

Preface
Genome maintenance is a constant problem in all cells and a coordinated response to DNA damage is required to maintain cellular viability and prevent disease. The ATR and ATM protein kinases are master regulators of the DNA damage response, signaling to control cell cycle transitions, DNA replication, DNA repair, and apoptosis. Recent studies have provided insights into the mechanisms controlling ATR activation, helped to explain the overlapping but non-redundant activities of ATR and ATM in DNA damage signaling, and clarified the critical functions of ATR in maintaining genome integrity.
Introduction
All cells have elaborate mechanisms to maintain their genomes. DNA can be damaged during replication, by reactive metabolic byproducts as well as environmental mutagens. Responding to and repairing DNA damage is critical for cell viability and disease prevention.
The DNA damage response (DDR) is a signal transduction pathway that coordinates cell cycle transitions, DNA replication, DNA repair and apoptosis. The major regulators of the DDR are the phosphoinositide 3-kinase related protein kinases (PIKKs), including ataxia-telangiectasia mutated (ATM) and ATM and Rad3 related (ATR). ATM and ATR share many biochemical and functional similarities. Both are large kinases with significant sequence homology and a strong preference to phosphorylate serine or threonine residues that are followed by glutamine. Both target an overlapping set of substrates that promote cell cycle arrest and DNA repair. However, ATR is essential for the viability of replicating human and mouse cells, whereas ATM is not1–3. ATM functions in response to rare occurrences of double strand breaks. By contrast, ATR is activated during every S-phase to regulate the firing of replication origins, the repair of damaged replication forks and to prevent the premature onset of mitosis4, 5 (Fig. 1).
Figure 1. Simple models for ATR and ATM activation.

(A) The ATR-ATRIP complex and the 9-1-1 complex are recruited to the ssDNA-5′ primer junction independently. RPA binds ATRIP and directs the Rad17-RFC complex to load the 9-1-1 checkpoint clamp at the 5′ primer junction. Loading of 9-1-1 brings the ATR activator TopBP1 to the damage site through an interaction involving two BRCT domains of TopBP1 and the phosphorylated C-terminal tail of Rad9 (see text). TopBP1 binds and activates ATR in an ATRIP-dependent manner, leading to phosphorylation of the downstream kinase Chk1 and other ATR effectors. In response to DNA damage or replication stress, ATR and its effectors ultimately slow origin firing and induce cell cycle arrest as well as stabilize and restart stalled replication forks. (B) Formation of a double-stranded DNA end leads to recruitment of the MRN complex and dissociation of the dimeric, inactive form of ATM to a monomeric, phosphorylated form. This monomeric form of ATM binds the MRN complex at the DSB, and is further activated by the DNA and MRN complex. Activated ATM then phosphorylates the carboxy terminal tail of the histone variant, H2AX. Phosphorylated H2AX (γ-H2AX) binds to MDC1 through the BRCT domains of MDC1, leading to recruitment of additional ATM/MRN complexes and further H2AX phosphorylation. The activated ATM also phosphorylates downstream targets, among which are Chk2. Phosphorylation of these proteins leads to cell cycle arrest, inhibition of oirgin firing in S phase and double-strand break repair.
Mutations in ATM predispose carriers to cancer and are found in approximately 0.5–1% of the population6, 7. People with mutations in both alleles of ATM suffer from the neurodegenerative and cancer predisposition disorder ataxia-telangiectasia 8. Mutations in ATR are rare and probably only compatible with viability when heterozygous or hypomorphic. While the only clear link between ATR gene mutation and disease is in a few patients with the rare Seckel syndrome (characterized by growth retardation and microcephaly)9, disruptions in the ATR pathway do cause genomic instability, and ATR is activated by most cancer chemotherapies. Furthermore, ATR signaling is a promising target for cancer drug development10, 11. This review will focus on ATR signaling in the DNA damage response, and compare and contrast it with the more specialized role of ATM.
Mechanisms of ATR Activation
The broad functions and physiological importance of ATR derive in large part from recognizing the signals that lead to its activation versus that of ATM. Thus, we will address the mechanism of activation in some detail.
Recognizing DNA damage
Although ATR is activated in response to many different types of DNA damage including double strand breaks (DSB), base adducts, crosslinks, as well as replication stress, a single DNA structure may be responsible. The majority of data suggest that this structure contains single-stranded DNA (ssDNA)12, 13. Replication protein A (RPA) coats most forms of ssDNA in the cell, including the ssDNA formed during DNA replication and DNA repair14. Consistent with this idea, a mutation in the large subunit of RPA in S. cerevisiae (rfa1-t11) supports replication but is partially compromised in DNA damage responses15, 16. Furthermore, depletion of RPA from Xenopus egg extracts reduces the association of ATR with chromatin12. Finally, RPA-coated ssDNA (hereafter RPA-ssDNA) is important to localize ATR to sites of DNA damage in both human and budding yeast systems13, although there are some indications of alternative mechanisms17–19.
ATR recognition of RPA-coated ssDNA depends upon another protein, ATRIP (ATR-interacting protein)1. The stability of ATR and ATRIP are linked and their association is not regulated. There are no known differences in the phenotypes that result upon loss of ATR or ATRIP in any organism, suggesting ATRIP should be considered an obligate subunit of the ATR kinase. Biochemical studies indicate that ATRIP binds RPA directly via evolutionarily conserved binding surfaces20. The primary interaction involves an acidic alpha-helix in ATRIP that binds in the basic cleft of the N-terminal OB-fold domain of the large RPA subunit20. The rfa1-t11 mutation causes a charge reversal mutation within this basic cleft and is impaired in binding and recruiting Ddc2/ATRIP to a double-strand break site in S. cerevisiae13.
Although RPA-coated ssDNA may be sufficient to localize the ATR-ATRIP complex, it is not sufficient for ATR activation21–24. ATR signaling is dependent on co-localization of the ATR-ATRIP complex with the Rad9-Rad1-Hus1 (9-1-1) complex, a heterotrimeric ring-shaped molecule related in structure and sequence to the replicative sliding clamp, PCNA25. Like PCNA, the 9-1-1 complex is loaded onto primer-template junctions in an ATP-dependent reaction that involves a damage-specific clamp loader26–28. In other words, the 9-1-1 complex recognizes a DNA end that is adjacent to a stretch of RPA-coated ssDNA. The presence of RPA is critical for this reaction as well, and imparts specificity in loading, creating a preference for the 5′-primer end26, 29. Indeed, the rfa1-t11 mutant is defective in loading the S. cerevisiae 9-1-1 complex30, suggesting RPA-coated ssDNA may be recognized by multiple checkpoint sensors.
So in most cases the structure that causes activation of the ATR checkpoint is single-stranded DNA bound to RPA with an adjacent stretch of double stranded DNA presenting a 5′ junction. Consistent with this idea, in Xenopus egg extracts ssDNA does not activate ATR but primed ssDNA with a free 5′-primer end is sufficient to promote ATR signaling24. This structure is generated during DNA replication when a polymerase stalls as a consequence of helicase and polymerase uncoupling, and in fact, is required for checkpoint activation23, 31, 32. It is also possible for the primed ssDNA structure to form during end resection at double-strand breaks, at telomeres, and even during nucleotide excision repair (Fig. 2).
Figure 2. A common DNA structure activates ATR.

ssDNA gaps with a 5′ primer end are formed during nucleic acid metabolism. Most noteably, helicase/polymerase uncoupling occurs at replication forks upon encountering lesions that stall the polymerase but not the more permissive helicase. A lesion on the lagging strand would immediately leave a gap with a 5′ primer end. Lesions on the leading strand would require repriming to generate the gapped structure. End resection of DSBs and intermediates in nucleotide excision DNA repair also form the ATR activating structure. Finally, telomere erosion such that the normal telomere capping mechanism is removed produces a recessed 5′ end adjacent to telomeric ssDNA.
TOPBP1: an ATR activator
How does the 9-1-1 complex stimulate ATR signaling? In Saccharomyces cerevisiae, the 9-1-1 complex has been reported to directly activate the ATR kinase33. The C-terminal tail of Ddc1 (the yeast orthologue of Rad9) stimulates Mec1/ATR (the yeast orthologue of ATR) under certain in vitro conditions. However, it is unclear whether this is the activation mechanism in yeast cells, and there is no evidence that this direct activation occurs in other organisms. Instead, the 9-1-1 complex brings to ATR a critical activator, TOPBP134–36. TOPBP1 is a BRCT domain-containing protein necessary for ATR activation that can dramatically stimulate ATR activity in vitro37. 9-1-1-mediated recruitment of TOPBP1 relies on a C-terminal tail in RAD9, which is phosphorylated at residue S387 (S373 in xRAD9)38. This phosphorylation creates a recognition site for BRCT domains I and II in TOPBP1, thereby bringing TOPBP1 to ATR34, 35.
TOPBP1 contains an ATR activation domain (AD) located between BRCT domains VI and VII, which interacts with and activates ATR-ATRIP complexes in vitro37. Indeed, overexpression of this domain by itself activates ATR-ATRIP37, and fusion of it to PCNA or histone H2B bypasses the need for the RAD17 clamp loader35. Although it may be an oversimplification to say that the only role of the 9-1-1 clamp is to bring TOPBP1 to ATR, these observations do suggest that it is the primary role in checkpoint activation. It is interesting to note, however, that the phenotype resulting from loss of ATR is more severe than that resulting from the loss of HUS1 or RAD91–3, 39, 40. This suggests that the 9-1-1 complex is not needed for all functions of ATR.
The mechanism by which TOPBP1 binding activates ATR is poorly defined. The primary binding site for the TOPBP1 AD on the ATR complex is within ATRIP, and mutations in this ATRIP region block activation41. In addition, activation involves amino acids in ATR located between the ATR kinase domain and the FATC domain41 (Box 1). Mutations in this region, recently named the ATR PRD (PIK-kinase regulatory domain), have no effect on the basal activity of ATR but prevent its activation by TOPBP1, cause checkpoint defects, and mimic a complete deletion of ATR in human somatic cells41. Interestingly, this region of ATM and mTOR (another member of the PIKK family) is targeted for post-translational modifications that regulate their activity42, 43. The PRD may be part of the conserved architecture of PIK family members that allows for the regulation of these kinases by distinct cellular events (Box 1).
Box 1. Functional domains of PIK Kinases
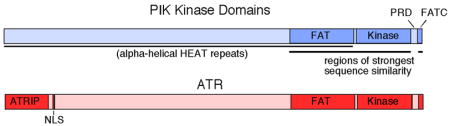
The PIK kinase family contains five active kinases, ATR, ATM, DNA-PKcs, mTOR, and SMG-1. Unfortunately, the lack of high-resolution structural information has prevented a mechanistic understanding of PIK kinase regulation. Low resolution, electron microscopy structures of DNA-PKcs and ATM show that these proteins fold into large globular structures suggesting that distant regions in the primary sequence may come together in space165–169. However, these structures do not yet provide enough information to recognize where individual domains reside or how the kinases are regulated. Nonetheless, the domain architecture of the PIK kinases is similar (see figure), suggesting that different regulatory inputs may alter the kinases in similar ways to cause activation. The kinase domain is flanked by two regions of high sequence similarity named the FAT and FATC domains in all the kinases. The FAT domain is part of a long N-terminal region predicted to fold into an extended alpha-helical HEAT repeat structure170. The small FATC domain of at least some of the kinases is interchangeable171.
The PIKK regions that lack sequence similarity among paralogs but retain sequence similarities in orthologs might provide opportunities for unique regulatory mechanisms. Indeed, the poorly conserved N-terminal regions of the kinases mediate interactions with the protein cofactors such as ATRIP, Nbs1, and Raptor54, 172–174. Furthermore, the PIK regulatory domain (PRD) between the kinase and FATC domains is poorly conserved between family members but highly conserved within orthologs in different organisms. This region is not essential for basal kinase activity but is a regulatory domain in at least ATM, mTOR, and ATR41–43.
