

Abstract
Silacarbonyl ylides, generated by metal-catalyzed silylene transfer to carbonyls, participate in formal intermolecular 1,3-dipolar cycloaddition reactions with carbonyl compounds and alkynes to form dioxasilacyclopentane acetals and oxasilacyclopentenes in an efficient, one-step process.
The reactions of silylenes with carbonyl compounds have produced a number of mechanistically and synthetically useful reactions. Silylenes (1) react with aldehydes and ketones (2) to give either oxasilacyclopropanes 3 or silacarbonyl ylides 4 (Scheme 1),1 and these isomeric products can be interconverted.2 The silacarbonyl ylide can be intercepted by π-bonds to form products formally arising from 1,3-dipolar cycloadditions.3 This reaction, which forms new carbon–carbon and carbon–silicon bonds, has not been found to be preparatively useful because yields are often low, even in intramolecular examples.1a In addition, the generation of silylenes requires forcing conditions such as extended photolysis with excess reagents.4
Scheme 1.

Silylene Transfer to Carbonyl Compounds
In this Letter, we report that metal-catalyzed silylene transfer to carbonyl compounds in the presence of an electron-deficient alkyne produced formal 1,3-dipolar cycloaddition products efficiently under mild conditions. The products of these reactions possess functional groups that can be functionalized with control of regioselectivity.
We discovered that silylene transfer to carbonyl compounds led to three-component cyclization reactions in our attempts to form oxasilacyclopropanes. Treatment of cyclohexene silacyclopropane 65 with two equivalents of benzaldehyde 5 and a catalytic amount of silver triflate did not yield the expected three-membered ring. Instead, it gave the product of silylene transfer to the C–O double bond and subsequent insertion of the second equivalent of aldehyde to form dioxasilacyclopentane 7 (Scheme 2).6 A screen of metal salts known to catalyze silylene transfer to alkenes7 did not lead to formation of oxasilacyclopropanes. When n-butyraldehyde (8) was subjected to the same catalyst screen, copper (II) bromide-catalyzed silylene transfer afforded the intermolecular 1,3-dipolar cycloaddition product, dioxasilacyclopentane acetal 9, in 89% yield (Scheme 2). A competition experiment between aldehydes 5 and 8 yielded dioxasilacyclopentane 7 as the sole product, suggesting that the difference in regioselectivity of the corresponding products 7 and 9 is caused by a divergence in the mechanistic pathways.
Scheme 2.

Silylene Transfer to Aldehydes
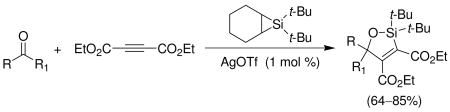
Silylene transfer to benzaldehyde in the presence of electron-deficient alkyne 10 led to a different three-component coupling reaction. When one equivalent of alkyne 10 and one equivalent of benzaldehyde (5) were treated under metal-catalyzed silylene transfer conditions, oxasilacyclopentene 11 was formed in 85% yield (Scheme 3). This reaction is complementary to our earlier synthesis of oxasilacyclopentenes.8 That method involved a two-step process whereupon silacyclopropenes, prepared from alkynes, underwent insertion reactions with carbonyl compounds. Electron-deficient alkynes, however, did not participate in that reaction because the silylenoid intermediate is electrophilic, not nucleophilic.9
Scheme 3.

Formation of Oxasilacyclopentene
A variety of carbonyl compounds participated in the cycloaddition employing alkyne 10 as the dipolarophile (Table 1). The cycloaddition reaction tolerated α,β–unsaturated aldehydes and formate esters (entries 1 and 2). Formamides also reacted to form N,O-acetal oxasilacyclopentene 13c, although the reaction was slower. Ketones underwent cyclization to form oxasilacyclopentenes containing tetrasubstituted carbon atoms (entry 4). α,β–Unsaturated ester 12e reacted with alkyne 10 to form oxasilacyclopentene 13e, although silylene transfer to the α,β–unsaturated ester was also observed (Scheme 4). 10 Excess ester and silacyclopropane were employed to optimize the formation of oxasilacyclopentene 13e (entry 5).
Table 1.
Silver-Catalyzed Silylcarbonyl Ylide Cycloaddition
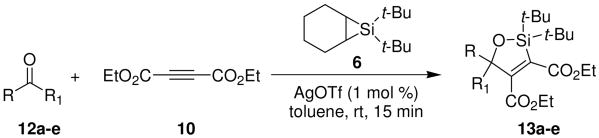
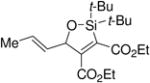
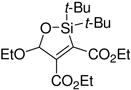
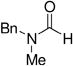
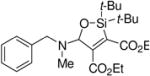
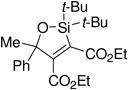
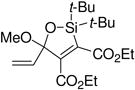
At 50 °C for 12 h.
For 3 h.
Four equivalents of 6 and four equivalents of 12e.
Scheme 4.

