

Abstract
The NMR structures of the recombinant human prion protein, hPrP(23–230), and two C-terminal fragments, hPrP(90–230) and hPrP(121–230), include a globular domain extending from residues 125–228, for which a detailed structure was obtained, and an N-terminal flexibly disordered “tail.” The globular domain contains three α-helices comprising the residues 144–154, 173–194, and 200–228 and a short anti-parallel β-sheet comprising the residues 128–131 and 161–164. Within the globular domain, three polypeptide segments show increased structural disorder: i.e., a loop of residues 167–171, the residues 187–194 at the end of helix 2, and the residues 219–228 in the C-terminal part of helix 3. The local conformational state of the polypeptide segments 187–193 in helix 2 and 219–226 in helix 3 is measurably influenced by the length of the N-terminal tail, with the helical states being most highly populated in hPrP(23–230). When compared with the previously reported structures of the murine and Syrian hamster prion proteins, the length of helix 3 coincides more closely with that in the Syrian hamster protein whereas the disordered loop 167–171 is shared with murine PrP. These species variations of local structure are in a surface area of the cellular form of PrP that has previously been implicated in intermolecular interactions related both to the species barrier for infectious transmission of prion disease and to immune reactions.
Prion proteins (PrP) are associated with transmissible spongiform encephalopathies (TSE), which are invariably fatal diseases characterized by loss of motor control, dementia, and paralysis wasting (1, 2). Human TSEs include Creutzfeldt-Jakob disease, fatal familial insomnia, the Gerstmann-Sträussler-Scheinker syndrome, and kuru, and there is bovine spongiform encephalopathy in cattle and scrapie in sheep. The “protein-only” hypothesis (3, 4) proposes that TSEs are caused by the conversion of a ubiquitous “cellular form” of PrP (PrPC) into an aggregated “scrapie form” (PrPSc). According to this model, the prion protein (PrP) would at the same time be target and infectious agent in TSEs, which could explain that this class of diseases can be traced to infectious, inherited, and spontaneous origins (2, 5). PrPSc is characterized by a high β-sheet content, insolubility in detergents, and resistance to proteolysis in its aggregated form (6–8) whereas PrPC is a soluble protein with a high content of α-helices (8, 9) and high susceptibility to proteolytic digestion. No chemical modifications have as yet been identified by which the two PrP forms would differ (10).
Considering that the protein-only hypothesis suggests a change of protein conformation as a possible cause of the onset of TSEs, the three-dimensional prion protein structures have attracted keen interest. So far, nuclear magnetic resonance (NMR) solution studies have been described for monomeric, cellular forms of PrP of the two most widely used laboratory animals in prion research, the mouse (m) and the Syrian hamster (sh), using the constructs mPrP(121–231) (9, 11), shPrP(90–231) (12, 13), mPrP(23–231) (14, 15), and shPrP(29–231) (16). Both of these prion proteins contain a globular domain that extends approximately from residues 125–228 and an N-terminal flexibly disordered “tail” (14–16). The globular domain contains a two-stranded anti-parallel β-sheet and three α-helices (9). In the shPrP structure, the third helix extends from residues 200–227 (13) whereas the corresponding helix in mPrP is well defined only up to residue 219 (11), and the loop of residues 167–171 between the second β-strand and helix 2, which is poorly defined in mPrP (9, 11), was reported to be well defined in shPrP (13).
The extensive sequence homology (17) indicates that the mammalian prion proteins should all have a common polypeptide fold. Nonetheless, in view of the high profile of the problems raised, we determined the NMR structures of the intact recombinant human prion protein, hPrP(23–230), and the two C-terminal fragments hPrP(90–230) and hPrP(121–230) to provide a direct basis for future structure–function arguments on hPrP. Special interest in this investigation focused on the molecular regions that showed differences between mPrP and shPrP, i.e., the loop 167–171 and helix 3, because these structure elements have been implicated in immune reactions of PrP (18) as well as in species-specific contacts with a so-far not-further-characterized “protein X,” which has been suggested to mediate the transition from PrPC to PrPSc (19–22).
Materials and Methods
For the cloning of the recombinant human PrP polypeptides, we followed a previously described strategy (23) in which an Escherichia coli expression plasmid codes for a 17-aa N-terminal histidine tail that contains an engineered thrombin cleavage site (24). Compared with the previous protocol (23), some modifications were introduced in the purification procedure: the expression of prion protein in Luria broth medium was induced at 30°C and at an OD600 of 0.6–1.0; the soluble protein fraction obtained after harvesting was added to 20 ml of nickel-nitrilotriacetic acid agarose resin and was stirred for 1 h at room temperature; to prevent unspecific nitrilotriacetic acid-binding of proteins devoid of histidine tails, 5 mM imidazole was added to the buffers during the purification of hPrP(121–230) and hPrP(90–230), and 10 mM imidazole for hPrP(23–230); oxidative refolding and imidazole elution were repeated up to five times to obtain additional batches of soluble prion protein, where each cycle was followed by a 40-ml washing step; the thrombin cleavage reaction was carried out at room temperature overnight in 5 mM Tris⋅HCl buffer at pH 8.3; the thrombin concentration was 1 unit/μmol of protein for hPrP(23–230) and hPrP(90–230), and 2 or 3 units/μmol of protein for hPrP(121–230); the pH of the buffer solution during anion/cation exchange was adjusted so that it was 1.3 units above/below the isoelectric point of the respective protein.