Localization and the two-man rule for ATR activation
The localization of ATR and ATM to sites of DNA damage is a key step in the regulation of these kinases. Sustained colocalization of the ATR-ATRIP and 9-1-1-TOPBP1 complexes at the DNA damage site may increase their local concentration such that a relatively weak interaction between TOPBP1 and ATR-ATRIP is promoted allowing activation (Fig. 1A). In the case of ATM, localization of ATM to the break by the MRN complex and further anchoring of these proteins to this region by additional checkpoint proteins contributes significantly to the damage response44 (Fig. 1B). Tethering and concentrating the MRN complex in a small region of chromatin is sufficient to activate ATM in the absence of DNA damage45. Similarly, in S. cerevisiae, artificially colocalizing and concentrating the Ddc1/RAD9 and Ddc2/ATRIP proteins can activate Mec1/ATR in the absence of any DNA damage46. Finally, overexpression of the TOPBP1 AD can cause phosphorylation of at least some ATR substrates20, 37 and induce cellular senescence47, bypassing the requirement for DNA damage. Thus, the double strand break and primed ssDNA structures may not be strictly required for checkpoint activation and may primarily serve as a scaffold to colocalize and concentrate the proteins needed for ATM or ATR activation respectively. Indeed, RPA-coated ssDNA does not increase the specific activity of ATR as is observed with TOPBP120, 48.
Recruitment of the 9-1-1-TOPBP1 and ATR-ATRIP complexes to sites of DNA damage or stalled replication forks are largely independent events49–53. This independent recruitment may be an important design element in the signaling pathway because it provides a mechanism to ensure that the checkpoint is only activated when needed by creating a molecular version of a “two-man rule”. (A two-man rule is a control system that prevents one person from performing an act such as launching a weapon). By requiring two receptors to sense the problem, inappropriate launching of the checkpoint may be prevented.
So how does this two-man mechanism work in checkpoint activation? We can propose at least two models. First, the requirement for two receptors may enforce a requirement for two signals. Each of the signals may have a chance of being present by itself in normal DNA metabolism, but having both signals simultaneously would be an unusual circumstance that requires checkpoint intervention. However, in some ways this is not a satisfactory explanation. To be most effective the signals should be independent of each other. ssDNA and the 5′ junction do not satisfy this requirement; for example, lagging strand replication produces both a 5′ junction and ssDNA. It is possible to hypothesize some fixes to this problem. Other proteins within the replisome may mask one or more of the signals, or the nature of the 5′ junction during lagging strand replication might not support 9-1-1 loading. (An RNA primer forms the 5′ junction, and it is not clear whether the RNA-DNA duplex is equivalent to a DNA-DNA duplex for 9-1-1 loading24). A second problem with the two-signal model is that loading of both ATR-ATRIP and the 9-1-1 complex require part of the same signal–RPA13, 26–29, 54.
A second model might be that the two receptors actually recognize one signal. If there is one signal with two necessary receptors, then this may force a situation in which the amount of signal becomes critical. For example, the more ssDNA-RPA the more chance both ATR-ATRIP and 9-1-1 will be recruited to the same site, bringing the TOPBP1 activator to ATR. Indeed, larger gaps are more efficient at generating checkpoint activation24. Of course, the situation is likely to be more complex than either of these alternative scenarios and could be a combination of both.
Post-translational modifications
Post-translational modifications may regulate the activity of PIKKs as well as localization. Indeed, activation of ATM involves autophosphorylation, an event that may help convert an inactive ATM dimer into active monomers55. Several phosphorylation sites have been mapped on ATR and ATRIP (Table 2), and like ATM, ATR-ATRIP also may form a higher order oligomeric complex56–62. However, as yet, none of the currently identified modifications has been demonstrated to be a reliable indicator of ATR activation, and there is no data to indicate that the oligomerization status of ATR-ATRIP is regulated. A clear and early mark of ATR activation would be enormously useful for the field, as current approaches to monitoring ATR signaling rely on analysis of downstream substrates. Because the phosphorylation and activation of these substrates requires several additional steps, these are indirect readouts.
Post-translationally modified kinases can often be purified in an activated state. Indeed, ATM isolated from irradiated cells is more activate than the ATM from untreated cells63, 64. However, there has been little success in isolating an activated ATR kinase. The Dunphy lab has shown that an activated form of ATR can be found on a DNA template containing single- and double-stranded regions65. This activation is salt sensitive, but is not sensitive to treatment with micrococcal nuclease, suggesting it relies upon a protein activator that is lost at higher salt concentrations. It seems likely that the activation observed on these DNA templates results from the ability of the template to bring in TOPBP1. Thus, there is no convincing evidence that a pure, activated form of ATR can be obtained. These observations may indicate that ATR activation is dependent upon continued stimulation by TOPBP1.
Signal amplification
Another theme in kinase signaling is feedback/amplification. Auto-amplification is important in the ATM response and is mediated through ATM-dependent phosphorylation of H2AX, a histone variant that is phosphorylated following DNA damage 44. MDC1, a BRCT-domain containing protein needed for ATM activation, binds to γH2AX (the phosphorylated form of H2AX) through its tandem BRCT domains and brings more ATM to the DNA damage site (Fig. 1B). In the ATR pathway, the interaction between ATR and TOPBP1 may provide a point for signal auto-amplification. Evidence for this comes from an experiment done in X. laevis egg extracts using DNA structures containing double-stranded DNA ends (annealed homopolymers of (dA)70 and (dT)70). These templates activate ATR in an ATM-dependent and RPA-independent manner66, and this activation requires the phosphorylation of TOPBP1 by ATM on residue S1131, an event that promotes the interaction of TOPBP1 with ATR-ATRIP. While it is unclear what types of DNA damage the (dA)70-(dT)70 polymers mimic in vivo, the phosphorylation of TOPBP1 by ATM could facilitate ATR activation at double-strand breaks induced by ionizing radiation (IR) and other agents. This induced interaction between TOPBP1 and ATR-ATRIP may reflect an important component of the ATR signaling response that helps to enhance ATR activation after replication stress as well. Residue S1131 of X. laevis TOPBP1 is phosphorylated after replication stress, presumably by ATR, and under physiological conditions or low levels of replication stress, this may provide positive feedback that enhances ATR activation.
Common themes and outstanding questions
Common regulatory themes for all of the PIK kinases continue to emerge, suggesting that we should look to translate discoveries about the regulation of one of the kinases into our research on the others (Box 2). Although a nice model for ATR activation has emerged from the current body of work, the picture is undoubtedly more complex. For example, ATR may also respond to stalled transcriptional complexes67, 68. Similarly, our understanding of TOPBP1 regulation and function is incomplete. TOPBP1 BRCT domains I and II bind phosphorylated RAD934, 35, but BRCT domain V is needed for the recruitment of TOPBP1 to nuclear foci69. In addition, there is evidence that BRCT domains VII and VIII are also important for checkpoint activation70 so the regulation of TOPBP1 may be complex. Finally, although TOPBP1 is needed for the phosphorylation of RAD1 by ATR in X. laevis egg extracts, the C-terminal tail of Rad9 is not, suggesting that the 9-1-1-TOPB1 interaction is not needed for all ATR-dependent phosphorylation events71
Box 2. Common Regulatory Themes of the PIK Family of Atypical Kinases
| Kinase | Partner | Nucleic Acid | Activator(s) | PTM Regulation |
|---|---|---|---|---|
| ATR | ATRIP | RPA-ssDNA | TOPBP1 | Phosphorylation |
| ATM | Nbs1 | DSB | Mre11/Rad50 and DNA | Phosphorylation and Acetylation |
| DNA-PKcs | Ku70/80 | DSB | Ku70/80 and DNA | Phosphorylation |
| SMG1 | UPF1 | mRNA | UPF2 and exon junction complex | ? |
| mTOR | Raptor/Rictor | - | mLST8, Rheb-GTP | Phosphorylation |
Since all PIK kinases share a similar domain architecture and sequence similarity, it might be expected that the mechanisms controlling their regulation would be similar. Indeed, there is increasing evidence that most if not all of these kinase are regulated by localization, a protein or protein/nucleic acid activator, and post-translational modifications (see Table).
DNA-PKcs, ATR, and ATM localize to DNA through their interactions with Ku70/80, ATRIP, and Nbs1 respectively13, 54, 175–179. SMG-1 localizes to mRNAs with premature termination codons to mediate non-sense mediated mRNA decay at least in part through an interaction with its major substrate UPF1180. mTOR also binds one of two protein cofactors (Raptor and Rictor) forming two signaling complexes mTORC1 and mTORC2 resepectively174, 181, 182. mTOR may signal from intracellular membranes, but it is unclear whether Raptor and Rictor regulate this localization. Each of the PIK kinases are also regulated by a protein or protein and nucleic acid activator (Mre11/Rad50 + DNA for ATM183, Ku + DNA for DNA-PKcs175, 176, TOPBP1 for ATR37, UPF2 and the exon-junction complex for SMG-1180, and mLST8 with Rheb-GTP for mTORC1184, 185). Post-translational modifications including phosphorylation and acetylation have been described on all of the kinases although in many cases the key sites and the mechanisms of regulation remain to be identified. Finally, there is at least one common regulator of all 5 PIK kinases as well as the related transformation/transcription domain-associated protein (TRRAP). Tel2 binds all six proteins and regulates their stability186. Common regulatory mechanisms for the PIK kinases suggests that advances in understanding the regulation of one of the kinases may be informative for the other family members.
In addition, several experimental observations do not yet fit the model that multiple lesions produce a single ATR-activating DNA structure. Several results indicate that there may be other means of activating ATR-ATRIP, which are secondary to or work with RPA-ssDNA. Surprisingly, deletion of the primary RPA-binding surface in human, X. laevis and S. cerevisiae ATRIP proteins yields only mild defects in ATR signaling20, 54, 59. The rfa1-t11 mutation (which alters the corresponding ATRIP-binding surface on RPA20) has no apparent effect on Ddc2/ATRIP recruitment to stalled replication forks and is largely proficient in supporting the replication checkoint30. Additional RPA-ssDNA interaction motifs on ATRIP72, direct DNA interactions, or even additional proteins may help explain these results. Indeed, in S. pombe, Cdc18 (the homologue of human CDC6, a protein required for initiation of DNA replication) anchors ATR-ATRIP to chromatin in the presence of replication stress, an event that is necessary for the long-term maintenance of checkpoint activation18. In human cells, a new checkpoint protein, Cep164, was shown to regulate ATR-ATRIP foci formation and signaling73, and mismatch repair proteins may recruit ATR some DNA adducts using an RPA-independent mechanism19. Even the lesion itself may contribute to activation. ATR can bind to UV-damaged DNA, and this DNA can stimulate ATR activity in a partially reconstituted system17, 48, 74. Finally, there are still a number of other proteins that have been implicated in ATR regulation through as yet undefined mechanisms including PP5, p18, and SNIP175–77. Some of these regulatory activities could help produce or maintain the checkpoint-activating nucleic acid structure78. Others may regulate ATR in response to specific DNA lesions. Fitting all of these observations into a unified ATR activation model will require much more research.
ATR Signaling
Once an active ATR complex is assembled at a DNA lesion or stalled replication fork, signaling to coordinate cell cycle, repair, and replication begins. The list of ATR substrates is rapidly expanding due to large-scale proteomic profiling methodologies79–82. The best studied is the serine-threonine kinase CHK1.
