

Abstract
Translocations involving c-myc and an Ig locus have been reported rarely in human multiple myeloma (MM). Using specific fluorescence in situ hybridization probes, we show complex karyotypic abnormalities of the c-myc or L-myc locus in 19 of 20 MM cell lines and approximately 50% of advanced primary MM tumors. These abnormalities include unusual and complex translocations and insertions that often juxtapose myc with an IgH or IgL locus. For two advanced primary MM tumors, some tumor cells contain a karyotypic abnormality of the c-myc locus, whereas other tumor cells do not, indicating that this karyotypic abnormality of c-myc occurs as a late event. All informative MM cell lines show monoallelic expression of c-myc. For Burkitt's lymphoma and mouse plasmacytoma tumors, balanced translocation that juxtaposes c-myc with one of the Ig loci is an early, invariant event that is mediated by B cell-specific DNA modification mechanisms. By contrast, for MM, dysregulation of c-myc apparently is caused principally by complex genomic rearrangements that occur during late stages of MM progression and do not involve B cell-specific DNA modification mechanisms.
Chromosomal translocations that juxtapose a c-myc locus with one of the Ig loci (IgH, IgLκ, or IgLλ) represent an essentially invariant, early oncogenetic event in human Burkitt's lymphoma and murine plasmacytoma (1, 2). These translocations are simple, reciprocal recombination events that appear to occur as a result of errors in V(D)J recombination, IgH switch recombination, or somatic hypermutation (3–6). Most involve the IgH locus at 14q32 (mouse chromosome 12), but a minority involves the Igκ locus at 2p11 (mouse chromosome 6) or the Igλ locus at 22q11 (mouse chromosome 16). The physiological consequence of these translocations is dysregulated, increased expression of c-myc as a result of its juxtaposition to intronic and/or 3′ IgH or IgL enhancers. The nontranslocated c-myc allele is not expressed or is expressed at very low levels, corresponding to the silent state of the c-myc gene in resting germinal center B cells and terminally differentiated plasma cells.
Human multiple myeloma (MM) is a tumor of a B lymphocyte that terminally differentiates into a long-lived plasma cell after being subjected to somatic hypermutation, antigen selection, and IgH switching in a germinal center. The similarities in phenotype of human MM and murine plasmacytoma prompted numerous studies to demonstrate Ig translocations and dysregulation of c-myc in human MM tumors and human MM cell lines. Recently, it has been determined that IgH translocations, mostly involving IgH switch regions, occur in most MM cell lines and tumors (7–11). However, conventional cytogenetics and DNA studies identified c-myc/Ig translocations, c-myc gene rearrangements, and c-myc amplification in less than 10% of MM tumors and MM cell lines analyzed (10, 12–16).
To overcome the difficulties of examining the c-myc locus in myeloma tumor cells with a low proliferative activity and myeloma cell lines with highly complex karyotypic abnormalities, we have used a molecular cytogenetic approach. Using specific fluorescence in situ hybridization (FISH) probes for c-myc and the three Ig loci, together with the relevant chromosome-painting probes, we found chromosomal abnormalities involving the c-myc locus in nearly all MM cell lines and a lower fraction of MM tumors. The karyotypic complexity and apparently late appearance of these c-myc abnormalities during the pathogenesis of MM suggest a different mechanism of c-myc dysregulation and a different role of c-myc in tumorigenesis of MM compared with murine plasmacytoma tumors.
Materials and Methods
Probes.
FISH probes.