Concentrated protein solutions for NMR spectroscopy were obtained by using Ultrafree-15 Centrifugal Filter Devices (Millipore). The Bruker (Fällanden, Switzerland) DRX 600 and DRX 750 spectrometers used for this study are equipped with 5-mm z-axis gradient/triple-resonance probeheads. All nuclear Overhauser enhancement (NOE) data were recorded at 750 MHz, the other experiments at 600 MHz. The 1H, 15N, and 13C chemical shifts are relative to 2,2-dimethyl-2-silapentane-5-sulfonate, sodium salt. For data processing and spectral analysis, we used the programs prosa (25) and xeasy (26), respectively.
Hydrogen–deuterium exchange rates of amide protons were obtained by measuring a series of two-dimensional [15N,1H]correlation spectroscopy spectra immediately after dissolving the lyophilized protein in D2O buffer. Each experiment was recorded for 15 min, and the study was pursued for 3 days. The decay curves of the volume integrals of the cross peaks were fitted to single exponential decays. Protection factors were calculated, taking into account the sequence effects on the random coil exchange rates (27).
Steady-state 15N{1H}-NOEs were measured following ref. 28, with a recovery delay of 3 s and a proton saturation period of 3 s, which was achieved by applying a cascade of 120-degree pulses in 10-ms intervals. The reference spectrum was recorded with a recovery delay of 6 s. The pulse sequence used for measurements of T1 of 15N has been adapted from ref. 29 by replacing the 1HN-selective 180-degree pulses during the 15N relaxation delay by 180-degree hard pulses to minimize the influence of the 1H carrier position on the suppression of the cross-correlation between 1H–15N dipolar interaction and 15N chemical shift anisotropy (30). The T1 relaxation delays were 31, 65, 102, 144, 190, 242, 302, 373, 460, 573, 731, 1,002, and 1,950 ms. Measurement of T1ρ of 15N was based on ref. 31, using a continuous spin-lock with a power of 1.45 kHz to suppress the line broadening contributions from chemical or conformational exchange (32), and a cascade of 180-degree 1H hard pulses with a 2-ms interval to suppress the cross-correlation between 1H–15N dipolar interaction and 15N chemical shift anisotropy (30). The relaxation delays used were 7, 15, 24, 33, 44, 56, 70, 86, 106, 132, 168, 231, and 450 ms. For all relaxation measurements, we used t1,max = 87.7 ms, t2,max = 142.5 ms, and a time domain data size of 128 × 1,024 complex points, and the data were analyzed with the program dasha (33).
For the structure determination of the polypeptide segment 121–230 in all three hPrP polypeptides, we used the tools of the program dyana (34), and in particular the module found was used to evaluate constraints on the torsion angles (35). The dyana conformers used to represent the NMR structure were refined with the program opal (36), using the amber force field (37), and were analyzed with the program molmol (38).
Results and Discussion
The following hPrP polypeptides were prepared for the present study: unlabeled, uniformly 15N-labeled, uniformly 13C/15N-labeled and 10% 13C-labeled hPrP(23–230), unlabeled, uniformly 15N-labeled and uniformly 13C/15N-labeled hPrP(90–230) and hPrP(121–230). This array of constructs enabled NMR structure determinations of the three proteins and investigations of possible influences of the overall chain length on the three-dimensional structure. The NMR measurements were all recorded in 1–2 mM protein solutions in 90% H2O/10% D2O or 99.9% D2O containing 10 mM sodium acetate and 0.05% sodium azide at pH 4.5 and 20°C.
The NMR structure of the intact recombinant human prion protein, hPrP(23–230), contains a globular domain that extends approximately from residues 125–228, a flexibly extended N-terminal tail of residues 23–124, and a short flexible chain end of residues 229–230 (Fig. 1a), which is similar to the previously described structure of mPrP(23–231) (14, 15) and the characterization of shPrP(29–231) (16). These global features are qualitatively manifested in the small dispersion of the 1H chemical shifts and negative values of the 15N{1H}-NOEs for the residues 23–124, as described in detail for mPrP(23–231) (14), which contrasts with the fact that the corresponding parameters for the residues 125–228 have typical values for a globular protein of the size of hPrP(23–230). In the remainder of this paper, we focus on the structure of the globular domain in the aforementioned constructs, and on comparisons with the globular domains of mPrP and shPrP.
Figure 1.

(a) Cartoon of the three-dimensional structure of the intact human prion protein, hPrP(23–230). The helices are orange, the β-strands cyan, the segments with nonregular secondary structure within the C-terminal domain yellow, and the flexibly disordered “tail” of residues 23–121 is represented by yellow dots. (b) Stereoview of an all-heavy atom presentation of the globular domain, with residues 125–228, in hPrP(23–230) in the same orientation as in a. The backbone is shown as a gray spline function through the Cα positions, hydrophobic side chains are yellow, and polar and charged side chains are orange. The figures were prepared with the program molmol (38).
