

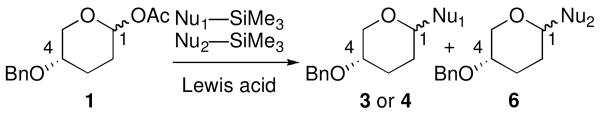
Abstract
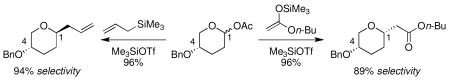
The effect of nucleophile strength on diastereoselectivity in the nucleophilic substitution of cyclic acetals was explored. Stereoselectivity remained constant and high as nucleophilicity increased until a threshold value was reached. Beyond this point, however, selection of Lewis acid determined whether stereochemical inversion or erosion was observed.
The development of stereocontrolled glycosylation reactions is complicated by the fact that these processes may proceed via SN1-like1-3 or SN2-like4-10 mechanisms. Changes in the glycosyl donor,11,12 nucleophile,13,14 activator,15 and solvent16 can alter selectivity unpredictably. This report documents the relationship between nucleophile strength and stereoselectivity for the substitution reactions of cyclic acetals; we describe dramatic changes in stereoselectivity and provide mechanistic rationales for these findings. This study provides insight applicable to the development of new stereoselective glycosylation reactions.
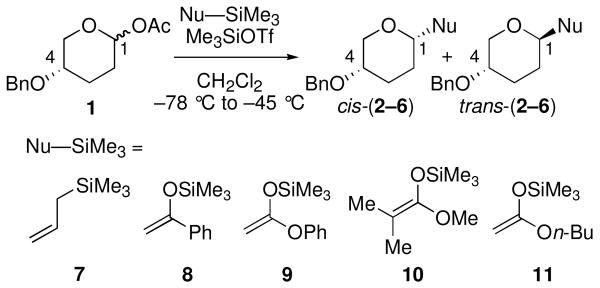
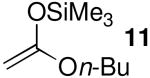
Acetal 1 was treated with a panel of nucleophiles having known nucleophilicity parameters (N)17 in the presence of Me3SiOTf (Table 1). A nucleophile's N value is a direct measure of reactivity: it correlates logarithmically with its rate of reaction with carbocationic electrophiles.17 Reactions with π-nucleophiles spanning more than four orders of magnitude of nucleophilicity led to selective formation of 1,4-trans products (entries 1 and 2). A roughly one hundred-fold further increase in N, however, associated with application of silylketene acetal nucleophiles 9–11, resulted in reversal of diastereoselectivity: 1,4-cis products were formed selectively (entries 3–5). This dichotomy in stereochemical outcomes suggests a change in reaction mechanism.18
Table 1.
Nucleophile Screen with Me3SiOTf
 | |||||
|---|---|---|---|---|---|
| entry | Nu–SiMe3 | Na | product | cis:transb,c | yield (%)d |
| 1 | 7 | 1.8 | 2 | 6:94 | 96 |
| 2 | 8 | 6.2 | 3 | 10:90 | 95 |
| 3 | 9 | 8.2 | 4 | 71:29 | 83 |
| 4 | 10 | 9.0 | 5 | 85:15 | 93 |
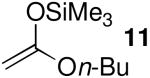
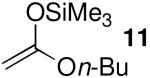
| 5 | 11 | 10.2 | 6 | 89:11 | 96 |
N = nucleophilicity parameter; see ref 17.
Determined by GC and 1H NMR spectroscopic analysis of the unpurified reaction mixture.
Diastereoselectivities were independent of starting anomer ratio.
Isolated yield.
We have reported previously an electrostatic model to explain the trans selectivities observed in the reactions of acetal 1 with weak nuceophiles (e.g., 7 and 8).19,20 These reactions occur by SN1-type mechanisms involving oxocarbenium ion intermediate I (Scheme 1).21 Axial attack on the electrostatically preferred axial conformer Iax affords trans products via a chair-like transition state. This model, however, does not account for the cis selectivities observed when strong nucleophiles 9–11 react with 1. It is unlikely that the 1,4-cis ester products cis-(4–6) arise from disfavored equatorial conformer Ieq because increased nucleophile strength should not alter the conformational equilibrium of the oxocarbenium ion. Moreover, the selectivities of reactions of Iax and Ieq should be independent of nucleophile reactivity unless reaction rates approach the diffusion limit.22
Scheme 1.