The plasmid c-myc probe contains a 12.5-kb EcoRI genomic fragment composed of 5′ flanking sequences and all three exons (Fig. 2). The CH bacterial artificial chromosome (BAC) probe (17) contains a 70-kb insert that starts within switch epsilon and extends beyond the 3′ α-2 enhancer region; it includes about 40 kb of sequence that cross-hybridizes with the region including ϕɛ and the 3′ α-1 enhancer (Fig. 2). The VH probe is cosmid yIgH6–9 (18), which is located at the telomeric end of the IgH locus, within 100 kb of the 14q telomere. Plasmids containing sequences (A2H10.5) from the α-2 enhancer (α-enhancer probe) or sequences (EG3–5) centromeric to the α-2 enhancer (centromeric IgH probe) were generously provided by F. Mills (Center for Biologic Evaluation and Research, Food and Drug Administration) (19). The Cλ BAC probe, isolated from a BAC library (Genome Systems, St. Louis), includes sequences that start about 40 kb upstream of Jλ1 and end about 50 kb downstream of the 3′ enhancer (Eλ in Fig. 2). The Cκ cosmid probe includes sequences that start in the Jk region and extend several kilobases downstream of the 3′ κ-deleting element (Fig. 2). The L-myc probe is a 10.6-kb L-myc genomic fragment in the pv11/1B vector that was generously provided by R. DePinho (Dana–Farber Cancer Center). A probe containing germ-line chromosome 21 sequences was selected from a P1 library (Genome Systems), with chromosome 21 sequences present in the 14;21 breakpoint cloned from KMM1.
Figure 2.

Ig and c-myc loci. (A) The c-myc gene (8q24.1) contains three exons (open box is noncoding), with the direction of transcription shown by an arrow; thick, horizontal line shows position of plasmid c-myc probe. (B) the Igλ locus (22q11) and Igκ locus (2p12) each include coding regions and a 3′ EL enhancer, with Igκ also including an intronic enhancer (dotted circle); thick horizontal lines indicate Cλ BAC probe and Cκ cosmid probe. (C) The 1-Mb IgH locus (14q32.3) includes coding regions, σμ and σδ 440-bp repeats, and Eμ and 3′ Eα enhancers; thick, horizontal lines indicate telomeric VH cosmid and centromeric CH BAC probes [dotted line indicates region of cross-hybridization of CH probe (17)]. (D) For KMM-1 HMCL, diagram of the complex insertion of chromosome 8 (including c-myc) and chromosome 21 into the IgH locus at a site just upstream of σδ. ▵, Deletion of about 10 kb of IgH locus starting in σμ. The germ-line chromosome 21 P1 clone was selected with 14;21 breakpoint sequences (7). The drawings are not to scale, but approximate positions of markers and probes are indicated. In each case, the telomeric end of the locus is to the left.
Northern blot probes.
The probes used for Northern blots included the following fragments: a 1.4-kb fragment containing exons 2 and 3 from c-myc cDNA, a 461-bp fragment containing exon 3 sequences from L-myc genomic DNA (20), and a 400-bp fragment containing exon 3 sequences from N-myc genomic DNA (21).
FISH.
Metaphase chromosomes were prepared by incubating exponentially growing cells with colcemid (0.08 mg/ml) for 0.5–2 hr and then in 0.075 M KCl for 25 min at 37°C. After being washed in prechilled fixative solution (3:1 volume of absolute methanol to glacial acetic acid) three times, the cells were dropped onto superfrost microscope slides (Fisher Scientific). Slides were aged, pretreated with RNase and pepsin, and denatured (70% formamide, 78°C for 2 min) before hybridization. The painting probes were directly labeled by PCR by using either Cy5–5-dUTP (Amersham Pharmacia) or Texas red-5-dUTP (DuPont). Other FISH probes were labeled with either biotin-16-dUTP or digoxigenin-11-dUTP (Boehringer Mannheim) by nick-translation. Labeled probes (250 ng each) were coprecipitated with 50-fold excess of Cot-1 DNA (GIBCO/BRL) and resuspended in 10 μl of hybridization solution (50% formamide and 10% dextran sulfate in 1× SSC). After denaturation (80°C for 5 min) and preannealing (37°C for 30 min), the hybridization mixture was placed on slides and incubated at 37°C for overnight. Slides were washed, incubated with avidin-FITC/rhodamine-antidigoxigenin dual-detection reagent (Vysis, Downers Grove, IL), and counterstained with 4′,6-diamidino-2-phenylindole (DAPI). FISH results were analyzed by a DM fluorescence microscope (Leica) equipped with DAPI, FITC, tetramethylrhodamine B isothiocyanate (TRITC), and Cy5 filters as well as a DAPI/FITC/TRITC triple band-pass filter (Chroma Technology, Brattleboro, VT). The Sensys charge-coupled device camera (Photometrics, Tucson, AZ) attached to the fluorescence microscope and qfish software (Leica) were used to capture and process images. At least five metaphases from each MM cell line and primary tumor were analyzed for each probe set. When two signals were found juxtaposed on metaphase chromosomes, at least 10 interphase nuclei in the same slide were examined for physical distance between these two signals in interphase. Before subjected to FISH analysis on MM samples, every probe was hybridized to a metaphase slide of normal blood T lymphocytes (Vysis).