Silylene Transfer to α-β-Unsaturated Ester
The highly electron-deficient alkyne 10 was the only alkyne that participated in these reactions. Treatment of silacyclopropane 6 and carbonyl compounds with other dipolarophiles led to either silylene transfer to the alkyne to form silacyclopropenes or dimerization with the aldehyde to form dioxasilacyclopentane 7 (Scheme 5).
Scheme 5.

Dipolarophile Screen
a As determined by 1H NMR spectroscopic analysis of the product relative to an internal standard (PhSiMe3).
Control experiments indicated that the formation of the oxasilacyclopentene did not involve a silacyclopropene intermediate. When electron-deficient alkyne 10 was treated with silacyclopropane 6, formation of silacyclopropene 19 was not detected (Scheme 6). The 29Si NMR spectrum of the reaction mixture revealed two peaks at δ 18.0 and 4.8 ppm, with no peaks around δ −70 ppm, which would be expected for a silacyclopropene.11 The silylene transfer product could not be indentified, but the chemical shifts were consistent with the presence of alkyl silanes.
Scheme 6.

Silacyclopropene Control Experiment
It has been previously reported that electrophilic carbenes react with aldehydes to form carbonyl ylides that then undergo 1,3-dipolar cycloadditions preferentially with electron-deficient alkynes.12 This precedent, suggests that the formation of the oxasilacyclopentenes occurs through an analogous concerted 1,3-dipolar cycloaddition pathway (Scheme 7).
Scheme 7.

Proposed Cycloaddition
The oxasilacyclopentenes that possessed acetal moieties could be elaborated by nucleophilic substitution reactions. Oxasilacyclopentene 13b was treated with allyltrimethylsilane and SnBr4 to afford allylated oxasilacyclopentene 21 (Scheme 8), although the modest stabilization of the oxocarbenium ion intermediate by the electron-deficient ester required long reaction times (40 hours).13 Oxasilacyclopentene 13e could be allylated under the same reaction conditions as 13b, but an unanticipated product was observed. Upon Lewis acid activation to form the oxocarbenium ion, allyltrimethylsilane added to the unsubstituted terminus of the conjugated system, resulting in oxasilacyclopentene 22.14
Scheme 8.

Nucleophilic Substitution to Oxasilacyclopentenes
The carbonyl groups can be functionalized and appear to be sterically differentiated. Oxasilacyclopentene 11 was reduced by i-Bu2AlH to form diol 23. When oxasilacyclopentene 11 was treated with two equivalents of methylmagnesium bromide, bicyclic α,β–unsaturated lactone 24 was formed (Scheme 9). Addition of one equivalent of a Grignard reagent resulted in a 1:1 mixture of starting material and lactone 24. These experiments reveal that the two carbonyls can be functionalized independently or simultaneously, and suggest that the dissimilarity is due to the steric hindrance imparted by the bulky tert-butyl groups attached to the silicon atom.
Scheme 9.

Functionalization of Carbonyls
Conversion of oxasilacyclopentene 13d to allylic alcohol 25 demonstrates the synthetic utility of the cycloaddition reaction. Initial attempts at removing the silicon atom through conventional protodesilylation methods were unsuccessful.15 The silicon atom could be removed to form allylic alcohol 25 utilizing palladium salts and Bu4NF as the fluoride source.16 Oxasilacyclopentene 11 also underwent protodesilylation, but allylic alcohol 26 isomerized to provide ketone 27.
In conclusion, silylene transfer to carbonyl compounds can be used to produce dioxasilacyclopentane acetals and oxasilacyclopentenes in a single step through formal intermolecular 1,3-dipolar cycloadditions. The oxasilacyclopentene products are useful intermediates that can be elaborated by substitution, addition, and protodesilylation reactions.
Supplementary Material
Supporting Information Available: Experimental procedures; spectroscopic, and analytical data for the products (PDF). This material is available free of charge via the Internet at http://pubs.acs.org.
Scheme 10.