ATR activation of CHK1
CHK1 activation requires phosphorylation by ATR on S317 and S345, which appears to be a reliable indicator of CHK1 activation83–85. Unlike many checkpoint proteins, which act at the sites of DNA damage, CHK1 functions to signal DNA damage to the rest of the nucleus. Presumably, it is transiently localized to sites of DNA damage, where it is activated. Claspin, an “adaptor” or “mediator” protein found at the replication fork, is critical for this activation, functioning to bring ATR and CHK1 together86. Claspin interacts with CHK1 in a damage-dependent manner, and this interaction requires the phosphorylation of Claspin on at least two sites (S864 and S895 in xClaspin)87. Though this phosphorylation is ATR-dependent, the responsible kinase may not be ATR itself since neither serine resides in a consensus ATR phosphorylation site. Unlike TOPBP1, which is needed for the phosphorylation of a number of ATR effectors, it is unclear whether any other ATR substrates require Claspin for their phosphorylation88. Claspin binds to phosphorylated Rad17 (a component of the 9-1-1 clamp loader) and this interaction is important for sustaining CHK1 phosphorylation89. Since Rad17 phosphorylation is ATR-dependent90, this may provide another mechanism for signal amplification. In addition to Claspin, a second replication fork associated complex, Tim/Tipin, may also mediate activation of CHK1 by ATR91, 92.
Once phosphorylated, CHK1 is released from chromatin to phosphorylate its substrates93. Key CHK1 targets for controlling cell cycle transitions are the CDC25 phosphatases94. Human cells have three CDC25 proteins that regulate cell cycle transitions by removing the inhibitory phosphorylations on cyclin-dependent kinases. CHK1 phosphorylation of the CDC25 proteins inhibits their activity and prevents CDK activation95–97. This is a major checkpoint mechanism that prevents entry into mitosis.
ATR signaling through CHK1 is also critical for regulating replication. ATR signaling slows DNA replication at least in part by inhibiting origin firing. ATR-dependent inhibition of origin firing is important even in the absence of added exogenous replication stress agents98, 99 and is critical to reduce the rate of DNA synthesis under DNA damaging conditions100–106. The precise mechanism by which ATR signaling regulates replication origins is not clear but may be at least partly through the regulation of CDK2-cyclin E kinases by the CHK1-CDC25 pathway. There is also some evidence that in mammalian cells and S. pombe systems ATR signaling slows replication fork elongation91, 107 but the precise mechanism is unknown.
ATR substrates at the replication fork
While ATR phosphorylation of CHK1 helps to spread the damage signal, many of the critical functions of ATR are on chromatin and more specifically at the site of the stalled replication fork (Fig. 3). These activities promote replication fork stability and recovery of stalled forks to ensure completion of replication.
Figure 3. ATR phosphorylates numerous substrates to regulate replication and cell cycle transitions.

A major ATR substrate is the checkpoint kinase CHK1. CHK1 phosphorylation releases it from chromatin and increases its kinase activity. CHK1 has numerous substrates some of which regulate cell cycle transitions and replication origin firing. Many ATR substrates are at the replication fork. In most cases, the consequences of phosphorylation remain unknown but likely contribute to fork stabilization. As discussed in the text, ATR-dependent MCM2 phosphorylation regulates binding to PLK1 which may promote the completion of DNA replication near the stalled fork.
The ATR-dependent substrates that maintain fork integrity are poorly understood. Evidence from a variety of systems suggests that replisome components such as Polε and PCNA dissociate from stalled forks in the absence of ATR signaling103, 108. Additional substrates include the replication factor C complex, RPA1 and RPA2, the MCM2-7 complex, MCM10, and several DNA polymerases79, 90, 109–114. The consequences of most of these phosphorylation events are unknown. However, by examining replication in Xenopus egg extracts under stressful conditions, the Costanzo group recently discovered a functional role for MCM2 phosphorylation115. MCM2 is a component of the MCM2-7 replicative helicase that unwinds the DNA duplex. ATR phosphorylates MCM2 on S108 and xMCM2 on the equivalent site, S92111, 112. MCM2 phosphorylation creates a docking site for Polo-like kinase (Plk) 1. PLK1 recruitment then promotes recovery of DNA replication in extracts treated with replication inhibitors115. Plk1 activity near a stalled replication fork may promote neighboring origins to fire through regulating the chromatin association of CDC45, association of which is a pre-requisite for unwinding and recruitment of DNA polymerase. By promoting origin firing, PLK1 overrides the ATR/CHK1-dependent inhibition of origin firing that is the major cause of decreased replication rates in damaged cells.
Why would ATR signaling both inhibit and promote origin firing? One possibility is suggested by the observation that dormant origins do fire under conditions of replication stress as a way of ensuring complete replication116, 117. It is possible the ATR-MCM2-PLK1 mechanism acts locally to promote replication near the stalled fork while an ATR-CHK1 signaling pathway operates throughout the nucleus to slow overall rates of DNA synthesis. Indeed, it is interesting to note that PLK1 suppresses CHK1 activation somewhat115. It may be that the local phosphorylation of MCM2 at stalled forks allows PLK1 to suppress CHK1 activation near these forks, further promoting the completion of replication in these problem areas. Although further testing of this idea is required, such a model would allow one to reconcile the apparently contradictory effects of ATR on origin firing and make use of both the global and local components of ATR signaling.
ATR and DNA repair
A final class of ATR substrates comprises those that regulate DNA repair. Specifically, ATR signaling may regulate recombination at stalled and collapsed replication forks. ATR phosphorylates several proteins that regulate recombination including BRCA1, WRN and BLM118–121. Recombination can be used to re-initiate replication but can also cause genome instability if not regulated properly. In S. pombe, deletion of the RecA-like, recombinase protein, RAD51 prevents aberrant strand-exchange events that occur at stalled replication forks of checkpoint-deficient cells122. It is still unclear how ATR-dependent phosphorylation of recombination proteins alters their activities; however, the effect may be to limit the types and frequencies of crossover events. ATR also targets the Fanconi-anemia protein FANCD2 to regulate inter-strand crosslink repair. ATR-dependent FANCD2 phosphorylation promotes its monoubiquitination and localization to damage foci123. Finally, ATR phosphorylates the nucleotide excision repair protein XPA to regulate its intracellular localization124. Thus, ATR signaling promotes repair of a variety of DNA lesions.
PIK kinase redundancy and crosstalk
Most ATR substrates can also be phosphorylated by ATM, and the major functions of ATR and ATM in cell cycle control are overlapping and redundant. So why is one kinase not sufficient? A simple but incomplete answer to this question is that ATM and ATR respond to different types of DNA damage, DSBs for ATM and replication stress for ATR. More specifically, the kinases sense different DNA structures–DNA ends [ATM] vs. ssDNA [ATR]. However, to think of ATM and ATR as interchangeable kinases that simply see different inputs ignores much of the complexity in the DNA damage response that is enabled by having two kinases. In particular, the ability of one DNA damage type to be converted into another, the kinetics of the ATM vs. ATR checkpoint responses, and crosstalk between the pathways suggests both unique and interdependent roles for these kinases.
ATR and the DSB response
Although ATR is primarily a replication stress response kinase, it is also activated by DSBs. Thus, agents such as ionizing radiation (IR) activate both ATM and ATR. However, ATM is activated rapidly irrespective of the cell cycle, whereas ATR is activated more slowly and predominantly in S and G2 phase cells125.
The slower and cell cycle-dependent ATR activation at a double-strand break site is a consequence of the need for CDK-dependent DSB end resection that reveals a larger single-stranded region. Importantly, several recent publications have demonstrated a requirement for ATM in end resection and recruitment of ATR125–128. A second level of crosstalk may occur at the level of TOPBP1. TOPBP1 is phosphorylated by ATM, and phosphorylated TOPBP1 is a more efficient activator of ATR66.
These data point to the importance of ATM in ATR activation. This is easily seen in experiments using cells from patients with ataxia-telangiectasia. In these cells, shared ATM and ATR substrates are not phosphorylated efficiently in response to IR because ATM is absent causing a delay in end resection. Eventually, ATM-independent end resection does occur allowing ATR to recognize the damage 125–127. The consequence of this interdependency is that ATM-deficient cells have severe cell cycle checkpoint defects in response to IR. The importance for active ATR signaling in the IR-induced checkpoint response can be visualized in systems where ATR is inactivated conditionally1, 129. In these circumstances, checkpoint responses to IR are compromised. ATM is often described as the kinase that initiates the checkpoint response and ATR is the kinase that maintains it. This is an oversimplification because in wild-type cells, end resection occurs rapidly and ATR is activated within 10–15 minutes.125, 126. Thus, even the initiation of the G2/M checkpoint is impaired in the absence of ATR129. Therefore, it is more appropriate to think of ATM and ATR as partners in the DSB response.
ATM and replication
If ATR partners with ATM to promote checkpoint signaling at a DSB site, then the obvious question to ask is whether ATM functions at a stalled replication fork? Certainly, ATM does signal at collapsed replication forks where DSBs are often formed. This is easiest to observe in cells in which ATR signaling has been inactivated resulting in the accumulation of collapsed forks, γH2AX phosphorylation, and ATM activation129. However, this ATM function is still a DSB response, leaving unanswered the question whether ATM functions at forks that stall without generating a DSB?
There are data that indicate that ATM and the ATM-activating MRE11, RAD50 NBS1 (MRN) complex does have important activities at replication forks. The most convincing data is from Xenopus egg extracts. xATM and xMRE11 are important to prevent double-strand breaks at stalled forks during replication108, 130. Inhibition of ATM increases the rate of DNA replication and ATM is transiently activated as DNA replication proceeds in the egg extract98. The data from human or mouse systems is less convincing although MRN components do localize to replication forks especially in response to agents that cause fork stalling131. It is formally possible that ATM and MRE11 prevent the accumulation of DSBs during replication by promoting rapid repair after they occur. In this scenario, ATM and MRE11 should still be thought of as DSB sensing proteins and their involvement in replication limited to when DSBs form at a collapsed fork. It is also possible that MRE11 has functions at replication forks independent of ATM. In contrast to ATM, MRE11, like ATR, is essential for life and all known mutations are hypomorphic132–134. The MRN complex has been found in some ATR-ATRIP complexes and may function upstream of ATR signaling events135, 136.
If ATM does function to regulate replication in the absence of DSBs, does its activity in these cases depend on ATR? The answer is unclear although there is one report that ATM may be directly phosphorylated by ATR137. Interestingly, ATR has also been reported to phosphorylate another PIK kinase, DNA-PKcs, in response to UV-induced replication stress138. Clearly, the role of ATR upstream of ATM and other PIK kinases needs further investigation.
Crosstalk downstream of ATR
The overlap in substrate specificity for ATM and ATR is also an important point of crosstalk. Most substrates that have been examined such as BRCA1 and p53 are phosphorylated by both kinases118, 139–141. However, there is evidence of some unique specificities. In particular, CHK1 and CHK2 may be exclusively ATR and ATM substrates respectively. Indications of crosstalk at the level of the CHK kinases (such as ATM-dependent CHK1 phosphorylation after IR142) are likely to be largely due to interconversion of the DNA lesions. End resection converts DSBs into ssDNA structures that activate ATR while nucleases can cleave ssDNA to yield DSBs that activate ATM (Fig. 4).
Figure 4. Inter-conversion of ATR- and ATM-activating DNA lesions.

(A) Stalled replication forks activate ATR. Nucleases can cleave stalled forks causing DSBs to form which activate ATM. The rate at which DSBs form at stalled forks is greatly increased in cells with defective ATR signaling. (B) DSBs activate ATM but will also activate ATR as a consequence of DNA end resection. This process is ATM- and cell cycle-dependent such that most ATR activation by double-strand breaks occurs in S and G2 phase cells. CHK1 and CHK2 are primarily ATR and ATM substrates respectively.
ATR’s Essential Function
Although ATM and ATR may collaborate in many ways, the organismal and cellular phenotypes that result from the loss of ATM and ATR are dramatically distinct: the loss of ATR causes rapid lethality at the earliest embryonic stages2, 3, whereas people with no functional ATM can live for decades. What then is the essential function of ATR? As already detailed, ATR responds primarily to primed ssDNA implying that the formation of this structure in unperturbed cell cycles is at the heart of the matter. One can thus envision at least two answers to this question.
