

Abstract
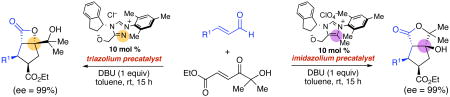
Chiral triazolium and imidazolium-derived N-heterocyclic carbene catalysts promote the direct annulation of α,β-unsaturated aldehydes and achiral α-hydroxyenones to afford cyclopentane-fused lactones with high enantioselectivity. Remarkably, otherwise structurally identical imidazolium and triazolium precatalysts afford different major products. These studies provide both an efficient entry to valuable chiral structures and a dramatic demonstration of stereodivergency of chiral imidazolium versus triazolium-derived N-heterocyclic carbene catalysts.
Intense interest in the use of azolium salts as precursors to N-heterocyclic carbene catalysts has led to a new generation of stereoselective transformations under exceptionally mild and convenient reaction conditions.1 Underlying this explosion of new methods are intricate and subtle effects of the reagents and catalysts in selecting discrete mechanistic manifolds,2 making possible the catalytic generation of acyl anions,3 homoenolates,4 enolates,5 and activated carboxylates6 from a common α,β-unsaturated aldehyde starting material.
As part of these efforts, we reported a cis-selective, highly enantioselective cyclopentene-forming annulation of enals and enones promoted by chiral triazolium precatalyst 1•Cl.7 This work complimented the elegant report of Nair on cyclopentene-forming annulations of α,β-unsaturated aldehydes and chalcones catalyzed by achiral imidazolium-derived carbenes.8 The unexpected formation of cyclopentenes was rationalized by spontaneous decarboxylation of a β-lactone intermediate,9 a facet of the reactions that initially appeared to limit them to substrates containing aromatic ketones; enones that would give stable lactone products were unsuccessful.10 In this report, we document the use of α-hydroxyenones11 as reactive substrates that trap the products at the lactone stage, thereby capturing the stereochemical and functional complexity of these previously transient intermediates (Scheme 1).12 Furthermore, we disclose a remarkable stereochemical divergence of reactions promoted by otherwise structurally identical chiral triazolium and imidazolium precatalysts that offers both synthetic utility and a window on the divergent reaction cascades of these two catalyst types.
Scheme 1.

Our attempts to trap the postulated activated carboxylate IV (see Scheme 2) with a pendant nucleophile began by investigating hydroxyenone 3a as an annulation substrate. Following reaction optimization we identified conditions that afforded high yields of a mixture of lactone products. Careful structure determination of the products by X-ray revealed that only three lactone products were formed, with the major component constituting >65% of the isolated material. To our surprise, this product was cis-substituted β-lactone 4a (Table 1). The minor products were trans-substituted γ-lactone 5a and a small amount of β-lactone 6a, with pseudoenantiomeric stereochemistry at the cyclopentane substitutents. The expected cis-substituted γ-lactone 7a (see Table 2) was not detected in reactions employing 1 as precatalyst. Although the product ratios varied somewhat depending on the cinnamaldehyde employed, annulations with 3a promoted by 1•Cl consistently provided the cis-substituted β-lactone as the major product in 99% ee (Table 1, entries 1–6). Interestingly, albeit in accord with our prior work,7 aryl substituted enones 3b and 3c afforded trans-substituted cyclopentane lactones (entries 7–8). In our prior studies, the trans products were formed in lower (∼60%) ee. In this case the enantiomeric, catalyst-bound intermediates partition at the lactone forming step into isomeric products 5 and 6, and each are formed with excellent enantioselectivity.
Scheme 2.

Table 1.