Electrostatic Stereochemical Model
The stereochemical inversion observed in Me3SiOTf-activated reactions of electrophile 1 with silylketene acetals 9–11 can be explained by SN2-like substitutions23-27 of triflate-trapped contact ion-pairs28 II via transition state III (Scheme 2). Transition state III is consistent with the electrostatic model. As the triflate group departs from the axial orientation, the transition state (III) would develop significant carbocationic character at C1.9,10 This accumulation of charge would cause the C4-benzyloxy group to adopt an axial orientation to stabilize the charge.19,20 Together, these explanations account for the observed selectivity.29
Scheme 2.

Proposed Transition State for SN2-like Pathway
In contrast to the results activated by Me3SiOTf, nucleophilic substitution reactions of acetal 1 mediated by BF3•OEt2 appeared to proceed via SN1-like mechanisms regardless of nucleophile strength (Table 2). As observed with Me3SiOTf, reactions of relatively weak π-nucleophiles 7 and 8 led to selective formation of 1,4-trans products with BF3•OEt2 (entries 1 and 2). Application of silylketene acetal nucleophiles 9–11 to the BF3•OEt2-mediated reactions of 1, however, led to loss of stereoselectivity (entries 3–5). It is possible that these low selectivities reflect competition between SN2-like and SN1-like reaction mechanisms. The borate anions formed in the BF3•OEt2-mediated reactions, however, are likely to coordinate quite poorly, disfavoring SN2-like processes.30 Further, unselective reactions were obtained with all three silylketene acetal nucleophiles (9–11) despite differences in steric bulk and nucleophilicity; this finding suggests a statistical process.31
Table 2.
Nucleophile screen with BF3•OEt2
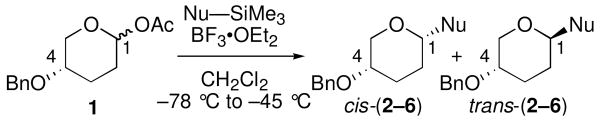 | |||||
|---|---|---|---|---|---|
| entry | Nu–SiMe3 | Na | product | cis:transb,c | yield (%)d |
| 1 | 7 | 1.8 | 2 | 8:92 | 82 |
| 2 | 8 | 6.2 | 3 | 8:92 | 87 |
| 3 | 9 | 8.2 | 4 | 50:50 | 88 |
| 4 | 10 | 9.0 | 5 | 58:42 | 80 |
| 5 | 11 | 10.2 | 6 | 60:40 | 86 |
N = nucleophilicity parameter; see ref 17.
Determined by GC and 1H NMR spectroscopic analysis of the unpurified reaction mixture.
Diastereoselectivities were independent of starting anomer ratio.
Isolated yield.
The loss of stereoselectivity in BF3•OEt2-mediated reactions of 1 with silylketene acetal nucleophiles 9–11 can be explained by SN1-like nucleophilic attack at the diffusion limit (Scheme 3).22,32 Encounter complexes24 IV and V are expected to form with no facial selectivity. If the rates of nucleophilic attack on the encounter complexes IV and V (k2 and k3) approach the rate of diffusion (k1 = k-1 ∼109 M−1s−1),17,33 product ratios will reflect the initial statistical mixture of IV and V.22,32 In this scenario, every nucleophile-electrophile collision will lead to product, so no selectivity is observed.34
Scheme 3.

Diffusion Limit Model
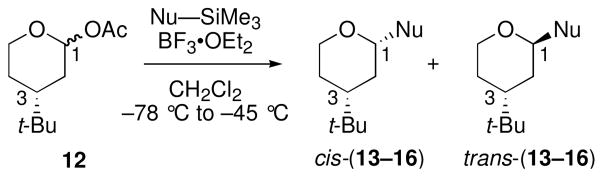
The model for loss of stereoselectivity depicted in Scheme 3 requires reaction via twist-boat intermediate VII. To test the viability of this intermediate, C3-tert-butyl acetal 12 was prepared and treated with a panel of nucleophiles under BF3•OEt2 activation (Table 3). As observed for acetal 1, stereoselectivities were high for relatively weak nucleophiles 7 and 8, but eroded with silylketene acetal nucleophiles 9 and 11.35
Table 3.