One possibility is that ATR is activated at low levels in every cell cycle as a result of normal Okazaki fragment-dependent lagging strand synthesis, during which the activating structure may be repeatedly produced. This activation may be required to enforce the order of S and M phase and to control origin timing5. In yeast, however, separation of function mutants indicate that Mec1/ATR’s essential function has little to do with cell cycle arrest or origin firing, but is related to its fork stabilization activity143.
The second possibility is that there are abundant sources of stalled forks in unperturbed cells due to endogenous damage and sequences that are difficult to replicate. ATR would thus be needed to stabilize these structures and promote their restart when collapse occurs. In effect then, the essential function of ATR may be to respond to DNA damage or replication stress, just as it does when cells are stressed by a DNA damaging agent or replication inhibitor. On this point, it should be noted that the essential function of Mec1/ATR can be rescued in unstressed cells by mutations that increase deoxynucleotide levels144, 145. Since Mec1/ATR regulates deoxynucleotide production by regulating ribonucleotide reductase, the loss of Mec1/ATR may create the very conditions (stalled forks) that require Mec1/ATR to be resolved.
Although these are not mutually exclusive models, it seems likely that the second possibility is true. After all, ATR did not evolve to respond to the doses of radiation and chemicals used in the laboratory to study signaling. DNA is a fairly stable molecule, but the large amount of it in the nucleus (3 billion base pairs for human cells) provides ample opportunities for chemical modifications such as nicks, base modifications and crosslinks146. Repair enzymes constantly correct these lesions but undoubtedly some are encountered by replication forks prior to repair. In addition, some DNA, DNA-RNA, and DNA-protein structures may stall replication even in the absence of any change in nucleic acid chemistry147–153. In some of these cases, the helicase may not uncouple from the polymerase so ATR would not be activated, but in many circumstances, uncoupling does occur and the checkpoint is required for fork stabilization. Even on the relatively small circular E. coli genome it is estimated that nearly all replication forks encounter lesions and must be repaired during every round of replication even in normal growth conditions154. For mammals, sister chromatid exchange rates suggest that approximately 10 DSBs per cell division form at replication forks155. The frequency of fork stalling is undoubtedly much higher.
Defects in ATR-dependent activities at stalled forks may be a major cause of genetic instability. Stalled forks are usually not problematic in eukaryotic cells with multiple replication origins since a fork converging from the other direction can complete replication. In the absence of ATR, however, a stalled fork will collapse and double-strand breaks accumulate156. Chromosomal fragile sites may be examples of this phenomenon. Elevated rates of fragile site breakage are observed when cells are treated with low doses of replication stress agents. Therefore, fragile sites are thought to represent chromosomal regions that are particularly difficult to replicate or perhaps are difficult to repair if replication forks collapse in these areas157. ATR-deficient cells have high levels of fragile site breakage158. Similar fragile sites are thought to exist in S. cerevisiae cells and are associated with slow replicating zones that have high rates of fork stalling especially in Mec1/ATR-deficient cells159. Thus, the consequences of defective ATR-dependent replication regulation are DSBs and, eventually, cell death.
Conclusions and Future Directions
The first described cell cycle checkpoint (the S-phase checkpoint) was discovered partly due to an analysis of cells from patients with ataxia-telangiectasia that had defects in ATM function160, 161. Cloning of the ATM gene opened an entire field of checkpoint signaling8. By contrast, the study of ATR lagged behind because ATR is essential for cell viability and the intimate connection between ATR and DNA replication make experimentation difficult. However, concerted efforts in multiple biological systems has yielded a good description of ATR loss of function phenotypes; the first outlines of the ATR activation mechanism; and rapidly increasing numbers of ATR substrates and signaling pathways.
Of course, there are still many unanswered questions. Many experimental observations suggest levels of complexity beyond the ssDNA-ATR activation model including additional protein regulators and even alternative mechanisms depending on the type of DNA lesion. Especially critical will be a better understanding of how post-translational modifications regulate ATR complexes and how TOPBP1 is regulated. A true mechanistic understanding will require structural information. The large size of the kinase may continue to foil attempts at crystallizing the entire complex, but domain mapping and mutagenesis studies have matured and may enable smaller sub-domain structures to be solved and interpreted.
Although genetic studies have yielded increasing evidence for critical ATR activities at replication forks and biochemical experiments have identified large numbers of substrates, we remain largely ignorant of the mechanisms by which this signaling translates into increased fork stability and how it regulates origin firing and fork elongation.
Finally, the relevance of ATR signaling to human disease will be an increasing area of research focus. Although homozygous loss of function mutations in ATR are likely to be exceedingly rare, mutations in ATR regulators and downstream substrates might be expected in cancer and other diseases linked to genome instability. The effects of heterozygous ATR mutations also need to be examined carefully since mouse studies suggest that ATR is a gene dosage-dependent tumor suppressor in specific genetic backgrounds162. The observation that the DNA damage response is activated in premalignant lesions at least in part due to replication stress163, 164 makes understanding how ATR promotes genome maintenance a critical research goal. ATR signaling may act as a barrier to cancer development by promoting DNA repair, cell cycle checkpoints, apoptosis and cellular senescence in hyperplastic lesions with DNA integrity challenges. Furthermore, the ATR pathway may be a useful target for new drug development10, 11, and it is important to remember that the effects of many current cancer treatments such as radiation and many chemotherapeutic agents are modulated by the DNA damage response. Thus, we should expect an increasing effort to translate our rapidly expanding knowledge of ATR signaling into the clinic.
Online Summary
ATR is a member of the PIK family of protein kinases that includes ATM, and regulates DNA damage responses to maintain genome integrity.
A common DNA structure–single stranded DNA (ssDNA) with a 5′ double stranded primer junction is responsible in most instances for ATR activation.
ATR binds to a protein co-factor, ATRIP, that regulates ATR localization and activation.
The TOPBP1 protein directly activates ATR-ATRIP complexes. Its recruitment to DNA lesions is promoted by the 9-1-1 checkpoint clamp.
ATR signals to regulate DNA replication, cell cycle transitions, and DNA repair through the phosphorylation of hundreds of substrates including CHK1 and the MCM helicase complex.
ATM and ATR have overlapping but non-redundant functions in the DNA damage response. Crosstalk between these pathways often occurs as a consequence of inter-conversion of the activating DNA lesions.
ATR is essential for the survival of most replicating cells perhaps due to the ubiquitous presence of DNA lesions and replication stress.
Table 1.
Phosphorylation sites on ATR-ATRIP
Acknowledgments
Research in the Cimprich lab is supported by the National Institute of Environmental Health Sciences, the American Cancer Society and the California Breast Cancer Research Program. KAC is a Leukemia and Lymphoma Scholar. The Cortez lab is supported by the National Cancer Institute, the Robert J. Kleberg Jr. and Helen C. Kleberg Foundation and the Ingram Charitable Fund. The authors apologize for the many important references that could not be discussed due to space limitations.
Glossary terms
- Seckel Syndrome
A rare autosomal recessive disorder characterized by microcephaly with mental retardation and growth retardation. One form is caused by mutations in ATR
- Replication stress
A problem during DNA replication caused by DNA lesions, inadequate deoxynucleotide supplies, or other difficulty that interferes with replication fork movement
- Replication protein A
A hetero-trimeric single-stranded DNA binding protein complex with multiple activities in nucleic acid metabolism
- Clamp loader
A protein complex that binds and then assembles a protein clamp onto the DNA at a 3′-OH primer end for DNA replication or 5′-P primer end for checkpoint signaling
- End resection
The nuclease dependent removal of base pairs at a double-strand break to leave an extended ssDNA end with a recessed 5′ end
- Nucleotide excision repair
A process in which a small region of the DNA strand that surrounds a bulky DNA lesion is removed from the DNA helix as an oligonucleotide
- BRCT domain
An evolutionarily conserved phospho-serine/threonine-interaction motif that was first identified in the carboxy-terminal part of BRCA1 and, subsequently, in several other checkpoint mediators
- Replisome
A multi-protein complex at the junction of the DNA replication fork that contains all the enzymes required for DNA replication
- Cellular senescence
A nearly irreversible stage of permanent G1 cell-cycle arrest, which is linked to morphological changes (flattening of the cells), metabolic changes and changes in gene expression
- Polo-like kinase
An evolutionarily conserved serine/threonine kinase with functions in mitosis and checkpoint signaling
- Collapsed replication fork
A blocked replication fork that has lost components of the replisome
- MCM Complex
A complex of 6 mini-chromosome maintenance proteins (MCM2-7) that functions together with accessory proteins as the replicative helicase to unwind double-stranded DNA
- MRN Complex
A DSB-sensing complex containing Mre11, Rad50, and Nbs1 that is important to recruit and activate ATM
- Hypomorphic
A mutation that reduces, but does not completely eliminate, the function of a gene
- HEAT repeat
A tandemly repeated, 37–47 amino acid long module that forms an extended alpha-helical structure. Named for four proteinshuntingtin, elongation factor 3 (EF3), protein phosphatase 2A (PP2A) and TOR1
- Nonsense-mediated mRNA decay
A pathway that ensures that mRNAs bearing premature stop codons are eliminated as templates for translation
- Exon-junction complex
A complex of proteins that is deposited as a consequence of pre-mRNA splicing 20–24 nucleotides upstream of splicing-generated exon–exon junctions of newly synthesized mRNA
Biographies
Karlene Cimprich has been a faculty member at Stanford University School of Medicine since 1998. She is currently an Associate Professor in the Department of Chemical and Systems Biology. She carried out her doctoral work with EJ Corey at Harvard University and her post-doctoral training with Stuart Schreiber, also at Harvard. Her laboratory is interested in how cells maintain genome integrity, particularly during S phase.
David Cortez has been a faculty member in the Department of Biochemistry at the Vanderbilt University School of Medicine since 2002. He is currently Ingram Associate Professor of Cancer Research and co-leader of the Genome Maintenance program in the Vanderbilt Ingram Cancer Center. He carried out his doctoral work with Ann Marie Pendergast at Duke University and his post-doctoral training with Stephen Elledge at the Baylor College of Medicine. His laboratory studies the DNA damage response and other genome maintenance activities.