Triazolium catalyzed annulations of enals and alpha-hydroxyenones.a
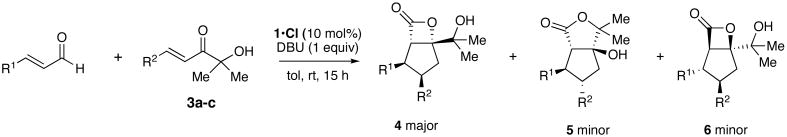 | |||||
|---|---|---|---|---|---|
| entry | R1 = | R2 = | % yieldb | 4:5:6c | % eed |
| 1 (a) | Ph | CO2Et (3a) | 95 (65) | 7e:2:1 | 99;88;99 |
| 2 (b) | p-MeOC6H4 | CO2Et (3a) | 90 (60) | 6:2:1 | 99 |
| 3 (c) | p-BrC6H4 | CO2Et (3a) | 91 (62) | 6:2f:1 | 99 |
| 4 (d) | p-CF3C6H4 | CO2Et (3a) | 89 (35) | 2:1:2 | 99 |
| 5 (e) | 1-naphthyl | CO2Et (3a) | (45) | nd | 99 |
| 6 (f) | 2-furyl | CO2Et (3a) | (40) | nd | 99 |
| 7 (g) | Ph | p-BrC6H4 (3b) | 73 (50) | –:3f:1f | –;93;99 |
| 8 (h) | Ph | p-CF3C6H4 (3c) | 91 (71) | –:4:1 | –;92;99 |
See Supporting Information for reaction details.
Total isolated yields of lactone products; yields of the major product in parenthesis
Determined by 1H NMR analysis of unpurified reaction mixtures.
Enantiomeric excesses of 4,5,6 as determined by SFC analysis on chiral columns. The ee of the minor products in entries 2–7 were not determined.
Structure determined by X-ray analysis of the corresponding methyl ester.
Structure determined by X-ray analysis.
Table 2.
Chiral imidazolium catalyzed annulations.a
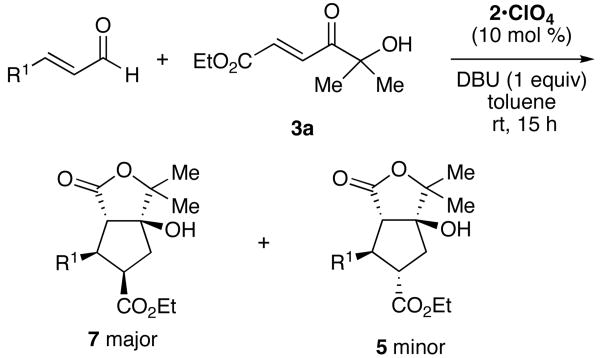 | ||||
|---|---|---|---|---|
| entry | R | % yieldb | 7:5c | % eed |
| 1 (a) | Ph | 85 | 5:1 | 99; 6 |
| 2 (b) | p-BrC6H4 | 76 | 3e:1 | 99; 4 |
| 3 (c) | 2-furyl | 80 | 3:1 | 99; 8 |
| 4 (d) | 2-thiophenyl | 68 | 3:1 | 99; 28 |
See Supporting Information for reaction details.
Total isolated yields of lactone products.
Determined by 1H NMR analysis of unpurified reaction mixtures.
Determined by SFC analysis on chiral columns.
Structure determined by X-ray analysis of the product from ent-2•ClO4.
In surprising contrast, the use of imidazolium-derived precatalyst 2•ClO413,14 resulted in preferential formation of cis-substituted γ-lactone 7, to the complete exclusion of cis-substituted β-lactone 4, which was the major product with triazolium 1 (Table 2). This is the first example of a high yielding and highly enantioselective annulation reaction catalyzed by a chiral imidazolium-derived carbene. The minor product of the reaction, 5, was identical to that from the triazolium catalyst reaction, but was formed in only 6% enantiomeric excess. The minor trans-substituted β-lactone 6 was not observed.
Subtle effects in the conditions or catalysts employed for NHC-catalyzed annulations can have dramatic effects on the products. In this case, an atomic substitution of carbon for nitrogen at a remote site of the precatalysts leads to a complete change in diastereoselectivity, resulting in the formation of the γ-lactone.