A superposition for best fit of the backbone atoms N, Cα and C′ of residues 125–228 of hPrP(23–230), hPrP(90–230), and hPrP(121–230) (Fig. 2) shows that the three-dimensional structure of the domain in the intact protein is very similar to that in the two C-terminal fragments. The regular secondary structure elements coincide identically in the three structures, with the residues 128–131 forming the β-strand 1, 144–154 the α-helix 1, 161–164 the β-strand 2, 173–194 the α-helix 2, and 200–228 the α-helix 3. As in mPrP(121–231) (11), there is a hydrogen bond from the amide proton of Met134 to the carbonyl oxygen of Asn159, which is reminiscent of an irregular, β-bulge-type elongation of the β-sheet toward the helix 1. The three proteins have nearly identical side chain conformations of the hydrophobic amino acids and the single disulfide bond Cys179–Cys214 (in Fig. 1b, only the all-atom presentation of one protein is shown, to prevent overcrowding.) In all three proteins, most of the regular secondary structures are well defined, with small backbone displacements. Increased disorder is seen in the loop of residues 167–171 between β-strand 2 and helix 2, at the end of helix 2 and the following loop, and for the last two turns of helix 3 (Fig. 2).
Figure 2.
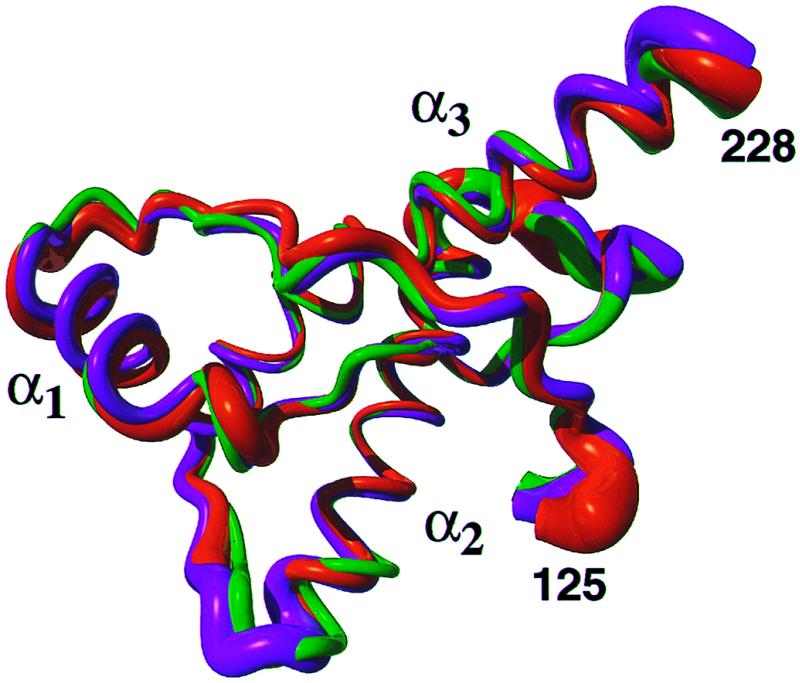
Comparison of the mean NMR structures of the polypeptide segments with residues 125 to 228 in hPrP(23–230) (green), hPrP(90–230) (orange), and hPrP(121–230) (violet). A spline function was drawn through the Cα positions. The variable radius of the cylindrical rods is proportional to the mean global backbone displacement per residue (39), as evaluated after superposition for best fit of the atoms N, Cα, and C′ of the residues 125–228 in the 20 energy-minimized conformers used to represent the solution structure.
Resonance Assignments and Structure Determination.
For the resonance assignments of the residues 121–230, which include the globular domain, we followed the standard triple-resonance strategy for 13C,15N-labeled proteins (40). For all three proteins, nearly complete assignments were obtained for the polypeptide backbone and the aliphatic CHn groups of the amino acid side chains, exceptions being the backbone amide protons of Tyr169, Ser170, Asn171, and Phe175, Hα and Hβ of Tyr169 and Phe175, and Hγ of Glu168. These resonances were not observed in any of the three polypeptides, presumably because of line broadening attributable to slow conformational exchange. The Xxx–Pro peptide bonds with the prolines 137, 158, and 165 are in the trans-conformation, as evidenced by the observation of strong dαδ NOEs (41). The sequence-specific assignments were independently confirmed by sequential and medium-range NOEs (41). At least one heteronuclear sequential scalar connectivity or a sequential NOE has been observed for each pair of neighboring residues, except for 168–169, 169–170, 170–171, and 174–175.
The methyl groups of the nine Val and two Leu were stereospecifically assigned with the use of biosynthetically directed fractional 13C labeling (42, 43). In the course of the structure calculations for hPrP(23–230), hPrP(90–230), and hPrP(121–230), additional stereospecific assignments were obtained for 31 βCH2, 32 γCH2, and 27 δCH2 groups, using the programs found (35) and glomsa (44) implemented in the dyana package (34).
Complete assignments were obtained for the 1H and 13C resonances of the 11 Tyr, 3 Phe, and 4 His rings, except for the ɛCH groups of His155 and His187, where the NOEs between the rings and the αCH–βCH2 moieties could not be detected. Among the labile side chain protons, the amide groups of all seven Asn and Gln residues and the ɛ-proton resonances of the eight Arg residues were assigned by intraresidual NOEs (41). Of the side chain hydroxyl protons of Ser, Thr, and Tyr, only the resonance of Thr183 could be observed and assigned.