References
- 1.Cortez D, Guntuku S, Qin J, Elledge SJ. ATR and ATRIP: partners in checkpoint signaling. Science. 2001;294:1713–6. doi: 10.1126/science.1065521. Identified ATRIP and along with reference 129 demonstrated that ATR is essential for cell viability. [DOI] [PubMed] [Google Scholar]
- 2.Brown EJ, Baltimore D. ATR disruption leads to chromosomal fragmentation and early embryonic lethality. Genes Dev. 2000;14:397–402. Together with reference 3, found that disruption of ATR in mice causes early embryonic lethality. [PMC free article] [PubMed] [Google Scholar]
- 3.de Klein A, et al. Targeted disruption of the cell-cycle checkpoint gene ATR leads to early embryonic lethality in mice. Curr Biol. 2000;10:479–82. doi: 10.1016/s0960-9822(00)00447-4. [DOI] [PubMed] [Google Scholar]
- 4.Nyberg KA, Michelson RJ, Putnam CW, Weinert TA. Toward maintaining the genome: DNA damage and replication checkpoints. Annu Rev Genet. 2002;36:617–56. doi: 10.1146/annurev.genet.36.060402.113540. [DOI] [PubMed] [Google Scholar]
- 5.Shechter D, Costanzo V, Gautier J. Regulation of DNA replication by ATR: signaling in response to DNA intermediates. DNA Repair (Amst) 2004;3:901–8. doi: 10.1016/j.dnarep.2004.03.020. [DOI] [PubMed] [Google Scholar]
- 6.Swift M, Reitnauer PJ, Morrell D, Chase CL. Breast and other cancers in families with ataxia-telangiectasia. N Engl J Med. 1987;316:1289–94. doi: 10.1056/NEJM198705213162101. [DOI] [PubMed] [Google Scholar]
- 7.Renwick A, et al. ATM mutations that cause ataxia-telangiectasia are breast cancer susceptibility alleles. Nat Genet. 2006;38:873–5. doi: 10.1038/ng1837. [DOI] [PubMed] [Google Scholar]
- 8.Savitsky K, et al. A single ataxia telangiectasia gene with a product similar to PI-3 kinase. Science. 1995;268:1749–53. doi: 10.1126/science.7792600. [DOI] [PubMed] [Google Scholar]
- 9.O’Driscoll M, Ruiz-Perez VL, Woods CG, Jeggo PA, Goodship JA. A splicing mutation affecting expression of ataxia-telangiectasia and Rad3-related protein (ATR) results in Seckel syndrome. Nat Genet. 2003;33:497–501. doi: 10.1038/ng1129. [DOI] [PubMed] [Google Scholar]
- 10.Collins I, Garrett MD. Targeting the cell division cycle in cancer: CDK and cell cycle checkpoint kinase inhibitors. Curr Opin Pharmacol. 2005;5:366–73. doi: 10.1016/j.coph.2005.04.009. [DOI] [PubMed] [Google Scholar]
- 11.Kaelin WG., Jr The concept of synthetic lethality in the context of anticancer therapy. Nat Rev Cancer. 2005;5:689–98. doi: 10.1038/nrc1691. [DOI] [PubMed] [Google Scholar]
- 12.Costanzo V, et al. An ATR- and Cdc7-dependent DNA damage checkpoint that inhibits initiation of DNA replication. Mol Cell. 2003;11:203–13. doi: 10.1016/s1097-2765(02)00799-2. [DOI] [PubMed] [Google Scholar]
- 13.Zou L, Elledge SJ. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science. 2003;300:1542–8. doi: 10.1126/science.1083430. Demonstrated that ATRIP promotes ATR binding to RPA-coated ssDNA. [DOI] [PubMed] [Google Scholar]
- 14.Fanning E, Klimovich V, Nager AR. A dynamic model for replication protein A (RPA) function in DNA processing pathways. Nucleic Acids Res. 2006;34:4126–37. doi: 10.1093/nar/gkl550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Umezu K, Sugawara N, Chen C, Haber JE, Kolodner RD. Genetic analysis of yeast RPA1 reveals its multiple functions in DNA metabolism. Genetics. 1998;148:989–1005. doi: 10.1093/genetics/148.3.989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee SE, et al. Saccharomyces Ku70, mre11/rad50 and RPA proteins regulate adaptation to G2/M arrest after DNA damage. Cell. 1998;94:399–409. doi: 10.1016/s0092-8674(00)81482-8. [DOI] [PubMed] [Google Scholar]
- 17.Jiang G, Sancar A. Recruitment of DNA damage checkpoint proteins to damage in transcribed and nontranscribed sequences. Mol Cell Biol. 2006;26:39–49. doi: 10.1128/MCB.26.1.39-49.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hermand D, Nurse P. Cdc18 enforces long-term maintenance of the S phase checkpoint by anchoring the Rad3-Rad26 complex to chromatin. Mol Cell. 2007;26:553–63. doi: 10.1016/j.molcel.2007.04.014. [DOI] [PubMed] [Google Scholar]
- 19.Yoshioka K, Yoshioka Y, Hsieh P. ATR kinase activation mediated by MutSalpha and MutLalpha in response to cytotoxic O6-methylguanine adducts. Mol Cell. 2006;22:501–10. doi: 10.1016/j.molcel.2006.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ball HL, et al. Function of a conserved checkpoint recruitment domain in ATRIP proteins. Mol Cell Biol. 2007;27:3367–77. doi: 10.1128/MCB.02238-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stokes MP, Van Hatten R, Lindsay HD, Michael WM. DNA replication is required for the checkpoint response to damaged DNA in Xenopus egg extracts. J Cell Biol. 2002;158:863–72. doi: 10.1083/jcb.200204127. Demonstrated that checkpoint activation in Xenopus egg extracts by agents that stall DNA replication forks requires uncoupling of helicase and polymerase activities to generate long stretches of ssDNA. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Michael WM, Ott R, Fanning E, Newport J. Activation of the DNA replication checkpoint through RNA synthesis by primase. Science. 2000;289:2133–7. doi: 10.1126/science.289.5487.2133. [DOI] [PubMed] [Google Scholar]
- 23.Byun TS, Pacek M, Yee MC, Walter JC, Cimprich KA. Functional uncoupling of MCM helicase and DNA polymerase activities activates the ATR-dependent checkpoint. Genes Dev. 2005;19:1040–52. doi: 10.1101/gad.1301205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.MacDougall CA, Byun TS, Van C, Yee MC, Cimprich KA. The structural determinants of checkpoint activation. Genes Dev. 2007;21:898–903. doi: 10.1101/gad.1522607. Used defined DNA structures in Xenopus egg extracts to show that both ssDNA and a 5′ junction is sufficient to activate ATR signaling. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Parrilla-Castellar ER, Arlander SJ, Karnitz L. Dial 9-1-1 for DNA damage: the Rad9-Hus1-Rad1 (9-1-1) clamp complex. DNA Repair (Amst) 2004;3:1009–14. doi: 10.1016/j.dnarep.2004.03.032. [DOI] [PubMed] [Google Scholar]
- 26.Ellison V, Stillman B. Biochemical characterization of DNA damage checkpoint complexes: clamp loader and clamp complexes with specificity for 5′ recessed DNA. PLoS Biol. 2003;1:E33. doi: 10.1371/journal.pbio.0000033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zou L, Liu D, Elledge SJ. Replication protein A-mediated recruitment and activation of Rad17 complexes. Proc Natl Acad Sci U S A. 2003;100:13827–32. doi: 10.1073/pnas.2336100100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bermudez VP, et al. Loading of the human 9-1-1 checkpoint complex onto DNA by the checkpoint clamp loader hRad17-replication factor C complex in vitro. Proc Natl Acad Sci U S A. 2003;100:1633–8. doi: 10.1073/pnas.0437927100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Majka J, Binz SK, Wold MS, Burgers PM. Replication protein A directs loading of the DNA damage checkpoint clamp to 5′-DNA junctions. J Biol Chem. 2006;281:27855–61. doi: 10.1074/jbc.M605176200. [DOI] [PubMed] [Google Scholar]
- 30.Kanoh Y, Tamai K, Shirahige K. Different requirements for the association of ATR-ATRIP and 9-1-1 to the stalled replication forks. Gene. 2006;377:88–95. doi: 10.1016/j.gene.2006.03.019. [DOI] [PubMed] [Google Scholar]
- 31.Pacek M, Walter JC. A requirement for MCM7 and Cdc45 in chromosome unwinding during eukaryotic DNA replication. Embo J. 2004;23:3667–76. doi: 10.1038/sj.emboj.7600369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nedelcheva MN, et al. Uncoupling of unwinding from DNA synthesis implies regulation of MCM helicase by Tof1/Mrc1/Csm3 checkpoint complex. J Mol Biol. 2005;347:509–21. doi: 10.1016/j.jmb.2005.01.041. [DOI] [PubMed] [Google Scholar]
- 33.Majka J, Niedziela-Majka A, Burgers PM. The Checkpoint Clamp Activates Mec1 Kinase during Initiation of the DNA Damage Checkpoint. Mol Cell. 2006;24:891–901. doi: 10.1016/j.molcel.2006.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee J, Kumagai A, Dunphy WG. The Rad9-Hus1-Rad1 checkpoint clamp regulates interaction of TopBP1 with ATR. J Biol Chem. 2007;282:28036–44. doi: 10.1074/jbc.M704635200. [DOI] [PubMed] [Google Scholar]
- 35.Delacroix S, Wagner JM, Kobayashi M, Yamamoto K, Karnitz LM. The Rad9-Hus1-Rad1 (9-1-1) clamp activates checkpoint signaling via TopBP1. Genes Dev. 2007;21:1472–7. doi: 10.1101/gad.1547007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Furuya K, Poitelea M, Guo L, Caspari T, Carr AM. Chk1 activation requires Rad9 S/TQ-site phosphorylation to promote association with C-terminal BRCT domains of Rad4TOPBP1. Genes Dev. 2004;18:1154–64. doi: 10.1101/gad.291104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kumagai A, Lee J, Yoo HY, Dunphy WG. TopBP1 activates the ATR-ATRIP complex. Cell. 2006;124:943–55. doi: 10.1016/j.cell.2005.12.041. Demonstrated that TopBP1 is a protein activator of ATR kinase activity. [DOI] [PubMed] [Google Scholar]
- 38.St Onge RP, Besley BD, Pelley JL, Davey S. A role for the phosphorylation of hRad9 in checkpoint signaling. J Biol Chem. 2003;278:26620–8. doi: 10.1074/jbc.M303134200. [DOI] [PubMed] [Google Scholar]
- 39.Weiss RS, Enoch T, Leder P. Inactivation of mouse Hus1 results in genomic instability and impaired responses to genotoxic stress. Genes Dev. 2000;14:1886–98. [PMC free article] [PubMed] [Google Scholar]
- 40.Hopkins KM, et al. Deletion of mouse rad9 causes abnormal cellular responses to DNA damage, genomic instability, and embryonic lethality. Mol Cell Biol. 2004;24:7235–48. doi: 10.1128/MCB.24.16.7235-7248.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mordes DA, Glick GG, Zhao R, Cortez D. TopBP1 activates ATR through ATRIP and a PIKK regulatory domain. Genes and Development. 2008 doi: 10.1101/gad.1666208. in press. Defined binding surfaces on ATR and ATRIP for TopBP1, and identified a common regulatory domain among PIK family members that provides specialized regulatory opportunities for these kinases. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sekulic A, et al. A direct linkage between the phosphoinositide 3-kinase-AKT signaling pathway and the mammalian target of rapamycin in mitogen-stimulated and transformed cells. Cancer Res. 2000;60:3504–13. [PubMed] [Google Scholar]
- 43.Sun Y, Xu Y, Roy K, Price BD. DNA damage induced acetylation of lysine 3016 of ATM activates ATM kinase activity. Mol Cell Biol. 2007;24:8502–8509. doi: 10.1128/MCB.01382-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stucki M, Jackson SP. gammaH2AX and MDC1: anchoring the DNA-damage-response machinery to broken chromosomes. DNA Repair (Amst) 2006;5:534–43. doi: 10.1016/j.dnarep.2006.01.012. [DOI] [PubMed] [Google Scholar]