Consideration of the reaction cascade leading to the formation of the lactone products can shed some light on the stereochemical divergence. The initial bond-forming event for the formation of cis-configured products 4 and 7 is likely to be a tandem or concerted benzoin–oxy-Cope reaction via a boat-like transition state (Scheme 2). This mechanism, rather than direct conjugate addition of an extended Breslow intermediate, provides a basis for both the observed cis-selectivity as well as a platform for the high levels of enantioinduction. The fate of resulting intermediate III appears to depend on the nature of the catalyst employed via differences in the facial selectivity of the intramolecular aldol reaction. We initially hypothesized that intermediate III, when formed with the imidazolium catalyst (cat = 2), was slow to undergo aldol reaction; however, all attempts to trap it by addition of nucleophiles failed. We therefore speculate that both catalysts initially provide product IV but the imidazolium-derived activated carboxylate (IV, cat = 2) is not competent for β-lactone formation. The poor enantioselectivity for the formation of 5 when the imidazolium precatalyst 2 is employed, in contrast to the high levels observed with triazolium precatalyst 1, reflects the inability of the imidazolium catalyst to form β-lactone 6, thereby funneling both diastereomers of VII to a nearly racemic mixture of γ-lactone 5.
These observations demonstrate a discrete difference in reactivity between otherwise identical imidazolium and triazolium-derived N-heterocyclic carbenes. Based on our current observation and understanding, we attribute this not, in this case, to a mechanistic difference between the reactions of the two catalysts or their interactions with the enal substrates but rather to a discrete difference in the reactivity of the acyl azolium species involved in the lactone-forming step. The fate of postulate tetrahedral intermediate A (Scheme 3), appears to be determined by the leaving group ability of the N-heterocyclic carbene. In the case of the triazolium catalyst, the NHC is a sufficiently good leaving group to afford β-lactones as the major products. In contrast, the identical intermediate derived from the imidazolilum catalyst prefers to eliminate alkoxide, leading to acyl imidazolium B, which undergoes a retro-aldol–aldol sequence mutating the stereochemistry and leading to the formation of the more stable γ-lactone product 7a.
Scheme 3.

In addition to serving as an auxiliary for enhancing the reactivity of enone 3 and, in the case of 7, trapping activated carboxylate VI (Scheme 2), the pendant alcohol offers a synthetic handle for product processing. For example, lactone 4 is readily transformed to enantiopure cyclopentanone 8 (eq 1).
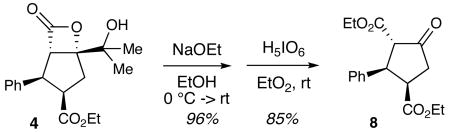 |
(1) |
Our findings establish that otherwise identical triazolium and imidazolium derived NHC-catalysts can effect stereodivergent transformations. These intriguing results make a compelling case for further design of chiral azolium salts and investigations into the remarkable properties of these catalysts and the powerful synthetic transformation they enable under simple, operationally friendly reaction conditions.
Supplementary Material
Experimental procedures and characterization data for all compounds. This material is available free of charge via the internet at http://pubs.acs.org.
Acknowledgments
We are grateful to the NIH (GM-079339) and generous gifts from Boehringer-Ingelheim, Bristol Myers Squibb, Eli Lilly, and Roche for support of this research program. J.K. is a predoctoral fellow of the Royal Thai Government. We thank Justin Struble for 1 and 2 and Guang Wu (UCSB) and Pat Sheppard (UPenn) for X-ray analyses.
References
- 1.(a) Enders D, Niemeier O, Henseler A. Chem Rev. 2007;107:5606–5655. doi: 10.1021/cr068372z. [DOI] [PubMed] [Google Scholar]; (b) Marion N, Díez-González S, Nolan SP. Angew Chem Int Ed. 2007;46:2988–3000. doi: 10.1002/anie.200603380. [DOI] [PubMed] [Google Scholar]
- 2.Zeitler K. Angew Chem Int Ed. 2005;44:7506–7510. doi: 10.1002/anie.200502617. [DOI] [PubMed] [Google Scholar]
- 3.(a) Enders D, Breuer K, Runsink J, Teles JH. Helv Chim Acta. 1996;79:1899–1902. [Google Scholar]; (b) Kerr MS, Read de Alaniz J, Rovis T. J Am Chem Soc. 2002;124:10298–10299. doi: 10.1021/ja027411v. [DOI] [PubMed] [Google Scholar]; (c) Read de Alaniz J, Rovis T. J Am Chem Soc. 2005;127:6284–6289. doi: 10.1021/ja0425132. [DOI] [PubMed] [Google Scholar]; (d) Johnson JS. Angew Chem Int Ed. 2004;43:1326–1328. doi: 10.1002/anie.200301702. [DOI] [PubMed] [Google Scholar]
- 4.For the first reports, see: Sohn SS, Rosen EL, Bode JW. J Am Chem Soc. 2004;126:14370–14371. doi: 10.1021/ja044714b.Burstein C, Glorius F. Angew Chem Int Ed. 2004;43:6205–6208. doi: 10.1002/anie.200461572.
