Soc 2005;127:7964–7965. [DOI] [PubMed] [Google Scholar]
- 24.Gow AJ, Stamler JS. Reactions between nitric oxide and haemoglobin under physiological conditions. Nature 1998;391:169–173. [DOI] [PubMed] [Google Scholar]
- 25.Lipton A, Johnson M, Macdonald T, Lieberman M, Gozal D, Gaston B. S-nitrosothiols signal the ventilatory response to hypoxia. Nature 2001;413:171–174. [DOI] [PubMed] [Google Scholar]
- 26.McMahon TJ, Moon RE, Luschinger BP, Carraway MS, Stone AE, Stolp BW, Gow AJ, Pawloski JR, Watke P, Singel DJ, et al. Nitric oxide in the human respiratory cycle. Nat Med 2002;8:711–717. [DOI] [PubMed] [Google Scholar]
- 27.Rogers SC, Khalatbari A, Gapper PW, Frenneaux MP, James PE. Detection of human red blood cell-bound nitric oxide. J Biol Chem 2005;280:26720–26728. [DOI] [PubMed] [Google Scholar]
- 28.Doctor A, Platt R, Sheram ML, Eischeid A, McMahon T, Maxey T, Doherty J, Axelrod M, Kline J, Gurka M, et al. Hemoglobin conformation couples S-nitrosothiol content in erythrocytes to oxygen gradients. Proc Natl Acad Sci USA 2005;102:5709–5714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Palmer LA, Chhabra P, Doctor A, Sheram ML, Laubach V, Gaston B. N acetyl cysteine induces pulmonary hypertension: Role of S-nitrosothiols [abstract]. Proc Am Thorac Soc 2005;2:A707. [Google Scholar]
- 30.Sonveaux P, Kaz AM, Snyder SA, Richardson RA, Cardenas-Navia LI, Braun RD, Pawloski JR, Tozer GM, Bonaventura J, McMahon TJ, et al. Oxygen regulation of tumor perfusion by S-nitrosohemoglobin reveals a pressor activity of nitric oxide. Circ Res 2005;96:1119–1126. [DOI] [PubMed] [Google Scholar]
- 31.Funai EF, Davidson A, Seligman SP, Finlay TH. S-nitrosohemoglobin in the fetal circulation may represent a cycle for blood pressure regulation. Biochem Biophys Res Commun 1997;239:875–877. [DOI] [PubMed] [Google Scholar]
- 32.Rassaf T, Bryan N, Maloney R, Specian V, Kelm M, Kalyanaraman B, Rodriquez J, Feelisch M. NO adducts in mammalian red blood cells: too much or too little? Nat Med 2003;9:481–482. [DOI] [PubMed] [Google Scholar]
- 33.McMahon TJ, Ahearn GS, Moya MP, Gow AJ, Huang YC, Luchsinger BP, Nudelman R, Yan Y, Krichman AD, Baashore TM, et al. A nitric oxide processing defect of red blood cells created by hypoxia: deficiency of S-nitrosohemoglobin in pulmonary hypertension. Proc Natl Acad Sci USA 2005;102:14801–14806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Palmer L, Gaston B, Johns R. Normoxic stabilization of hypoxia-inducible factor 1 expression and activity: redox-dependent effect of nitrogen oxides. Mol Pharmacol 2000;58:1197–1203. [DOI] [PubMed] [Google Scholar]
- 35.Pawloski JR, Hess DT, Stamler JS. Impaired vasodilation by red blood cells in sickle cell disease. Proc Natl Acad Sci USA 2005;102:2531–2536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chan N, Rogers PH, Arnone A. Crystal structure of the S-nitroso form of liganded human hemoglobin. J Biochem 1998;37:16459–16464. [DOI] [PubMed] [Google Scholar]
- 37.Jeffers A, Xu X, Huang K, Cho M, Hogg N, Patel RP, Kim-Shapiro DB. Hemoglobin mediated nitrite activation of soluble guanylyl cyclase. Compar Biochem Physiol 2005;142:130–135. [DOI] [PubMed] [Google Scholar]