- 45.Soutoglou E, Misteli T. Activation of the Cellular DNA Damage Response in the Absence of DNA Lesions. Science. 2008 doi: 10.1126/science.1159051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bonilla CY, Melo JA, Toczyski DP. Colocalization of sensors is sufficient to activate the DNA damage checkpoint in the absence of damage. Mol Cell. 2008;30:267–76. doi: 10.1016/j.molcel.2008.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Toledo LI, Murga M, Gutierrez-Martinez P, Soria R, Fernandez-Capetillo O. ATR signaling can drive cells into senescence in the absence of DNA breaks. Genes Dev. 2008;22:297–302. doi: 10.1101/gad.452308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Choi JH, Lindsey-Boltz LA, Sancar A. Reconstitution of a human ATR-mediated checkpoint response to damaged DNA. Proc Natl Acad Sci U S A. 2007;104:13301–6. doi: 10.1073/pnas.0706013104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kondo T, Wakayama T, Naiki T, Matsumoto K, Sugimoto K. Recruitment of Mec1 and Ddc1 checkpoint proteins to double-strand breaks through distinct mechanisms. Science. 2001;294:867–70. doi: 10.1126/science.1063827. [DOI] [PubMed] [Google Scholar]
- 50.Melo JA, Cohen J, Toczyski DP. Two checkpoint complexes are independently recruited to sites of DNA damage in vivo. Genes Dev. 2001;15:2809–21. doi: 10.1101/gad.903501. References 49 and 50 provided the first evidence that the checkpoint clamp and the ATR kinase are recruited to sites of DNA lesions independently. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zou L, Cortez D, Elledge SJ. Regulation of ATR substrate selection by Rad17-dependent loading of Rad9 complexes onto chromatin. Genes Dev. 2002;16:198–208. doi: 10.1101/gad.950302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.You Z, Kong L, Newport J. The role of single-stranded DNA and polymerase alpha in establishing the ATR, Hus1 DNA replication checkpoint. J Biol Chem. 2002;277:27088–93. doi: 10.1074/jbc.M204120200. [DOI] [PubMed] [Google Scholar]
- 53.Lee J, Kumagai A, Dunphy WG. Claspin, a Chk1-regulatory protein, monitors DNA replication on chromatin independently of RPA, ATR, and Rad17. Mol Cell. 2003;11:329–40. doi: 10.1016/s1097-2765(03)00045-5. [DOI] [PubMed] [Google Scholar]
- 54.Ball HL, Myers JS, Cortez D. ATRIP Binding to RPA-ssDNA Promotes ATR-ATRIP Localization but Is Dispensable for Chk1 Phosphorylation. Mol Biol Cell. 2005;16:2372–2381. doi: 10.1091/mbc.E04-11-1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bakkenist CJ, Kastan MB. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature. 2003;421:499–506. doi: 10.1038/nature01368. [DOI] [PubMed] [Google Scholar]
- 56.Ball HL, Cortez D. ATRIP oligomerization is required for ATR-dependent checkpoint signaling. J Biol Chem. 2005;280:31390–31396. doi: 10.1074/jbc.M504961200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Itakura E, Sawada I, Matsuura A. Dimerization of the ATRIP Protein through the Coiled-Coil Motif and Its Implication to the Maintenance of Stalled Replication Forks. Mol Biol Cell. 2005;16:5551–62. doi: 10.1091/mbc.E05-05-0427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lee SJ, Duong JK, Stern DF. A Ddc2-Rad53 fusion protein can bypass the requirements for RAD9 and MRC1 in Rad53 activation. Mol Biol Cell. 2004;15:5443–55. doi: 10.1091/mbc.E04-07-0608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kim SM, Kumagai A, Lee J, Dunphy WG. Phosphorylation of Chk1 by ATM- and Rad3-related (ATR) in xenopus egg extracts requires binding of ATRIP to ATR but not the stable DNA-binding or coiled-coil domains of ATRIP. J Biol Chem. 2005;280:38355–64. doi: 10.1074/jbc.M508673200. [DOI] [PubMed] [Google Scholar]
- 60.Bentley NJ, et al. The Schizosaccharomyces pombe rad3 checkpoint gene. Embo J. 1996;15:6641–51. [PMC free article] [PubMed] [Google Scholar]
- 61.Lindsay HD, et al. S-phase-specific activation of Cds1 kinase defines a subpathway of the checkpoint response in Schizosaccharomyces pombe. Genes Dev. 1998;12:382–95. doi: 10.1101/gad.12.3.382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Paciotti V, Clerici M, Scotti M, Lucchini G, Longhese MP. Characterization of mec1 kinase-deficient mutants and of new hypomorphic mec1 alleles impairing subsets of the DNA damage response pathway. Mol Cell Biol. 2001;21:3913–25. doi: 10.1128/MCB.21.12.3913-3925.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Canman CE, et al. Activation of the ATM kinase by ionizing radiation and phosphorylation of p53. Science. 1998;281:1677–9. doi: 10.1126/science.281.5383.1677. [DOI] [PubMed] [Google Scholar]
- 64.Banin S, et al. Enhanced phosphorylation of p53 by ATM in response to DNA damage. Science. 1998;281:1674–7. doi: 10.1126/science.281.5383.1674. [DOI] [PubMed] [Google Scholar]
- 65.Kumagai A, Kim SM, Dunphy WG. Claspin and the activated form of ATR-ATRIP collaborate in the activation of Chk1. J Biol Chem. 2004 doi: 10.1074/jbc.M408353200. [DOI] [PubMed] [Google Scholar]
- 66.Yoo HY, Kumagai A, Shevchenko A, Shevchenko A, Dunphy WG. Ataxia-telangiectasia mutated (ATM)-dependent activation of ATR occurs through phosphorylation of TopBP1 by ATM. J Biol Chem. 2007;282:17501–6. doi: 10.1074/jbc.M701770200. [DOI] [PubMed] [Google Scholar]
- 67.Buchmann AM, Skaar JR, DeCaprio JA. Activation of a DNA damage checkpoint response in a TAF1-defective cell line. Mol Cell Biol. 2004;24:5332–9. doi: 10.1128/MCB.24.12.5332-5339.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Derheimer FA, et al. RPA and ATR link transcriptional stress to p53. Proc Natl Acad Sci U S A. 2007;104:12778–83. doi: 10.1073/pnas.0705317104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yamane K, Wu X, Chen J. A DNA damage-regulated BRCT-containing protein, TopBP1, is required for cell survival. Mol Cell Biol. 2002;22:555–66. doi: 10.1128/MCB.22.2.555-566.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yan S, Lindsay HD, Michael WM. Direct requirement for Xmus101 in ATR-mediated phosphorylation of Claspin bound Chk1 during checkpoint signaling. J Cell Biol. 2006;173:181–6. doi: 10.1083/jcb.200601076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lupardus PJ, Cimprich KA. Phosphorylation of Xenopus Rad1 and Hus1 defines a readout for ATR activation that is independent of Claspin and the Rad9 carboxy terminus. Mol Biol Cell. 2006;17:1559–69. doi: 10.1091/mbc.E05-09-0865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Namiki Y, Zou L. ATRIP associates with replication protein A-coated ssDNA through multiple interactions. Proc Natl Acad Sci U S A. 2006;103:580–5. doi: 10.1073/pnas.0510223103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sivasubramaniam S, Sun X, Pan YR, Wang S, Lee EY. Cep164 is a mediator protein required for the maintenance of genomic stability through modulation of MDC1, RPA, and CHK1. Genes Dev. 2008;22:587–600. doi: 10.1101/gad.1627708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Unsal-Kacmaz K, Makhov AM, Griffith JD, Sancar A. Preferential binding of ATR protein to UV-damaged DNA. Proc Natl Acad Sci U S A. 2002;99:6673–8. doi: 10.1073/pnas.102167799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Roche KC, Rocha S, Bracken CP, Perkins ND. Regulation of ATR-dependent pathways by the FHA domain containing protein SNIP1. Oncogene. 2007;26:4523–30. doi: 10.1038/sj.onc.1210233. [DOI] [PubMed] [Google Scholar]
- 76.Park BJ, et al. The haploinsufficient tumor suppressor p18 upregulates p53 via interactions with ATM/ATR. Cell. 2005;120:209–21. doi: 10.1016/j.cell.2004.11.054. [DOI] [PubMed] [Google Scholar]
- 77.Zhang J, et al. Protein phosphatase 5 is required for ATR-mediated checkpoint activation. Mol Cell Biol. 2005;25:9910–9. doi: 10.1128/MCB.25.22.9910-9919.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zachos G, Rainey MD, Gillespie DA. Chk1-dependent S-M checkpoint delay in vertebrate cells is linked to maintenance of viable replication structures. Mol Cell Biol. 2005;25:563–74. doi: 10.1128/MCB.25.2.563-574.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Matsuoka S, et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science. 2007;316:1160–6. doi: 10.1126/science.1140321. [DOI] [PubMed] [Google Scholar]
- 80.Mu JJ, et al. A proteomic analysis of ataxia telangiectasia-mutated (ATM)/ATM-Rad3-related (ATR) substrates identifies the ubiquitin-proteasome system as a regulator for DNA damage checkpoints. J Biol Chem. 2007;282:17330–4. doi: 10.1074/jbc.C700079200. [DOI] [PubMed] [Google Scholar]
- 81.Stokes MP, et al. Profiling of UV-induced ATM/ATR signaling pathways. Proc Natl Acad Sci U S A. 2007;104:19855–60. doi: 10.1073/pnas.0707579104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Smolka MB, Albuquerque CP, Chen SH, Zhou H. Proteome-wide identification of in vivo targets of DNA damage checkpoint kinases. Proc Natl Acad Sci U S A. 2007;104:10364–9. doi: 10.1073/pnas.0701622104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Walworth NC, Bernards R. rad-dependent response of the chk1-encoded protein kinase at the DNA damage checkpoint. Science. 1996;271:353–6. doi: 10.1126/science.271.5247.353. [DOI] [PubMed] [Google Scholar]
- 84.Liu Q, et al. Chk1 is an essential kinase that is regulated by Atr and required for the G(2)/M DNA damage checkpoint. Genes Dev. 2000;14:1448–59. [PMC free article] [PubMed] [Google Scholar]
- 85.Lopez-Girona A, et al. Serine-345 is required for Rad3-dependent phosphorylation and function of checkpoint kinase Chk1 in fission yeast. Proc Natl Acad Sci U S A. 2001;98:11289–94. doi: 10.1073/pnas.191557598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kumagai A, Dunphy WG. Claspin, a novel protein required for the activation of Chk1 during a DNA replication checkpoint response in Xenopus egg extracts. Mol Cell. 2000;6:839–49. doi: 10.1016/s1097-2765(05)00092-4. [DOI] [PubMed] [Google Scholar]
- 87.Kumagai A, Dunphy WG. Repeated phosphopeptide motifs in Claspin mediate the regulated binding of Chk1. Nat Cell Biol. 2003;5:161–5. doi: 10.1038/ncb921. [DOI] [PubMed] [Google Scholar]
- 88.Liu S, et al. Claspin operates downstream of TopBP1 to direct ATR signaling towards Chk1 activation. Mol Cell Biol. 2006;26:6056–64. doi: 10.1128/MCB.00492-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wang X, et al. Rad17 phosphorylation is required for claspin recruitment and Chk1 activation in response to replication stress. Mol Cell. 2006;23:331–41. doi: 10.1016/j.molcel.2006.06.022. [DOI] [PubMed] [Google Scholar]
- 90.Bao S, et al. ATR/ATM-mediated phosphorylation of human Rad17 is required for genotoxic stress responses. Nature. 2001;411:969–74. doi: 10.1038/35082110. [DOI] [PubMed] [Google Scholar]
- 91.Unsal-Kacmaz K, et al. The human Tim/Tipin complex coordinates an Intra-S checkpoint response to UV that slows replication fork displacement. Mol Cell Biol. 2007;27:3131–42. doi: 10.1128/MCB.02190-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Errico A, Costanzo V, Hunt T. Tipin is required for stalled replication forks to resume DNA replication after removal of aphidicolin in Xenopus egg extracts. Proc Natl Acad Sci U S A. 2007;104:14929–34. doi: 10.1073/pnas.0706347104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Smits VA, Reaper PM, Jackson SP. Rapid PIKK-dependent release of Chk1 from chromatin promotes the DNA-damage checkpoint response. Curr Biol. 2006;16:150–9. doi: 10.1016/j.cub.2005.11.066. [DOI] [PubMed] [Google Scholar]