Except for the central three octapeptide repeats, nearly complete resonance assignments were obtained for the residues 23–120 in the flexible tail of hPrP(23–230). Nonstandard approaches were applied, which have been described in detail elsewhere (45). The chemical shift lists for the three proteins have been deposited with the Biological Magnetic Resonance Bank, files 4402, 4434, and 4379.
In each of the three proteins, ≈2,500 NOE cross-peaks were assigned for the polypeptide segment of residues 121–230 and were used for the generation of the input of upper-limit distance constraints for the structure calculation. As supplementary conformational constraints, all residues with 13Cα chemical shifts deviating from the random coil values by more than 1.5 parts per million were subjected to the following bounds of torsion angles: −120° < Φ < −20° and −100° < Ψ < 0° for deviations >1.5 parts per million; −200° < Φ < −80° and 40° < Ψ < 220° for deviations < −1.5 parts per million (46). Using these constraints together with the intraresidual and sequential NOE distance constraints as input, the program found (35) yielded constraints on the dihedral angles φ, ψ, χ1, and χ2. Three upper and three lower distance limits were used to enforce the disulfide bond Cys179–Cys214 (47). For each of the three constructs, the input for the final structure calculations for the segment 121–230 contained ≈1,700 NOE upper distances limits and ≈440 dihedral angle constrains (Table 1). The final dyana calculation was performed with 100 randomized starting structures, and the 20 best dyana conformers, which are used to represent the NMR structure, were further energy-refined with the program opal. The high quality of the structure determinations is reflected by the average global rms deviation values relative to the mean coordinates of 0.7–0.8 Å for the backbone of residues 125–228, and 1.1–1.2 Å for all heavy atoms of the same polypeptide segment. For each of the three hPrP polypeptides, the atomic coordinates of the bundle of 20 conformers and of the best conformer have been deposited with the Protein Data Bank, accession codes 1QLX, 1QLZ, 1QM0, 1QM1, 1QM2, and 1QM3.
Table 1.
Collection of the input for the structure calculation and characterization of the energy-minimized NMR structures of the polypeptide segment 121–230 in different human PrP constructs
| Quantity* | hPrP(23–230) | hPrP(90–230) | hPrP(121–230) |
|---|---|---|---|
| NOE upper distance limits | 1,732 | 1,705 | 1,752 |
| Dihedral angle constraints | 429 | 453 | 436 |
| Residual target function value, Å2 | 0.25 ± 0.06 | 0.34 ± 0.08 | 0.39 ± 0.05 |
| Residual distance constraint violations | |||
| Number ≥ 0.1, Å | 0.3 ± 0.5 | 0.7 ± 0.9 | 0.1 ± 0.3 |
| Maximum, Å | 0.10 ± 0.01 | 0.11 ± 0.01 | 0.10 ± 0.00 |
| Residual dihedral angle constraint violations | |||
| Number ≥ 2.0 degrees | 1.8 ± 1.0 | 1.5 ± 1.0 | 5.6 ± 2.0 |
| Maximum, degrees | 2.9 ± 0.8 | 3.0 ± 1.2 | 3.7 ± 0.9 |
| amber energies, kcal/mol | |||
| Total | −4824 ± 85 | −4533 ± 79 | −4698 ± 83 |
| Van der Waals | −352 ± 16 | −315 ± 15 | −325 ± 16 |
| Electrostatic | −5398 ± 84 | −5164 ± 71 | −5283 ± 67 |
| rms deviation from ideal geometry | |||
| Bond lengths, Å | 0.0084 ± 0.0002 | 0.0089 ± 0.0002 | 0.0084 ± 0.0003 |
| Bond angles, degrees | 2.25 ± 0.04 | 2.41 ± 0.04 | 2.29 ± 0.04 |
| rms deviation to the averaged coordinates, Å | |||
| N, Cα, C′ (125–228) | 0.65 ± 0.10 | 0.79 ± 0.11 | 0.81 ± 0.11 |
| All heavy atoms (125–228) | 1.06 ± 0.09 | 1.27 ± 0.10 | 1.26 ± 0.13 |
| N, Cα, C′ of regular secondary structures† | 0.51 ± 0.12 | 0.60 ± 0.12 | 0.68 ± 0.13 |
Except for the top two entries, the data characterize the group of 20 conformers that is used to represent the NMR structure; the mean value and the standard deviation are given.
† Secondary structure elements are formed by residues 128–131 (β-strand 1), 144–154 (α-helix 1), 161–164 (β-strand 2), 173–194 (α-helix 2), and 200–228 (α-helix 3).
Conformational Equilibria in the Globular Domain of hPrP.
In the preceding sections of the text, in Table 1, and in Figs. 1 and 2, we evaluated the data on the globular domain in hPrP that are obtained with the standard protocol for NMR structure determination and that correspond to a static description of the molecule as it would, in a qualitative sense, also be obtained by x-ray crystallography (48). Here, we now make further use of the fact that some of the NMR parameters collected during the structure determination undergo different spatial and temporal averaging in a dynamic molecular structure, and that additional NMR experiments can provide supplementary data on conformational equilibria and on the rate processes that lead to transitions between the equilibrium states. The focus of the discussion is on the polypeptide segments that show reduced precision of the structure determination: i.e., the C-terminal parts of the helices 2 and 3, and the loop of residues 167–171 between the β-strand 2 and the helix 2.