- 38.Zaman K, Palmer LA, Doctor A, Hunt JF, Gaston B. Concentration-dependent effects of endogenous S-nitrosoglutathione on gene regulation by specificity proteins Sp3 and Sp1. Biochem J 2004;380:67–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kluge I, Gutleck-Amsler U, Zollinger M, Do KQ. S- nitrosoglutathione in rat cerebellum: identification and quantification by liquid chromatography-mass spectrometry. J Neurochem 1997;69:2599–2607. [DOI] [PubMed] [Google Scholar]
- 40.Hildebrandt W, Alexander S, Bärtsch P, Dröge W. Effect of N-acetyl-cysteine on the hypoxic ventilatory response and erythropoietin production: linkage between plasma thiol redox state and O2 chemosensitivity. Blood 2002;99:1552–1555. [DOI] [PubMed] [Google Scholar]
- 41.Demedts M, Behr J, Buhl R, Costabel U, Dekhuijzen R, Jansen H, MacNee W, Thomeer M, Wallaert B, Laurent F, et al. High-dose acetylcysteine in idiopathic pulmonary fibrosis. N Engl J Med 2005;353:2229–2242. [DOI] [PubMed] [Google Scholar]
- 42.Moya MP, Gow AJ, Califf RM, Goldberg RN, Stamler JS. Inhaled ethyl nitrite gas for persistent pulmonary hypertension of the newborn. Lancet 2002;360:141–143. [DOI] [PubMed] [Google Scholar]
- 43.Atz AM, Wessel DL. Inhaled nitric oxide in sickle cell disease with acute chest syndrome. Anesthesiology 1997;87:988–990. [DOI] [PubMed] [Google Scholar]
- 44.Gaston B, Reilly J, Drazen JM, Fackler J, Ramdev P, Arnelle D, Mullins M, Sugarbaker D, Chee C, Singel D, et al. Endogenous nitrogen oxides and bronchodilator S-nitrosothiols in human airways. Proc Natl Acad Sci USA 1993;90:10957–10961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li D, Shirakami G, Zhan X, Johns R. Regulation of ciliary beat frequency by the nitric oxide-cyclic guanosine monophosphate signaling pathway in rat airway epithelial cells. Am J Respir Cell Mol Biol 2000;23:175–181. [DOI] [PubMed] [Google Scholar]
- 46.Fortenberry JD, Owens ML, Brown LA. S-nitrosoglutathione enhances neutrophil DNA fragmentation and cell death. Am J Physiol 1999;276:435–442. [DOI] [PubMed] [Google Scholar]
- 47.Janssen LJ, Premji M, Hwa L, Cox G, Kshavjee S. NO+ but not NO radical relaxes airway smooth muscle via cGMP-independent release of internal Ca2+. Am J Physiol Lung Cell Mol Physiol 2000;278:899–905. [DOI] [PubMed] [Google Scholar]
- 48.White TA, Walseth TF, Kannan MS. Nitric oxide inhibits ADP-ribosyl cyclase through a cGMP-independent pathway in airway smooth muscle. Am J Physiol Lung Cell Mol Physiol 2002;283:1065–1071. [DOI] [PubMed] [Google Scholar]
- 49.Matalon S, Hardiman KM, Jain L, Eaton DC, Kotlikoff M, Eu JP, Sun J, Meissner G, Stamler JS. Regulation of ion channel structure and function by reactive oxygen-nitrogen species. Am J Physiol Lung Cell Mol Physiol 2003;285:1184–1189. [DOI] [PubMed] [Google Scholar]
- 50.Snyder A, McPherson ME, Hunt JF, Johnson M, Stamler JS, Gaston B. Acute effects of aerosolized S-nitrosoglutathione in cystic fibrosis. Am J Respir Crit Care Med 2002;165:922–926. [DOI] [PubMed] [Google Scholar]
- 51.Nozik-Grayck E, Whalen E, Stamler JS, McMahon TJ, Chitano P, Piantadosi CA. S-nitrosoglutathione inhibits α1-adrenergic receptor-mediated vasoconstriction and ligand binding in pulmonary artery. Am J Physiol Lung Cell Mol Physiol 2006;290:136–143. [DOI] [PubMed] [Google Scholar]