- 94.Boutros R, Dozier C, Ducommun B. The when and wheres of CDC25 phosphatases. Curr Opin Cell Biol. 2006;18:185–91. doi: 10.1016/j.ceb.2006.02.003. [DOI] [PubMed] [Google Scholar]
- 95.Furnari B, Rhind N, Russell P. Cdc25 mitotic inducer targeted by chk1 DNA damage checkpoint kinase. Science. 1997;277:1495–7. doi: 10.1126/science.277.5331.1495. [DOI] [PubMed] [Google Scholar]
- 96.Peng CY, et al. Mitotic and G2 checkpoint control: regulation of 14-3-3 protein binding by phosphorylation of Cdc25C on serine-216. Science. 1997;277:1501–5. doi: 10.1126/science.277.5331.1501. [DOI] [PubMed] [Google Scholar]
- 97.Sanchez Y, et al. Conservation of the Chk1 checkpoint pathway in mammals: linkage of DNA damage to Cdk regulation through Cdc25. Science. 1997;277:1497–501. doi: 10.1126/science.277.5331.1497. [DOI] [PubMed] [Google Scholar]
- 98.Shechter D, Costanzo V, Gautier J. ATR and ATM regulate the timing of DNA replication origin firing. Nat Cell Biol. 2004;6:648–55. doi: 10.1038/ncb1145. [DOI] [PubMed] [Google Scholar]
- 99.Maya-Mendoza A, Petermann E, Gillespie DA, Caldecott KW, Jackson DA. Chk1 regulates the density of active replication origins during the vertebrate S phase. Embo J. 2007;26:2719–31. doi: 10.1038/sj.emboj.7601714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Shirahige K, et al. Regulation of DNA-replication origins during cell-cycle progression. Nature. 1998;395:618–21. doi: 10.1038/27007. [DOI] [PubMed] [Google Scholar]
- 101.Tercero JA, Diffley JF. Regulation of DNA replication fork progression through damaged DNA by the Mec1/Rad53 checkpoint. Nature. 2001;412:553–7. doi: 10.1038/35087607. Used the S. cerevisiae system to show that the ATR checkpoint pathway regulates both fork stability and origin firing. [DOI] [PubMed] [Google Scholar]
- 102.Merrick CJ, Jackson D, Diffley JF. Visualization of altered replication dynamics after DNA damage in human cells. J Biol Chem. 2004;279:20067–75. doi: 10.1074/jbc.M400022200. [DOI] [PubMed] [Google Scholar]
- 103.Dimitrova DS, Gilbert DM. Temporally coordinated assembly and disassembly of replication factories in the absence of DNA synthesis. Nat Cell Biol. 2000;2:686–94. doi: 10.1038/35036309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Feijoo C, et al. Activation of mammalian Chk1 during DNA replication arrest: a role for Chk1 in the intra-S phase checkpoint monitoring replication origin firing. J Cell Biol. 2001;154:913–23. doi: 10.1083/jcb.200104099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Alvino GM, et al. Replication in hydroxyurea: it’s a matter of time. Mol Cell Biol. 2007;27:6396–406. doi: 10.1128/MCB.00719-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Heffernan TP, et al. An ATR- and Chk1-dependent S checkpoint inhibits replicon initiation following UVC-induced DNA damage. Mol Cell Biol. 2002;22:8552–61. doi: 10.1128/MCB.22.24.8552-8561.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Mickle KL, et al. Checkpoint independence of most DNA replication origins in fission yeast. BMC Mol Biol. 2007;8:112. doi: 10.1186/1471-2199-8-112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Trenz K, Smith E, Smith S, Costanzo V. ATM and ATR promote Mre11 dependent restart of collapsed replication forks and prevent accumulation of DNA breaks. Embo J. 2006;25:1764–74. doi: 10.1038/sj.emboj.7601045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Brush GS, Morrow DM, Hieter P, Kelly TJ. The ATM homologue MEC1 is required for phosphorylation of replication protein A in yeast. Proc Natl Acad Sci U S A. 1996;93:15075–80. doi: 10.1073/pnas.93.26.15075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Wang H, Guan J, Perrault AR, Wang Y, Iliakis G. Replication protein A2 phosphorylation after DNA damage by the coordinated action of ataxia telangiectasia-mutated and DNA-dependent protein kinase. Cancer Res. 2001;61:8554–63. [PubMed] [Google Scholar]
- 111.Cortez D, Glick G, Elledge SJ. Minichromosome maintenance proteins are direct targets of the ATM and ATR checkpoint kinases. Proc Natl Acad Sci U S A. 2004;101:10078–83. doi: 10.1073/pnas.0403410101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Yoo HY, Shevchenko A, Dunphy WG. Mcm2 is a direct substrate of ATM and ATR during DNA damage and DNA replication checkpoint responses. J Biol Chem. 2004;279:53353–64. doi: 10.1074/jbc.M408026200. [DOI] [PubMed] [Google Scholar]
- 113.Liu JS, Kuo SR, Melendy T. Phosphorylation of replication protein A by S-phase checkpoint kinases. DNA Repair (Amst) 2006;5:369–80. doi: 10.1016/j.dnarep.2005.11.007. [DOI] [PubMed] [Google Scholar]
- 114.Oakley GG, et al. UV-induced hyperphosphorylation of replication protein a depends on DNA replication and expression of ATM protein. Mol Biol Cell. 2001;12:1199–213. doi: 10.1091/mbc.12.5.1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Trenz K, Errico A, Costanzo V. Plx1 is required for chromosomal DNA replication under stressful conditions. Embo J. 2008;27:876–85. doi: 10.1038/emboj.2008.29. Determined that the ATR-dependent phosphorylation of MCM2 (previously identified in references 111 and 112) recruits the Polo-like kinase to stalled replication forks to promote completion of DNA synthesis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Ge XQ, Jackson DA, Blow JJ. Dormant origins licensed by excess Mcm2-7 are required for human cells to survive replicative stress. Genes Dev. 2007;21:3331–41. doi: 10.1101/gad.457807. Determined that excess MCM complex promotes the firing of additional origins of replication when replication is stalled. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Woodward AM, et al. Excess Mcm2-7 license dormant origins of replication that can be used under conditions of replicative stress. J Cell Biol. 2006;173:673–83. doi: 10.1083/jcb.200602108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Tibbetts RS, et al. Functional interactions between BRCA1 and the checkpoint kinase ATR during genotoxic stress. Genes Dev. 2000;14:2989–3002. doi: 10.1101/gad.851000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Pichierri P, Rosselli F, Franchitto A. Werner’s syndrome protein is phosphorylated in an ATR/ATM-dependent manner following replication arrest and DNA damage induced during the S phase of the cell cycle. Oncogene. 2003;22:1491–500. doi: 10.1038/sj.onc.1206169. [DOI] [PubMed] [Google Scholar]
- 120.Davies SL, North PS, Dart A, Lakin ND, Hickson ID. Phosphorylation of the Bloom’s syndrome helicase and its role in recovery from S-phase arrest. Mol Cell Biol. 2004;24:1279–91. doi: 10.1128/MCB.24.3.1279-1291.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Li W, Kim SM, Lee J, Dunphy WG. Absence of BLM leads to accumulation of chromosomal DNA breaks during both unperturbed and disrupted S phases. J Cell Biol. 2004;165:801–12. doi: 10.1083/jcb.200402095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Meister P, et al. Temporal separation of replication and recombination requires the intra-S checkpoint. J Cell Biol. 2005;168:537–44. doi: 10.1083/jcb.200410006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Andreassen PR, D’Andrea AD, Taniguchi T. ATR couples FANCD2 monoubiquitination to the DNA-damage response. Genes Dev. 2004;18:1958–63. doi: 10.1101/gad.1196104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Wu X, Shell SM, Liu Y, Zou Y. ATR-dependent checkpoint modulates XPA nuclear import in response to UV irradiation. Oncogene. 2007;26:757–64. doi: 10.1038/sj.onc.1209828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Jazayeri A, et al. ATM- and cell cycle-dependent regulation of ATR in response to DNA double-strand breaks. Nat Cell Biol. 2006;8:37–45. doi: 10.1038/ncb1337. Together with references 126–128 demonstrated a function of ATM upstream of ATR activation due to ATM-dependent end-resection of the DSBs. [DOI] [PubMed] [Google Scholar]
- 126.Myers JS, Cortez D. Rapid activation of ATR by ionizing radiation requires ATM and Mre11. J Biol Chem. 2006;281:9346–50. doi: 10.1074/jbc.M513265200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Adams KE, Medhurst AL, Dart DA, Lakin ND. Recruitment of ATR to sites of ionising radiation-induced DNA damage requires ATM and components of the MRN protein complex. Oncogene. 2006;25:3894–904. doi: 10.1038/sj.onc.1209426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Cuadrado M, et al. ATM regulates ATR chromatin loading in response to DNA double-strand breaks. J Exp Med. 2006;203:297–303. doi: 10.1084/jem.20051923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Brown EJ, Baltimore D. Essential and dispensable roles of ATR in cell cycle arrest and genome maintenance. Genes Dev. 2003;17:615–28. doi: 10.1101/gad.1067403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Costanzo V, et al. Mre11 protein complex prevents double-strand break accumulation during chromosomal DNA replication. Mol Cell. 2001;8:137–47. doi: 10.1016/s1097-2765(01)00294-5. [DOI] [PubMed] [Google Scholar]
- 131.Mirzoeva OK, Petrini JH. DNA replication-dependent nuclear dynamics of the Mre11 complex. Mol Cancer Res. 2003;1:207–18. [PubMed] [Google Scholar]
- 132.Xiao Y, Weaver DT. Conditional gene targeted deletion by Cre recombinase demonstrates the requirement for the double-strand break repair Mre11 protein in murine embryonic stem cells. Nucleic Acids Res. 1997;25:2985–91. doi: 10.1093/nar/25.15.2985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Luo G, et al. Disruption of mRad50 causes embryonic stem cell lethality, abnormal embryonic development, and sensitivity to ionizing radiation [In Process Citation] Proc Natl Acad Sci U S A. 1999;96:7376–81. doi: 10.1073/pnas.96.13.7376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Kang J, Bronson RT, Xu Y. Targeted disruption of NBS1 reveals its roles in mouse development and DNA repair. Embo J. 2002;21:1447–55. doi: 10.1093/emboj/21.6.1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Stiff T, et al. Nbs1 is required for ATR-dependent phosphorylation events. Embo J. 2005;24:199–208. doi: 10.1038/sj.emboj.7600504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Olson E, Nievera CJ, Lee AY, Chen L, Wu X. The Mre11-Rad50-Nbs1 complex acts both upstream and downstream of ataxia telangiectasia mutated and Rad3-related protein (ATR) to regulate the S-phase checkpoint following UV treatment. J Biol Chem. 2007;282:22939–52. doi: 10.1074/jbc.M702162200. [DOI] [PubMed] [Google Scholar]
- 137.Stiff T, et al. ATR-dependent phosphorylation and activation of ATM in response to UV treatment or replication fork stalling. Embo J. 2006;25:5775–82. doi: 10.1038/sj.emboj.7601446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Yajima H, Lee KJ, Chen BP. ATR-dependent phosphorylation of DNA-dependent protein kinase catalytic subunit in response to UV-induced replication stress. Mol Cell Biol. 2006;26:7520–8. doi: 10.1128/MCB.00048-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Siliciano JD, et al. DNA damage induces phosphorylation of the amino terminus of p53. Genes Dev. 1997;11:3471–81. doi: 10.1101/gad.11.24.3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Cortez D, Wang Y, Qin J, Elledge SJ. Requirement of ATM-dependent phosphorylation of brca1 in the DNA damage response to double-strand breaks. Science. 1999;286:1162–6. doi: 10.1126/science.286.5442.1162. [DOI] [PubMed] [Google Scholar]