Removal of Silicon
Acknowledgments
This research was supported by the National Institute of General Medical Sciences of the National Institutes of Health (GM-54909). L.E.B. thanks the Department of Education (GAANN) for a predoctoral fellowship. K.A.W. thanks Amgen and Lilly for awards to support research. We thank Dr. Phil Dennison (UCI) for assistance with NMR spectroscopy and Dr. John Greaves and Ms. Shirin Sorooshian (UCI) for mass spectrometry.
References
- 1.(a) Ando W, Ikeno M, Sekiguchi A. J Am Chem Soc. 1978;100:3613–3615. [Google Scholar]; (b) Ando W, Hagiwara K, Sekiguchi A. Organometallics. 1987;6:2270–2271. [Google Scholar]; (c) Sakai N, Fukushima I, Minakata S, Ryu I, Komatsu M. Chem Commun. 1999:1857–1858. [Google Scholar]
- 2.(a) Gordon MS, George C. J Am Chem Soc. 1984;106:609–611. [Google Scholar]; (b) Becerra R, Cannady JP, Walsh R. J Phys Chem. 1999;103:4457–4464. [Google Scholar]; (c) Becerra R, Cannady JP, Walsh R. Phys Chem Chem Phys. 2001;3:2343–2351. [Google Scholar]
- 3.Padwa A, Weingarten MD. Chem Rev. 1996;96:223–270. doi: 10.1021/cr950022h. [DOI] [PubMed] [Google Scholar]
- 4.(a) Sakai N, Fukushima T, Okada A, Ohashi S, Minakata S, Komatsu M. J Organomet Chem. 2003;686:368–372. [Google Scholar]; (b) Minakata S, Ohashi S, Amano Y, Oderaotoshi Y, Komatsu M. Synthesis. 2007:2481–2484. [Google Scholar]
- 5.Boudjouk P, Black E, Kumarathasan R. Organometallics. 1991;10:2095–2096. [Google Scholar]
- 6.(a) Jutzi P, Eikenberg D, Bunte EA, Möhrke A, Neumann B, Stammler HG. Organometallics. 1996;15:1930–1934. [Google Scholar]; (b) Belzner J, Ihmels H, Pauletto L, Noltemeyer M. J Org Chem. 1996;61:3315–3319. [Google Scholar]; (c) Franz AK, Woerpel KA. J Am Chem Soc. 1999;121:949–957. [Google Scholar]
- 7.Ćiraković J, Driver TG, Woerpel KA. J Org Chem. 2004;69:4007–4012. doi: 10.1021/jo0355505. [DOI] [PubMed] [Google Scholar]
- 8.(a) Seyferth D, Vick SC, Shannon ML. Organometallics. 1984;3:1897–1905. [Google Scholar]; (b) Clark TB, Woerpel KA. J Am Chem Soc. 2004;126:9522–9523. doi: 10.1021/ja047498f. [DOI] [PubMed] [Google Scholar]
- 9.Driver TG, Woerpel KA. J Am Chem Soc. 2004;126:9993–10002. doi: 10.1021/ja0306563. [DOI] [PubMed] [Google Scholar]
- 10.Calad SA, Woerpel KA. J Am Chem Soc. 2005;127:2046–2047. doi: 10.1021/ja042893r. [DOI] [PubMed] [Google Scholar]
- 11.Palmer WS, Woerpel KA. Organometallics. 1997;16:4824–4827. [Google Scholar]
- 12.(a) Hodgson DM, Glen R, Grant GH, Redgrave AJ. J Org Chem. 2003;68:581–586. doi: 10.1021/jo026307t. [DOI] [PubMed] [Google Scholar]; (b) Russell AE, Brekan J, Gronenberg L, Doyle MP. J Org Chem. 2004;69:5269–5274. doi: 10.1021/jo049403y. [DOI] [PubMed] [Google Scholar]
- 13.Creary X, Geiger CC. J Am Chem Soc. 1982;104:4151–4162. [Google Scholar]
- 14.One alkene isomer was observed. We have assigned the product as the Z-alkene isomer because this configuration should minimize steric interactions. However, spectroscopic experiments showed no NOE between the vinyl hydrogen and the carboethoxy groups.
- 15.Heitzman CL, Lambert WT, Mertz E, Shotwell JB, Tinsley JM, Va P, Roush WR. Org Lett. 2005;7:2405–2408. doi: 10.1021/ol0506821. and references therein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Copper salts have been employed in protodesilylation reactions: Trost BM, Ball ZT, Jöge T. J Am Chem Soc. 2002;124:7922–7923. doi: 10.1021/ja026457l.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information Available: Experimental procedures; spectroscopic, and analytical data for the products (PDF). This material is available free of charge via the Internet at http://pubs.acs.org.