- 52.Cao W, Baniecki ML, McGrath WJ, Bao C, Deming CB, Rade JJ, Lowenstein CJ, Mangel WF. Nitric oxide inhibits the adenovirus proteinase in vitro and viral infectivity in vivo. FASEB J 2003;17:2345–2346. [DOI] [PubMed] [Google Scholar]
- 53.Venketaraman V, Dayaram YK, Talaue MT, Connell ND. Glutathione and nitrosoglutathione in macrophage defense against Mycobactrerium tuberculosis. Infect Immun 2005;73:1886–1889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hara MR, Agrawal N, Kim SF, Cascio MB, Fujimuro M, Ozeki Y, Takahashi M, Cheah JH, Tankou SK, Hester KD, et al. S-nitrosylated GAPDH initiates apoptotic cell death by nuclear translocation following Siahi binding. Nat Cell Biol 2005;7:665–674. [DOI] [PubMed] [Google Scholar]
- 55.Reynaert NL, Ckless K, Korn SH, Vos N, Guala AS, Wouters EF, van der Vliet A, Janssen-Heininger YM. Nitric oxide represses inhibitory kappaB kinase through S-nitrosylation. Proc Natl Acad Sci USA 2004;101:8945–8950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ricciardolo F, Sterk P, Gaston B, Folkerts G. Nitric oxide in health and disease of the respiratory system. Physiol Rev 2004;84:731–765. [DOI] [PubMed] [Google Scholar]
- 57.Gaston B, Sears S, Woods J, Hunt J, Ponaman M, McMahon T, Stamler JS. Bronchodilator S-nitrosothiol deficiency in asthmatic respiratory failure. Lancet 1998;351:1317–1319. [DOI] [PubMed] [Google Scholar]
- 58.Dweik R, Comhair S, Gaston B, Thunnissen F, Farver C, Thomassen M, Kavuru M, Hammel J, Abu-Soud H, Erzurum S. NO chemical events in the human airway during the immediate and late antigen-induced asthmatic response. Proc Natl Acad Sci USA 2001;98:2622–2627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Grasemann H, Gaston B, Fang K, Paul K, Ratjen F. Decreased levels of nitrosothiols in the lower airways of patients with cystic fibrosis and normal pulmonary function. J Pediatr 1999;135:770–772. [DOI] [PubMed] [Google Scholar]
- 60.Kelley TJ, Drumm ML. Inducible nitric oxide synthase expression is reduced in cystic fibrosis murine and human airway epithelial cells. J Clin Invest 1998;102:1200–1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Howard M, Fischer H, Roux J, Santos BC, Gullans SR, Yancey PH, Welch WJ. Mammalian osmolytes and S-nitrosoglutathione promote ΔF508 cystic fibrosis transmembrane conductance regulator (CFTR) protein maturation and function. J Biol Chem 2003;278:35159–35167. [DOI] [PubMed] [Google Scholar]
- 62.Jilling T, Haddad I, Cheng S, Matalon S. Nitric oxide inhibits heterologous CFTR expression in polarized epithelial cells. Am J Physiol 1999;277:89–96. [DOI] [PubMed] [Google Scholar]
- 63.Wang W, Oliva C, Li G, Holmgren A, Lillig CH, Kirk KL. Reversible silencing of CFTR chloride channels by glutathionylation. J Gen Physiol 2005;125:127–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Eu JP, Hare JM, Hess DT, Skaf M, Sun J, Cardenas-Navina I, Sun QA, Dewhirst M, Meissner G, Stamler JS. Concerted regulation of skeletal muscle contractility by oxygen tension and endogenous nitric oxide. Proc Natl Acad Sci USA 2003;100:15229–15234. [DOI] [PMC free article] [PubMed] [Google Scholar]