Nucleophile Screen with Acetal 12.
 | |||||
|---|---|---|---|---|---|
| entry | Nu–SiMe3 | Na | product | cis:transb,c | yieldd |
| 1 | 7 | 1.8 | 13 | 1:99 | 67 |
| 2 | 8 | 6.2 | 14 | 2:98 | 83 |
| 3 | 9 | 8.2 | 15 | 17:83 | 93 |
| 4 | 11 | 10.2 | 16 | 34:66 | 69 |
N = nucleophilicity parameter; see ref 17.
Determined by GC and 1H NMR spectroscopic analysis of the unpurified reaction mixture.
Diastereoselectivities were independent of starting anomer ratio.
Isolated yield.
The results in Table 3 are consistent with reaction via a twist-boat intermediate. Formation of the major 1,3-trans products arises from axial attack on equatorial conformer VIIIeq through a chair-like transition state (path a, Scheme 4). The minor 1,3-cis product is unlikely to arise by axial attack on minor conformer VIIIax through a chair-like transition state (path c). Not only should the VIIIax/VIIIeq conformational equilibrium favor equatorial conformer VIIIeq,36 but developing 1,3-diaxial interactions between the incoming nucleophile and the axial C3-tert-butyl group of VIIIax should also block substitution by path c. Consequently, cis products are more likely formed by path b, which involves a twist-boat intermediate.
Scheme 4.

C3 t-Bu Stereochemical Model
We next sought further evidence to support the stereochemical models developed for C4-benzyloxy acetal 1. A series of competition experiments between nucleophiles was used to probe both the diffusion-limited rate hypothesis developed for reactions involving BF3•OEt2 (Scheme 3) and the SN2-like pathway invoked in the case of Me3SiOTf (Scheme 2). If at least one of the two nucleophiles involved in a competition experiment reacts with a rate below the diffusion limit, chemoselectivity should be observed.22 This condition should obtain for all reactions of nucleophiles 7 and 8 and for the reactions that proceed by SN2-like reaction paths. Conversely, if both nucleophiles in a competition experiment react with rates at or near the diffusion limit, as proposed in the case of 9–11 with BF3•OEt2, no chemoselectivity should be observed.22
As a control experiment, acetal 1 was treated with an equimolar mixture of enoxysilane nucleophile 8 (N = 6.2) and silylketene acetal 11 (N = 10.2) in the presence of BF3•OEt2 (Table 4, entry 1).37 As expected, a small fraction of 3 was formed, indicating that the enoxysilane 8 did not react with the electrophile at a rate near the diffusion limit.
Table 4.
Effect of Nucleophile on Chemoselectivity
 | ||||
|---|---|---|---|---|
| entry | Nu1–SiMe3a | Nu2–SiMe3a | Lewis acid | product ratiob |
| 1 | 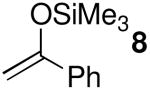 |
 |
BF3•OEt2 | 3(4):6(96) |
| 2 | 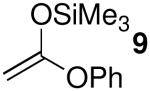 |
 |
BF3•OEt2 | 4(25):6(75) |
| 3 | 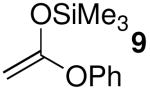 |
 |
Me3SiOTf | 4(13):6(87) |
Five equivalents of nucleophile.
Determined by GC spectroscopic analysis of the unpurified reaction mixture
The chemoselectivity observed when silylketene acetals 9 (N = 8.2) and 11 (N = 10.2) reacted with 1 in the presence of Me3SiOTf (Table 4, entry 3) also implies non-diffusion-limited rates of nucleophilic attack. Triflate-trapped species II should be less electrophilic than oxocarbenium ion encounter complexes IV/V.9,10 Consequently, SN2-type nucleophilic substitution reactions in the presence of triflate anion should occur with rates below the diffusion limit. As noted previously, the stereochemical results with these nucleophiles (Table 1, entries 3 and 5) are consistent with reaction through transition state III (vide supra).
The results of treatment of 1 with silylketene acetals 9 and 11 in the presence of BF3•OEt2 suggested simultaneous operation of both SN2-type and diffusion-limited SN1-type paths. In this reaction, products 4 and 6 were formed in a 25:75 ratio (Table 2, entry 2), suggesting participation of a [BF3-OAc]− counterion in the reaction with 11.