In an α-helix, the medium-range NOE distance constraints dαN (i, i + 3), dαN (i, i + 4), and dαβ (i, i + 3) have a dominant weight in the structure calculation (41). Because of the dependence of the NOE intensity on the inverse sixth power of the distance d, only the folded forms of a polypeptide, with short values of d, contribute significantly to the NOE, so that even in the presence of conformational equilibria only one structure type is usually obtained in a standard structure determination (41). In contrast, the differences between observed and random coil 13Cα chemical shifts, Δδ(13Cα), are qualitatively related to the population of regular secondary structures (46, 49, 50). The Fig. 3b shows that, although for all 13Cα atoms located within the α-helices the resonances are shifted downfield relative to the random coil shifts, the smaller values of Δδ(13Cα) for all residues in the C-terminal two turns of the helices 2 and 3 indicate that, in these segments, the α-helical structure is in equilibrium with unfolded forms of the polypeptide. This conclusion from the 13Cα shifts coincides with the amide proton exchange rates measured by using two-dimensional [15N,1H]-correlation spectroscopy (Fig. 3a). Most of the amide protons in the hydrogen bonds of the regular secondary structures are measurably protected against exchange, the exceptions being residues 187–194 in helix 2 and residues 225–228 in helix 3. In helix 3, one observes further a gradual decrease in amide proton protection from residues 219–224.
Figure 3.

(a) Logarithmic plot of the amide proton exchange protection factors (PF) of residues 121–230 in hPrP(23–230) versus the sequence. Hydrogen–deuterium exchange was measured at 20°C in 99.9% D2O containing 10 mM sodium acetate and 0.05% sodium azide at pH 4.5. (b) 13Cα chemical shift differences, Δδ(13Cα), between hPrP(23–230) and the random coil shifts (51). (c) Δδ(13Cα) between hPrP(23–230) and hPrP(121–230) at pH 4.5 (Δδ = δ[hPrP(23–230)] − δ[hPrP(121–230)]). The locations of the regular secondary structure elements are given in a.
In an attempt to correlate the conformational equilibria manifested by Fig. 3 a and b with intramolecular rate processes, we measured the heteronuclear 15N{1H}-NOEs, the longitudinal relaxation time (T1), and the relaxation time in the rotating frame (T1ρ). For hPrP(121–230), these parameters show a uniform distribution over most of the amino acid sequence, with typical values for a globular protein with the size of PrP. Only for the residues 191–198, which form the C-terminal end of helix 2 and the subsequent loop, slightly decreased 15N{1H}-NOEs and increased T1ρ values (data not shown) indicate somewhat increased flexibility. Near the disordered loop of residues 167–171 (Fig. 2), the segment 165–168 displays 15N{1H}-NOEs close to the mean values for the domain whereas for the residues 169–171 the NMR lines of the amide groups were not observed. The reduced precision of the structure determination of the polypeptide segment 167–171 (Fig. 2) thus seems to arise from slow exchange between two or more polypeptide backbone conformations in the millisecond time-range and concomitant line broadening, which limited the collection of conformational constraints.
The 13Cα chemical shifts provide indications for transient contacts between the flexibly disordered tail of PrP and the globular domain (Fig. 3c). The chemical shift differences between hPrP(23–230) and hPrP(121–230) are small but have the same sign for seven successive 13Cα atoms in the C-terminal part of helix 2, i.e., residues 187–193, and for eight successive residues in the C-terminal part of helix 3, i.e., 219–226. The observation on helix 2 is in agreement with NMR investigations of shPrP(29–231) and shPrP(90–231), where differences in 13Cα chemical shifts were found for the residues 187–193 (16) but were eventually attributed to small pH differences between the two protein samples (13). In the present study, the pH-values were carefully monitored, and pH variation can be excluded as the cause of the shifts in Fig. 3c. Because identical patterns of 13Cα shifts were obtained at three different protein concentrations between 0.l and 1 mM (data not shown), we conclude that the observed variations are attributable to transient intramolecular interactions of the globular domain with the flexible tail. The interaction sites coincide with the aforementioned disordered ends of the helices 2 and 3, which appear to be slightly stabilized in the presence of the intact tail. The implicated increase of helix population amounts to only a few percent (Fig. 3c), which would not be reliably observable, either in the amide proton exchange data or the amide proton–α-proton scalar coupling constants.
Comparison of hPrP, mPrP, and shPrP.
As expected from the high sequence identity between hPrP, mPrP, and shPrP, the three-dimensional structures of the C-terminal domain are very similar. Local differences between the backbone conformations of the three proteins are manifest in helix 3 and the nearby loop between the β-strand 2 and helix 2. In mPrP, the helix 3 ends at residue 219, and, after a “kink” in the segment 219–222, the chain forms a helix-like turn (11). In contrast, a straight, mostly regular α-helix is observed in hPrP (Fig. 1a) and shPrP (13). The helix 3 in hPrP contains a nearly continuous pattern of medium-range NOE cross peaks dαN(i, i + 3) and dαβ(i, i + 3) for the residues 217–221 (Fig. 4). Only for Tyr218, no medium-range NOE cross peak was observed. In mPrP(121–231), the corresponding medium-range NOE cross peaks are either absent or correspond to distances >5 Å, so that the polypeptide chain is not restricted to form an α-helical secondary structure for the residues 220–224 (41).