Expression of myc.
Total RNA was isolated from each human myeloma cell line (HMCL) by using Trizol reagent (GIBCO/BRL), fractionated on 1% agarose-formaldehyde gels, blotted to GeneScreen Plus filters (DuPont), and hybridized with random-primed probes described above (8). Methods for identification of c-myc mutations and determining which allele(s) are expressed were described previously (22). The predominant (“normal”) c-myc allele and nucleotide positions of sequence variants are from c-myc genomic DNA (GenBank accession no. X00364).
Other Procedures.
Results
Either c-myc or L-myc Is Expressed in All Human MM Cell Lines Examined.
We analyzed the expression of c-myc in 20 HMCL (7, 23–27). Northern blots demonstrated substantial expression of the normal 2.4-kb c-myc mRNA in 18 HMCL (JJN3 and MM.1 in Fig. 1A, plus 16 other HMCL not shown) (21). Two HMCL did not express the normal 2.4-kb mRNA species despite the presence of at least one apparently normal c-myc allele (Southern blot analysis, not shown). H929 expresses a heterogeneous mixture of c-myc mRNA species (Fig. 1A) from a c-myc allele that is altered by a complex translocation just upstream of the normal polyadenylation site (28). The U266 HMCL is the only one that expresses no c-myc mRNA, but does express L-myc as a 3.6-kb mRNA species (Fig. 1A) (29). With the exception of U266, no expression of L-myc or N-myc was detected in any of the other 19 HMCL (not shown).
Figure 1.

Expression of c-myc and L-myc in MM cell lines. (A) A Northern blot containing 5 μg of total RNA from four HMCL was probed sequentially with c-myc (Top) and L-myc (Middle). The normal 2.4-kb c-myc mRNA is seen in JJN3 and MM-S1, and the normal 3.6-kb L-myc is seen in mRNA in U266. Ethidium bromide staining of the 5-kb rRNA is shown for each HMCL (Bottom). Arrows indicate the positions of the 2- and 5-kb rRNAs. (B) Sequencing gels of polymorphic regions of c-myc are shown for PCR-amplified genomic DNA and cDNA fragments in the FR4 and FLAM-76 HMCL. Arrows indicate the position of the polymorphic nucleotides.
Only One c-myc Allele Is Expressed in Eight Informative MM Cell Lines.
Previously, we showed that the JJN3 and OPM-2 HMCL had genetically distinguishable c-myc alleles, with selective expression of one allele, suggesting tumor-specific dysregulation of one allele (22, 30). Genomic DNA from the other 18 HMCL was screened for exon mutations that might distinguish the two c-myc alleles. In total, seven HMCL other than H929 had two genetically distinguishable c-myc alleles: G→A in the 5′ untranslated region (nucleotide 2397) in JJN3, C→T in the 5′ untranslated region (nucleotide 2510) in KMS-11 and FLAM-76, CGG(Arg)→CGC(Arg) in codon 83 (nucleotide 4769) in MM.1, and CCG(Pro)→CCA(Pro) in codon 231(nucleotide 5213) in OPM-2, MM-S1, and FR4. None of these mutations are missense, and at least three (nucleotides 2397, 2510, and 5213) are germ-line polymorphisms. For each of these seven informative HMCL, there is selective expression of either the predominant (“normal”) c-myc allele (FR4, MM.1) or the variant/polymorphic c-myc allele (FLAM-76, KMS-11, OPM-2, MM-S1, JJN3 and corresponding primary tumor). For example, paired genomic DNA and expressed cDNA sequences illustrate selective expression of the “normal” c-myc allele in FR4 and the polymorphic c-myc allele in FLAM-76 (Fig. 1B). These seven HMCL plus H929 (above) express one of two c-myc alleles, whereas U266 expresses L-myc and no c-myc. The results are consistent with tumor-specific dysregulation of L-myc or one c-myc allele in all nine informative HMCL.