To eliminate the potential for ion pairing, acetal 17, bearing a pivaloate group, was examined. The steric bulk of the pivaloate group should decrease its ability to coordinate, thereby pushing the reaction completely to an SN1-type mechanism. The reaction of 17 with 9 and 11 was unselective: a 56:44 ratio of 4 to 6 (eq 1) was obtained.38 This result confirms the exclusive operation of a diffusion-limited SN1-type mechanism when 9 and 11 react with the free oxocarbenium ion IV/V derived from pivaloate 17.
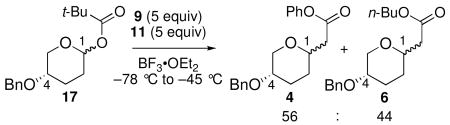 |
(1) |
We have described the effects of varying nucleophile strength on the stereochemical outcomes of acetal substitution reactions. Stereoselective SN1-type mechanisms occur with weak and moderate nucleophiles and poor leaving groups, and unselective diffusion-limited SN1 mechanisms and stereoselective SN2 reaction pathways emerge with strong nucleophiles.
Supplementary Material
Experimental procedures and compound characterization is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
This research was supported by the National Institutes of Health, National Institute of General Medical Sciences (Grant GM-61066 to K.A.W. and Grant GM-081996 to J.R.K). W.A.S. thanks the ARCS foundation for a fellowship. K.A.W. thanks Amgen and Lilly for generous support of research. We thank Dr. Phil Dennison (UCI) for assistance with NMR spectroscopy, and Dr. John Greaves and Ms. Shirin Sorooshian (UCI) for mass spectrometry.
References Cited
- 1.Garcia BA, Gin DY. J Am Chem Soc. 2000;122:4269–4279. [Google Scholar]
- 2.Sammakia T, Smith RS. J Am Chem Soc. 1994;116:7915–7916. [Google Scholar]
- 3.Boebel TA, Gin DY. J Org Chem. 2005;70:5818–5826. doi: 10.1021/jo050294c. [DOI] [PubMed] [Google Scholar]
- 4.Kim JH, Yang H, Park J, Boons GJ. J Am Chem Soc. 2005;127:12090–12097. doi: 10.1021/ja052548h. [DOI] [PubMed] [Google Scholar]
- 5.Nukada T, Berces A, Zgierski MZ, Whitfield DM. J Am Chem Soc. 1998;120:13291–13295. [Google Scholar]
- 6.Berces A, Enright G, Nukada T, Whitfield DM. J Am Chem Soc. 2001;123:5460–5464. doi: 10.1021/ja001194l. [DOI] [PubMed] [Google Scholar]
- 7.Fan E, Shi W, Lowary TL. J Org Chem. 2007;72:2917–2928. doi: 10.1021/jo062542q. [DOI] [PubMed] [Google Scholar]
- 8.Banait NS, Jencks WP. J Am Chem Soc. 1991;113:7951–7958. [Google Scholar]
- 9.Crich D, Chandrasekera NS. Angew Chem Int Ed. 2004;43:5386–5389. doi: 10.1002/anie.200453688. [DOI] [PubMed] [Google Scholar]
- 10.El-Badri MH, Willenbring D, Tantillo DJ, Gervay-Hague J. J Org Chem. 2007;72:4663–4672. doi: 10.1021/jo070229y. [DOI] [PubMed] [Google Scholar]
- 11.Crich D, Sun S. J Org Chem. 1996;61:4506–4507. doi: 10.1021/jo9606517. [DOI] [PubMed] [Google Scholar]
- 12.Crich D, Li L. J Org Chem. 2007;72:1681–1690. doi: 10.1021/jo062294y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Minehan TG, Kishi Y. Tetrahedron Lett. 1997;38:6815–6818. [Google Scholar]
- 14.Brown DS, Bruno M, Davenport RJ, Ley SV. Tetrahedron. 1989;45:4293–4308. [Google Scholar]
- 15.Crich D, Pedersen CM, Bowers AA, Wink DJ. J Org Chem. 2007;72:1553–1565. doi: 10.1021/jo061440x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vankar YD, Vankar PS, Behrendt M, Schmidt RR. Tetrahedron. 1991;47:9985–9992. [Google Scholar]
- 17.Mayr H, Kempf B, Ofial AR. Acc Chem Res. 2003;36:66–77. doi: 10.1021/ar020094c. [DOI] [PubMed] [Google Scholar]
- 18.A control experiment was performed to verify that diastereomer ratios resulted from kinetic product distributions under these reaction conditions. An isolated sample of trans-4 was treated with silylketene acetal 11 (4.0 equiv) and Me3SiOTf (1.6 equiv) under standard reaction conditions. The ester trans-4 did not react under these conditions: neither stereochemical inversion to cis-4 nor chemical exchange to form cis- or trans-6 was observed.