Figure 4.
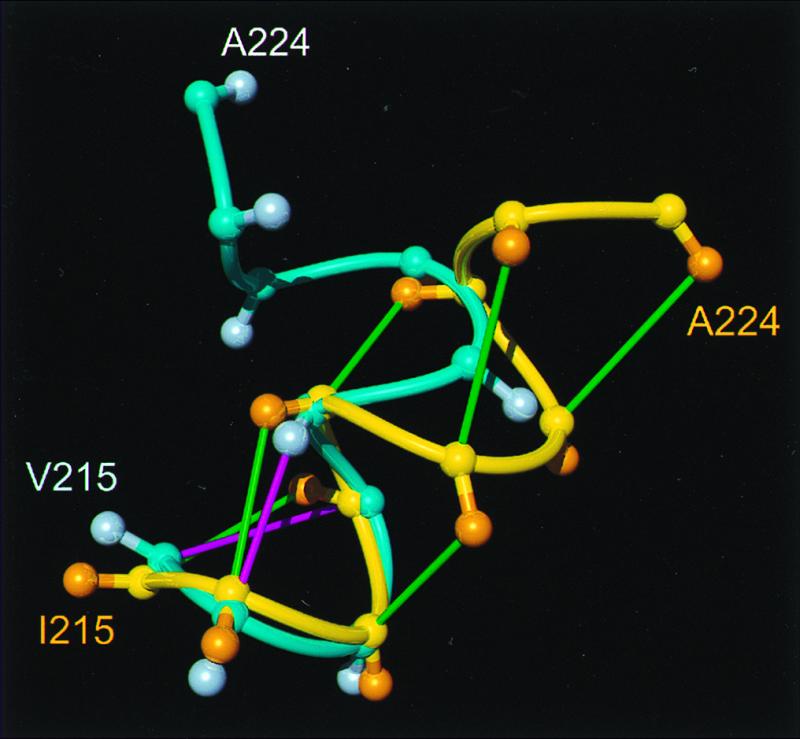
Comparison of helix 3 in hPrP(121–230) and mPrP(121–231), where the backbone of the polypeptide segment 215–224 is represented as a spline function drawn through the Cα positions. The figure results from a global superposition of the two proteins for best fit of the backbone atoms of the residues 144–154, 175–193, and 200–219, which correspond to the α-helices in mPrP. The following color code was used: yellow and orange, backbone and Cβ atoms of hPrP(121–230), respectively; cyan and light blue, backbone and Cβ atoms of mPrP(121–231), respectively; green, dαβ(i, i + 3) and dαN (i, i + 3) NOE distance constraints observed in hPrP; magenta, same types of NOE constraints observed for mPrP (11).
The loop 167–171 has been reported to be well defined in shPrP(90–231), where complete resonance assignments could be obtained for this segment (12, 13). For hPrP and mPrP, some resonance peaks of the loop have not been observed, either at pH 4.5 or at pH 7.0, and, because of the resulting scarcity of conformational constraints, the structure of this peptide segment is not precisely defined (Fig. 2).
The aforementioned species variations of the three-dimensional structure in the region of helix 3 and the loop 167–171 are intriguing because this surface area was previously suggested to be a binding epitope for a putative protein X, which would promote the transition from PrPC to PrPSc (19, 20, 22). It has also been known since the structure determination of mPrP(121–231) (9) that this surface area of the molecular structure is formed by two polypeptide segments with high frequency of species-dependent amino acid exchanges, which are otherwise well separated in the PrP polypeptide chain (21). To gain more direct insight into the apparent correlations between amino acid sequence and local three-dimensional structure in this part of the molecule, we have started structural studies of a selection of variant proteins with single amino acid exchanges relative to wild-type hPrP(121–230).
Acknowledgments
We thank Mrs. M. Geier and Mrs. E. Ulrich for the careful processing of the manuscript. Financial support was obtained from the Schweizerischer Nationalfonds (Projects 31.49047.96 and 438+050287).
Abbreviations
- NOE
nuclear Overhauser enhancement
- 15N{1H}-NOE
heteronuclear Overhauser enhancement of 15N after saturation of 1H
- PrP
prion protein
- PrPC
cellular form of PrP
- PrPSc
scrapie form of PrP
- hPrP(23–230)
complete polypeptide chain of the mature human PrP
- hPrP(90–230) and hPrP(121–230)
fragments of human PrP comprising, respectively, residues 90–230 and 121–230
- mPrP(121–231)
fragment of mouse PrP comprising residues 121–231
- shPrP(90–231) and shPrP(29–231)
fragments of Syrian hamster PrP comprising, respectively, residues 90–231 and 29–231
- TSE
transmissible spongiform encephalopathy
Footnotes
Data deposition: The atomic coordinates have been deposited in the Protein Data Bank, www.rcsb.org (PDB ID codes 1QLX, 1QLZ, 1QM0, 1QM1, 1QM2, and 1QM3).