Karyotypic Abnormalities of the c-myc (or L-myc) Locus Are Present in 19 of 20 HMCL.
Excepting H929, none of the other eight informative HMCL showed rearrangement or amplification of the c-myc or L-myc gene by Southern blot analysis (not shown). In addition, with the exception of FR4, which was reported to have a classical t(8;14) translocation (24), none of the other eight lines had a c-myc/Ig translocation demonstrated by conventional cytogenetic analyses. Advanced MM tumors and HMCL generally have extensive chromosomal rearrangements so that karyotypic abnormalities of the c-myc locus may be difficult to detect by conventional cytogenetic analyses. Therefore, we subjected the 20 HMCL to specific FISH analyses, by using chromosome-painting probes, together with a c-myc probe, Ig CH, Cλ, and Cκ probes that include strong enhancer sequences, and a VH probe that is located within 100 kb of the 14q telomere (Fig. 2) (17, 31).
As an example of the FISH analyses, consider the results for ARK, which has a classical t(8;14)(q24.1;q32.3) translocation resulting in separation between CH and VH sequences and juxtaposition of c-myc and CH sequences (diagram in Fig. 3A1). The same der(14) t(8;14) chromosome from two metaphases of ARK is shown in Fig. 3B1 (Upper and Lower). Fig. 3B1 Upper shows hybridization with a chromosome 8-painting probe (red) and a c-myc probe (green but yellow on a red background) and counterstain with DAPI (blue). A portion of chromosome 8 (red) is translocated to another chromosome (blue), with two green/yellow c-myc signals (one for each chromatid) positioned at the breakpoint. Fig. 3B1 Lower shows hybridization with a chromosome 14-painting probe (purple), a CH probe (green but blue on a purple background), and a c-myc probe (red). Chromosome 14 (blue in Upper) is now purple but the telomeric piece of chromosome 8 (counterstained blue) is not visualized on this exposure. Both CH (two blue signals on purple background) and c-myc (two red signals) overlap (white signals) at the 8;14 breakpoint.
Figure 3.

FISH analyses of c-myc and Ig loci in MM cell lines. (A) Examples of chromosomal translocations and rearrangements that juxtapose c-myc and CH sequences, with relative positions of c-myc, CH, and VH sequences indicated. In a reciprocal translocation, the two derivative chromosomes are denoted by the source of the centromere, so that for t(8;14), der (8) and der (14) contain the centromeres of 8 and 14, respectively. (B) Metaphase and interphase chromosomes of 20 HMCL were examined by FISH by using chromosome-specific painting probes, c-myc probes (L-myc probe in case of U266), and various Ig probes (Fig. 2) in different combinations and labeled with up to three different fluorochromes (FITC, green; Texas red or rhodamine, red; and Cy5, purple) (17, 31). The type of probe and color are indicated to the left of each group of images. Metaphase chromosomes were counterstained with DAPI. Note that overlapping signals generate composite colors, so that red + green = yellow, purple + green = blue, and purple + red + green = white. Examples of chromosomes with a karyotypic abnormality of the c-myc locus are shown for each HMCL. In B1-4, Upper and Lower represent the same abnormal chromosome from two different metaphases, but hybridized with different combinations of probes (note that JJN3 and Karpas 620 each have two kinds of abnormal chromosomes). Normal T cell chromosomes 8 and 14 (B1) and 1 (B5b) are included as controls. The results are organized in accordance with the five groups described in the text.