- 141.Tibbetts RS, et al. A role for ATR in the DNA damage-induced phosphorylation of p53. Genes Dev. 1999;13:152–7. doi: 10.1101/gad.13.2.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Gatei M, et al. Ataxia-telangiectasia-mutated (ATM) and NBS1-dependent phosphorylation of Chk1 on Ser-317 in response to ionizing radiation. J Biol Chem. 2003;278:14806–11. doi: 10.1074/jbc.M210862200. [DOI] [PubMed] [Google Scholar]
- 143.Tercero JA, Longhese MP, Diffley JF. A central role for DNA replication forks in checkpoint activation and response. Mol Cell. 2003;11:1323–36. doi: 10.1016/s1097-2765(03)00169-2. [DOI] [PubMed] [Google Scholar]
- 144.Zhao X, Muller EG, Rothstein R. A suppressor of two essential checkpoint genes identifies a novel protein that negatively affects dNTP pools. Mol Cell. 1998;2:329–40. doi: 10.1016/s1097-2765(00)80277-4. [DOI] [PubMed] [Google Scholar]
- 145.Desany BA, Alcasabas AA, Bachant JB, Elledge SJ. Recovery from DNA replicational stress is the essential function of the S-phase checkpoint pathway. Genes Dev. 1998;12:2956–70. doi: 10.1101/gad.12.18.2956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Lindahl T. Instability and decay of the primary structure of DNA. Nature. 1993;362:709–15. doi: 10.1038/362709a0. [DOI] [PubMed] [Google Scholar]
- 147.Mirkin EV, Mirkin SM. Replication fork stalling at natural impediments. Microbiol Mol Biol Rev. 2007;71:13–35. doi: 10.1128/MMBR.00030-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Gruber M, Wellinger RE, Sogo JM. Architecture of the replication fork stalled at the 3′ end of yeast ribosomal genes. Mol Cell Biol. 2000;20:5777–87. doi: 10.1128/mcb.20.15.5777-5787.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Ivessa AS, et al. The Saccharomyces cerevisiae helicase Rrm3p facilitates replication past nonhistone protein-DNA complexes. Mol Cell. 2003;12:1525–36. doi: 10.1016/s1097-2765(03)00456-8. [DOI] [PubMed] [Google Scholar]
- 150.Lenzmeier BA, Freudenreich CH. Trinucleotide repeat instability: a hairpin curve at the crossroads of replication, recombination, and repair. Cytogenet Genome Res. 2003;100:7–24. doi: 10.1159/000072836. [DOI] [PubMed] [Google Scholar]
- 151.Arlt MF, Durkin SG, Ragland RL, Glover TW. Common fragile sites as targets for chromosome rearrangements. DNA Repair (Amst) 2006;5:1126–35. doi: 10.1016/j.dnarep.2006.05.010. [DOI] [PubMed] [Google Scholar]
- 152.Lambert S, Watson A, Sheedy DM, Martin B, Carr AM. Gross chromosomal rearrangements and elevated recombination at an inducible site-specific replication fork barrier. Cell. 2005;121:689–702. doi: 10.1016/j.cell.2005.03.022. [DOI] [PubMed] [Google Scholar]
- 153.Calzada A, Hodgson B, Kanemaki M, Bueno A, Labib K. Molecular anatomy and regulation of a stable replisome at a paused eukaryotic DNA replication fork. Genes Dev. 2005;19:1905–19. doi: 10.1101/gad.337205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Cox MM, et al. The importance of repairing stalled replication forks. Nature. 2000;404:37–41. doi: 10.1038/35003501. [DOI] [PubMed] [Google Scholar]
- 155.Haber JE. DNA recombination: the replication connection. Trends Biochem Sci. 1999;24:271–5. doi: 10.1016/s0968-0004(99)01413-9. [DOI] [PubMed] [Google Scholar]
- 156.Paulsen RD, Cimprich KA. The ATR pathway: fine-tuning the fork. DNA Repair (Amst) 2007;6:953–66. doi: 10.1016/j.dnarep.2007.02.015. [DOI] [PubMed] [Google Scholar]
- 157.Glover TW, Arlt MF, Casper AM, Durkin SG. Mechanisms of common fragile site instability. Hum Mol Genet. 2005;14(Spec No 2):R197–205. doi: 10.1093/hmg/ddi265. [DOI] [PubMed] [Google Scholar]
- 158.Casper AM, Nghiem P, Arlt MF, Glover TW. ATR regulates fragile site stability. Cell. 2002;111:779–89. doi: 10.1016/s0092-8674(02)01113-3. [DOI] [PubMed] [Google Scholar]
- 159.Cha RS, Kleckner N. ATR homolog Mec1 promotes fork progression, thus averting breaks in replication slow zones. Science. 2002;297:602–6. doi: 10.1126/science.1071398. [DOI] [PubMed] [Google Scholar]
- 160.Painter RB, Young BR. Radiosensitivity in ataxia-telangiectasia: a new explanation. Proc Natl Acad Sci U S A. 1980;77:7315–7. doi: 10.1073/pnas.77.12.7315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Houldsworth J, Lavin MF. Effect of ionizing radiation on DNA synthesis in ataxia telangiectasia cells. Nucleic Acids Res. 1980;8:3709–20. doi: 10.1093/nar/8.16.3709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Fang Y, et al. ATR functions as a gene dosage-dependent tumor suppressor on a mismatch repair-deficient background. Embo J. 2004;23:3164–74. doi: 10.1038/sj.emboj.7600315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Bartkova J, et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature. 2005;434:864–70. doi: 10.1038/nature03482. [DOI] [PubMed] [Google Scholar]
- 164.Gorgoulis VG, et al. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature. 2005;434:907–13. doi: 10.1038/nature03485. References 163 and 164 demonstrated that the DNA damage response is activated in premalignant lesions as a consequence of replication stress. [DOI] [PubMed] [Google Scholar]
- 165.Llorca O, Rivera-Calzada A, Grantham J, Willison KR. Electron microscopy and 3D reconstructions reveal that human ATM kinase uses an arm-like domain to clamp around double-stranded DNA. Oncogene. 2003;22:3867–74. doi: 10.1038/sj.onc.1206649. [DOI] [PubMed] [Google Scholar]
- 166.Leuther KK, Hammarsten O, Kornberg RD, Chu G. Structure of DNA-dependent protein kinase: implications for its regulation by DNA. Embo J. 1999;18:1114–23. doi: 10.1093/emboj/18.5.1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Rivera-Calzada A, Maman JD, Spagnolo L, Pearl LH, Llorca O. Three-dimensional structure and regulation of the DNA-dependent protein kinase catalytic subunit (DNA-PKcs) Structure. 2005;13:243–55. doi: 10.1016/j.str.2004.12.006. [DOI] [PubMed] [Google Scholar]
- 168.Chiu CY, Cary RB, Chen DJ, Peterson SR, Stewart PL. Cryo-EM imaging of the catalytic subunit of the DNA-dependent protein kinase. J Mol Biol. 1998;284:1075–81. doi: 10.1006/jmbi.1998.2212. [DOI] [PubMed] [Google Scholar]
- 169.Williams DR, Lee KJ, Shi J, Chen DJ, Stewart PL. Cryo-EM Structure of the DNA-Dependent Protein Kinase Catalytic Subunit at Subnanometer Resolution Reveals alpha Helices and Insight into DNA Binding. Structure. 2008;16:468–77. doi: 10.1016/j.str.2007.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Perry J, Kleckner N. The ATRs, ATMs, and TORs are giant HEAT repeat proteins. Cell. 2003;112:151–5. doi: 10.1016/s0092-8674(03)00033-3. [DOI] [PubMed] [Google Scholar]
- 171.Jiang X, Sun Y, Chen S, Roy K, Price BD. The FATC domains of PIKK proteins are functionally equivalent and participate in the Tip60-dependent activation of DNA-PKcs and ATM. J Biol Chem. 2006;281:15741–6. doi: 10.1074/jbc.M513172200. [DOI] [PubMed] [Google Scholar]
- 172.Chen X, Zhao R, Glick GG, Cortez D. Function of the ATR N-terminal domain revealed by an ATM/ATR chimera. Exp Cell Res. 2007;313:1667–74. doi: 10.1016/j.yexcr.2007.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.You Z, Chahwan C, Bailis J, Hunter T, Russell P. ATM activation and its recruitment to damaged DNA require binding to the C terminus of Nbs1. Mol Cell Biol. 2005;25:5363–79. doi: 10.1128/MCB.25.13.5363-5379.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Kim DH, et al. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell. 2002;110:163–75. doi: 10.1016/s0092-8674(02)00808-5. [DOI] [PubMed] [Google Scholar]
- 175.Dvir A, Peterson SR, Knuth MW, Lu H, Dynan WS. Ku autoantigen is the regulatory component of a template-associated protein kinase that phosphorylates RNA polymerase II. Proc Natl Acad Sci U S A. 1992;89:11920–4. doi: 10.1073/pnas.89.24.11920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Gottlieb TM, Jackson SP. The DNA-dependent protein kinase: requirement for DNA ends and association with Ku antigen. Cell. 1993;72:131–42. doi: 10.1016/0092-8674(93)90057-w. [DOI] [PubMed] [Google Scholar]
- 177.Falck J, Coates J, Jackson SP. Conserved modes of recruitment of ATM, ATR and DNA-PKcs to sites of DNA damage. Nature. 2005;434:605–11. doi: 10.1038/nature03442. [DOI] [PubMed] [Google Scholar]
- 178.Andegeko Y, et al. Nuclear retention of ATM at sites of DNA double strand breaks. J Biol Chem. 2001;276:38224–30. doi: 10.1074/jbc.M102986200. [DOI] [PubMed] [Google Scholar]
- 179.Kitagawa R, Bakkenist CJ, McKinnon PJ, Kastan MB. Phosphorylation of SMC1 is a critical downstream event in the ATM-NBS1-BRCA1 pathway. Genes Dev. 2004;18:1423–38. doi: 10.1101/gad.1200304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Chang YF, Imam JS, Wilkinson MF. The nonsense-mediated decay RNA surveillance pathway. Annu Rev Biochem. 2007;76:51–74. doi: 10.1146/annurev.biochem.76.050106.093909. [DOI] [PubMed] [Google Scholar]
- 181.Sarbassov DD, et al. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr Biol. 2004;14:1296–302. doi: 10.1016/j.cub.2004.06.054. [DOI] [PubMed] [Google Scholar]
- 182.Hara K, et al. Raptor, a binding partner of target of rapamycin (TOR), mediates TOR action. Cell. 2002;110:177–89. doi: 10.1016/s0092-8674(02)00833-4. [DOI] [PubMed] [Google Scholar]
- 183.Lee JH, Paull TT. ATM Activation by DNA Double-Strand Breaks Through the Mre11-Rad50-Nbs1 Complex. Science. 2005;308:551–4. doi: 10.1126/science.1108297. [DOI] [PubMed] [Google Scholar]
- 184.Kim DH, et al. GbetaL, a positive regulator of the rapamycin-sensitive pathway required for the nutrient-sensitive interaction between raptor and mTOR. Mol Cell. 2003;11:895–904. doi: 10.1016/s1097-2765(03)00114-x. [DOI] [PubMed] [Google Scholar]
- 185.Long X, Lin Y, Ortiz-Vega S, Yonezawa K, Avruch J. Rheb binds and regulates the mTOR kinase. Curr Biol. 2005;15:702–13. doi: 10.1016/j.cub.2005.02.053. [DOI] [PubMed] [Google Scholar]
- 186.Takai H, Wang RC, Takai KK, Yang H, de Lange T. Tel2 regulates the stability of PI3K-related protein kinases. Cell. 2007;131:1248–59. doi: 10.1016/j.cell.2007.10.052. [DOI] [PubMed] [Google Scholar]
- 187.Itakura E, et al. ATR-dependent phosphorylation of ATRIP in response to genotoxic stress. Biochem Biophys Res Commun. 2004;323:1197–202. doi: 10.1016/j.bbrc.2004.08.228. [DOI] [PubMed] [Google Scholar]
- 188.Myers JS, Zhao R, Xu X, Ham AJ, Cortez D. Cyclin-dependent kinase 2 dependent phosphorylation of ATRIP regulates the G2-M checkpoint response to DNA damage. Cancer Res. 2007;67:6685–90. doi: 10.1158/0008-5472.CAN-07-0495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Venere M, Snyder A, Zgheib O, Halazonetis TD. Phosphorylation of ATR-interacting protein on Ser239 mediates an interaction with breast-ovarian cancer susceptibility 1 and checkpoint function. Cancer Res. 2007;67:6100–5. doi: 10.1158/0008-5472.CAN-07-0369. [DOI] [PubMed] [Google Scholar]