References
- 1.Prusiner S B. Proc Natl Acad Sci USA. 1998;95:13363–13383. doi: 10.1073/pnas.95.23.13363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Weissmann C. FEBS Lett. 1996;389:3–11. doi: 10.1016/0014-5793(96)00610-2. [DOI] [PubMed] [Google Scholar]
- 3.Alper T, Cramp W A, Haig D A, Clarke M C. Nature (London) 1967;214:764–766. doi: 10.1038/214764a0. [DOI] [PubMed] [Google Scholar]
- 4.Griffith J S. Nature (London) 1967;215:1043–1044. doi: 10.1038/2151043a0. [DOI] [PubMed] [Google Scholar]
- 5.Prusiner S B. Trends Biochem Sci. 1996;21:482–487. doi: 10.1016/s0968-0004(96)10063-3. [DOI] [PubMed] [Google Scholar]
- 6.Oesch B, Westaway D, Wälchli M, McKinley M P, Kent S B H, Aebersold R, Barry R A, Tempst P, Teplow D B, Hood L E, et al. Cell. 1985;40:735–746. doi: 10.1016/0092-8674(85)90333-2. [DOI] [PubMed] [Google Scholar]
- 7.Meyer R K, McKinley M P, Bowman K A, Braunfeld M B, Barry R A, Prusiner S B. Proc Natl Acad Sci USA. 1986;83:2310–2314. doi: 10.1073/pnas.83.8.2310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pan K-M, Baldwin M, Nguyen J, Gasset M, Serban A, Groth D, Mehlhorn I, Huang Z, Fletterick R J, Cohen F E, Prusiner S B. Proc Natl Acad Sci USA. 1993;90:10962–10966. doi: 10.1073/pnas.90.23.10962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Riek R, Hornemann S, Wider G, Billeter M, Glockshuber R, Wüthrich K. Nature (London) 1996;382:180–182. doi: 10.1038/382180a0. [DOI] [PubMed] [Google Scholar]
- 10.Stahl N, Baldwin M A, Teplow D B, Hood L, Gibson B W, Burlingame A L, Prusiner S B. Biochemistry. 1993;32:1991–2002. doi: 10.1021/bi00059a016. [DOI] [PubMed] [Google Scholar]
- 11.Riek R, Wider G, Billeter M, Hornemann S, Glockshuber R, Wüthrich K. Proc Natl Acad Sci USA. 1998;95:11667–11672. doi: 10.1073/pnas.95.20.11667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.James T L, Liu H, Ulyanov N B, Farr-Jones S, Zhang H, Donne D G, Kaneko K, Groth D, Mehlhorn I, Prusiner S B, Cohen F E. Proc Natl Acad Sci USA. 1997;94:10086–10091. doi: 10.1073/pnas.94.19.10086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu H, Farr-Jones S, Ulyanov N B, Llinas M, Marqusee S, Groth D, Cohen F E, Prusiner S B, James T L. Biochemistry. 1999;38:5362–5377. doi: 10.1021/bi982878x. [DOI] [PubMed] [Google Scholar]
- 14.Riek R, Hornemann S, Wider G, Glockshuber R, Wüthrich K. FEBS Lett. 1997;413:282–288. doi: 10.1016/s0014-5793(97)00920-4. [DOI] [PubMed] [Google Scholar]
- 15.Riek R. Ph. D. thesis. Zürich: Eidgenössische Technische Hochschule; 1998. [Google Scholar]
- 16.Donne D G, Viles J H, Groth D, Mehlhorn I, James T L, Cohen F E, Prusiner S B, Wright P E, Dyson H J. Proc Natl Acad Sci USA. 1997;94:13452–13457. doi: 10.1073/pnas.94.25.13452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schätzl H M, Da Costa M, Taylor L, Cohen F E, Prusiner S B. J Mol Biol. 1995;245:362–374. doi: 10.1006/jmbi.1994.0030. [DOI] [PubMed] [Google Scholar]
- 18.Korth C, Stierli B, Streit P, Moser M, Schaller O, Fischer R, Schulz-Schaeffer W, Kretzschmar H, Raeber A, Braun U, et al. Nature (London) 1997;390:74–77. doi: 10.1038/36337. [DOI] [PubMed] [Google Scholar]
- 19.Telling G C, Scott M, Hsiao K K, Foster D, Yang S-L, Torchia M, Sidle K C L, Collinge J, DeArmond S J, Prusiner S B. Proc Natl Acad Sci USA. 1994;91:9936–9940. doi: 10.1073/pnas.91.21.9936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Telling G C, Scott M, Mastrianni J, Gabizon R, Torchia M, Cohen F E, DeArmond S J, Prusiner S B. Cell. 1995;83:79–90. doi: 10.1016/0092-8674(95)90236-8. [DOI] [PubMed] [Google Scholar]
- 21.Billeter M, Riek R, Wider G, Hornemann S, Glockshuber R, Wüthrich K. Proc Natl Acad Sci USA. 1997;94:7281–7285. doi: 10.1073/pnas.94.14.7281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kaneko K, Zulianello L, Scott M, Cooper C M, Wallace A C, James T L, Cohen F E, Prusiner S B. Proc Natl Acad Sci USA. 1997;94:10069–10074. doi: 10.1073/pnas.94.19.10069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zahn R, von Schroetter C, Wüthrich K. FEBS Lett. 1997;417:400–404. doi: 10.1016/s0014-5793(97)01330-6. [DOI] [PubMed] [Google Scholar]