- 5.(a) He M, Struble JR, Bode JW. J Am Chem Soc. 2006;128:8418–8420. doi: 10.1021/ja062707c. [DOI] [PubMed] [Google Scholar]; (b) He M, Uc GJ, Bode JW. J Am Chem Soc. 2006;128:15088–15089. doi: 10.1021/ja066380r. [DOI] [PubMed] [Google Scholar]; (c) Phillips EM, Wadamoto M, Chan A, Scheidt KA. Angew Chem Int Ed. 2007;46:3107–3110. doi: 10.1002/anie.200605235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.(a) Sohn SS, Bode JW. Org Lett. 2005;7:3873–3876. doi: 10.1021/ol051269w. [DOI] [PubMed] [Google Scholar]; (b) Chan A, Scheidt KA. Org Lett. 2005;7:905–908. doi: 10.1021/ol050100f. [DOI] [PubMed] [Google Scholar]
- 7.Chiang PC, Kaeobamrung J, Bode JW. J Am Chem Soc. 2007;129:3520–3521. doi: 10.1021/ja0705543. [DOI] [PubMed] [Google Scholar]
- 8.Nair V, Vellalath S, Poonoth M, Suresh E. J Am Chem Soc. 2006;128:8736–8737. doi: 10.1021/ja0625677. [DOI] [PubMed] [Google Scholar]
- 9.For reviews on β-lactone chemistry: Pommier A, Pons JM. Synthesis. 1993:441–459.Yang HW, Romo D. Tetrahedron. 1999;55:6403–6434.Henry-Riyad H, Lee C, Purohit VC, Romo D. Org Lett. 2006;8:4363–4366. doi: 10.1021/ol061816t. Romo has reported that [3.2.0]-bicyclic β-lactone bearing aliphatic substituents do not undergo spontaneous decarboxylation.
- 10.This limitation can be overcome if the reaction is run in an intramolecular fashion: Wadamoto M, Phillips EM, Reynolds TE, Scheidt KA. J Am Chem Soc. 2007;129:10098–10099. doi: 10.1021/ja073987e.
- 11.For the use of achiral α-hydroxyenones in asymmetric catalysis, see: Palomo C, Oiarbide M, García JM, González A, Arceo E. J Am Chem Soc. 2003;125:13942–13943. doi: 10.1021/ja0368002.Palomo C, Oiarbide M, Halder R, Keiso M, Gómez-Bengoa E, García JM. J Am Chem Soc. 2004;126:9188–9189. doi: 10.1021/ja047004e.
- 12.We have reported the synthesis of β-lactams from chalcone-derived imines: He M, Bode J. J Am Chem Soc. 2008;130:418–419. doi: 10.1021/ja0778592. Imidazolium 2•ClO4 is ineffective for these reactions.
- 13.Due to availability at the onset of our studies, ent-2•ClO4 was used for some entries (see Supporting Information). Identical results were obtained with 2 for the preparation of the other enantiomers. No effect of the counterion was detected, and the change in counterion between the catalysts types does not affect the stereochemical outcome.
- 14.For the synthesis of 2•ClO4, see: Struble JR, Kaeobamrung J, Bode JW. Org Lett. 2008;10:957–960. doi: 10.1021/ol800006m.Struble JR, Bode JW. Tetrahedron. 2008;64:6961–6972.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Experimental procedures and characterization data for all compounds. This material is available free of charge via the internet at http://pubs.acs.org.