- 19.Romero JAC, Tabacco SA, Woerpel KA. J Am Chem Soc. 2000;122:168–169. [Google Scholar]
- 20.Ayala L, Lucero CG, Romero JAC, Tabacco SA, Woerpel KA. J Am Chem Soc. 2003;125:15521–15528. doi: 10.1021/ja037935a. [DOI] [PubMed] [Google Scholar]
- 21.Shenoy S, Woerpel KA. Org Lett. 2005;7:1157–1160. doi: 10.1021/ol0500620. [DOI] [PubMed] [Google Scholar]
- 22.Mayr H, Ofial AR. Angew Chem Int Ed. 2006;45:1844–1854. doi: 10.1002/anie.200503273. [DOI] [PubMed] [Google Scholar]
- 23.Winstein S, Grunwald E, Jones HW. J Am Chem Soc. 1951;73:2700–2707. [Google Scholar]
- 24.Jencks WP. Acc Chem Res. 1980;13:161–169. [Google Scholar]
- 25.Richard JP, Jencks WP. J Am Chem Soc. 1982;104:4689–4691. [Google Scholar]
- 26.Ta-Shma R, Rappoport Z. J Am Chem Soc. 1983;105:6082–6095. [Google Scholar]
- 27.Denmark SE, Almstead NG. J Org Chem. 1991;56:6485–6487. [Google Scholar]
- 28.Crich D, Sun S. J Am Chem Soc. 1997;119:11217–11223. [Google Scholar]
- 29.This argument implies that one of two possible scenarios obtain: a) The formation of anomeric triflates II occurs irreversibly to favor the trans isomer diastereoselectively, as observed in analogous nucleophilic additions to oxocarbenium ions (references 19 and 20). The resulting dominant trans triflate isomer, then, undergoes stereospecific SN2 substitution to form cis products. b) Anomeric isomers of triflate II are formed reversibly and interconvert rapidly via an oxocarbenium ion intermediate, presumably in a solvent cage with the triflate counterion. Cis selectivity, in this case, arises from a preference for reaction through trans diaxial triflate II. Attempts to observe triflate II at low temperature have been unsuccessful thus far.
- 30.Suzuki S, Matsumoto K, Kawamura K, Suga S, Yoshida J. Org Lett. 2004;6:3755–3758. doi: 10.1021/ol048524h. [DOI] [PubMed] [Google Scholar]
- 31.A control experiment was performed to verify that diastereomer ratios resulted from kinetic product distributions under these reaction conditions. An isolated sample of cis-4 was treated with silylketene acetal 11 (4.0 equiv) and BF3•OEt2 (1.6 equiv) under standard reaction conditions. The ester cis-4 did not react under these conditions: neither stereochemical inversion to trans-4 nor chemical exchange to form cis- or trans-6 was observed.
- 32.Shenoy S, Smith DM, Woerpel KA. J Am Chem Soc. 2006;128:8671–8677. doi: 10.1021/ja061110u. [DOI] [PubMed] [Google Scholar]
- 33.Bartl J, Steenken S, Mayr H, McClelland RA. J Am Chem Soc. 1990;112:6918–6928. [Google Scholar]
- 34.We attribute the slight cis selectivity observed in the reaction of 10 and 11 with 1 under BF3•OEt2–mediated conditions to ion pairing with the acetate counterion. This hypothesis is buttressed by our results with the pivaloate substrate 17 (vide infra).
- 35.We hypothesize that in for the reactions of silylketene acetals with oxocarbenium ion VIIIeq, addition to the top face (pathway a) is at the diffusion limit, while addition to the bottom face (pathway b) occurs with a rate that is slightly below the diffusion limit. This scenario leads to a reduced, but not completely eroded selectivity.
- 36.Manoharan M, Eliel EL. Tetrahedron Lett. 1984;25:3267–3268. [Google Scholar]
- 37.Diastereoselectivities were consistent with the data in Table 1 and Table 2.
- 38.All substitutions of pivaloate 17 gave diastereoselectivities similar to those of acetate 1, as detailed in the Supporting Information.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Experimental procedures and compound characterization is available free of charge via the Internet at http://pubs.acs.org.