- 24.Zahn R, Buckle A M, Perrett S, Johnson C M, Corrales F J, Golbik R, Fersht A R. Proc Natl Acad Sci USA. 1996;93:15024–15029. doi: 10.1073/pnas.93.26.15024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Güntert P, Dötsch V, Wider G, Wüthrich K. J Biomol NMR. 1992;2:619–629. [Google Scholar]
- 26.Bartels C, Xia T, Billeter M, Güntert P, Wüthrich K. J Biomol NMR. 1995;6:1–10. doi: 10.1007/BF00417486. [DOI] [PubMed] [Google Scholar]
- 27.Bai Y, Milne J S, Mayne L, Englander S W. Proteins. 1993;17:75–86. doi: 10.1002/prot.340170110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dayie K T, Wagner G. J Magn Reson A. 1994;111:121–126. [Google Scholar]
- 29.Farrow N A, Muhandiram R, Singer A U, Pascal S M, Kay C M, Gish G, Shoelson S E, Pawson T, Forman-Kay J D, Kay L E. Biochemistry. 1994;33:5984–6003. doi: 10.1021/bi00185a040. [DOI] [PubMed] [Google Scholar]
- 30.Palmer A G, Skelton N J, Chazin W J, Wright P E, Rance M. Mol Phys. 1992;75:699–711. [Google Scholar]
- 31.Peng J W, Thanabal V, Wagner G. J Magn Reson. 1991;94:82–100. [Google Scholar]
- 32.Szyperski T, Luginbühl P, Otting G, Güntert P, Wüthrich K. J Biomol NMR. 1993;3:151–164. doi: 10.1007/BF00178259. [DOI] [PubMed] [Google Scholar]
- 33.Orekhov V Yu, Orekhov V Yu, Nolde D E, Golovanov A P, Korzhnev D M, Arseniev A S. Appl Magn Reson. 1995;9:581–588. [Google Scholar]
- 34.Güntert P, Mumenthaler C, Wüthrich K. J Mol Biol. 1997;273:283–298. doi: 10.1006/jmbi.1997.1284. [DOI] [PubMed] [Google Scholar]
- 35.Güntert P, Billeter M, Ohlenschläger O, Brown L R, Wüthrich K. J Biomol NMR. 1998;12:543–548. doi: 10.1023/A:1008391403193. [DOI] [PubMed] [Google Scholar]
- 36.Luginbühl P, Güntert P, Billeter M, Wüthrich K. J Biomol NMR. 1996;8:136–146. doi: 10.1007/BF00211160. [DOI] [PubMed] [Google Scholar]
- 37.Cornell W D, Cieplak P, Bayly C I, Gould I R, Merz K M, Ferguson D M, Spellmeyer D C, Fox T, Caldwell J W, Kollman P A. J Am Chem Soc. 1995;117:5179–5197. [Google Scholar]
- 38.Koradi R, Billeter M, Wüthrich K. J Mol Graphics. 1996;14:51–55. doi: 10.1016/0263-7855(96)00009-4. [DOI] [PubMed] [Google Scholar]
- 39.Billeter M, Kline A D, Braun W, Huber R, Wüthrich K. J Mol Biol. 1989;206:677–687. doi: 10.1016/0022-2836(89)90575-5. [DOI] [PubMed] [Google Scholar]
- 40.Bax A, Grzesiek S. Acc Chem Res. 1993;26:131–138. [Google Scholar]
- 41.Wüthrich K. NMR of Proteins and Nucleic Acids. New York: Wiley; 1986. [Google Scholar]
- 42.Senn H, Werner B, Messerle B A, Weber C, Traber R, Wüthrich K. FEBS Lett. 1989;249:113–118. [Google Scholar]
- 43.Neri D, Szyperski T, Otting G, Senn H, Wüthrich K. Biochemistry. 1989;28:7510–7516. doi: 10.1021/bi00445a003. [DOI] [PubMed] [Google Scholar]
- 44.Güntert P, Braun W, Wüthrich K. J Mol Biol. 1991;217:517–530. doi: 10.1016/0022-2836(91)90754-t. [DOI] [PubMed] [Google Scholar]
- 45.Liu, A., Riek, R., Wider, G., von Schroetter, C., Zahn, R. & Wüthrich, K. (2000) J. Biomol. NMR, in press. [DOI] [PubMed]
- 46.Luginbühl P, Szyperski T, Wüthrich K. J Magn Reson B. 1995;109:229–233. [Google Scholar]
- 47.Williamson M P, Havel T F, Wüthrich K. J Mol Biol. 1985;182:295–315. doi: 10.1016/0022-2836(85)90347-x. [DOI] [PubMed] [Google Scholar]
- 48.Wüthrich K. Acta Crystallogr D. 1995;51:249–270. doi: 10.1107/S0907444994010188. [DOI] [PubMed] [Google Scholar]
- 49.Spera S, Bax A. J Am Chem Soc. 1991;113:5490–5492. [Google Scholar]
- 50.Wishart D S, Sykes B D. J Biomol NMR. 1994;4:171–180. doi: 10.1007/BF00175245. [DOI] [PubMed] [Google Scholar]
- 51.Richarz R, Wüthrich K. Biopolymers. 1978;17:2133–2141. [Google Scholar]