From the FISH analyses, we were able to detect complex structural abnormalities of the c-myc (or L-myc) locus in 19 of 20 HMCL, which we have divided into the five groups described below. This is summarized in Table 1 and illustrated by diagrams (Fig. 3A) and FISH results (Fig. 3B).
Table 1.
Karyotypic abnormalities of myc in MM cell lines
| Cell line | Chr | A | N | Abnormality |
|---|---|---|---|---|
| ARK | 46 | 1 | 1 | t(8;14) classic:der(14) and der(8) |
| *FR4 | 100 | 2 | 3 | t(8;14) classic;der(14) and der(8) |
| LP1 | 80 | 5 | 1 | t(8;14) variant:der(8) with VH |
| SK-MM1 | 79 | 4 | 0 | t(8;14) variant: der(8) without VH |
| KMS-12PE | 47 | 2† | 1 | CH insertion with duplication of c-myc and CH on 8 |
| KMS-12BM | 77 | 8† | 3 | CH insertion with duplication of c-myc and CH on 8 |
| *MM-S1 | 47 | 6† | 1 | CH insertion with duplication of c-myc and CH on 8 |
| *MM.1 | 44 | 1 | 1 | CH insertion on t(3;8)(q21;q24):der(3) |
| *OPM-1 | 74 | 2 | 2 | CH insertion on t(1;8)(q12?;q24):der(8) |
| *OPM-2 | 67 | 2 | 2 | CH insertion on t(1;8)(q12?;q24):der(8) |
| *KMS-11 | 70 | 2† | 3 | CH+c-myc insertion and duplication on ? |
| OCI-MY5 | 46 | 2† | 1 | CH+c-myc insertion and duplication on ? |
| KMM-1 | 80 | 2 | 3 | c-myc insertion on 14 |
| *JJN3 | 60 | 3 | 2 | CH insertion on 8 and CH insertion on t(8;14)(q24;p?):der(14) |
| KARPAS 620 | 68 | 4 | 2 | t(8;14)classic:der(14) and CH insertion on t(8;11):der(8) |
| 8226 | 60 | 1 | 3 | c-myc insertion on t(16;22)(q23;q11):der(16) |
| DELTA-47 | 45 | 2 | 2 | Cλ insertion on t(?;8):der(?) |
| *H929 | 45 | 2 | 1 | t(8;20)(q24;?):der(8) |
| L363 | 46 | 2 | 1 | t(5;8)(q11.2;q32):der(8) |
| *FLAM-76 | 42 | 1 | 1 | c-myc insertion on ? |
| ‡U266 | 39 | 0 | 2 | None |
| L-myc | 1 | 1 | inv(1)(p13;p34) | |
| H112 | 46 | 0 | 2 | None |
The cell lines are separated in accord with the five groups of abnormalities described in the text. For KMS-12 and OPM, there are two independent cell lines from the same tumor. Total chromosome number (Chr) and the number of karyotypically abnormal (A) and normal (N) c-myc alleles are listed (L-myc alleles also are listed for U266 only). For translocations, all derivative chromosomes that were detected are noted. If both derivative chromosomes were detected, the one containing c-myc is listed first. Karyotypic abnormalities that are far from c-myc are not described here or in the text.
Selective expression of one c-myc allele.
Includes duplication of c-myc and CH on each abnormal chromosome.
Expression of L-myc only.
Group 1 (Fig. 3B1) includes four HMCL that have a t(8;14) translocation. This was detected by conventional cytogenetics in only one HMCL (FR4) (24). Two HMCL (ARK and FR4) have classical t(8;14) translocations (diagram in Fig. 3A1 and results described above for ARK). Two other HMCL (LP1 and SK-MM1) have variant t(8;14) translocations. In LP1, 14q telomeric sequences, including CH and VH (VH not shown), are translocated telomeric to c-myc on chromosome 8 (diagram in Fig. 3A2). Note that the 14q sequences are too small to be detected by a chromosome 14-painting probe. SK-MM1 is a more complex variant in which a small piece of 14 that has CH sequences at each end replaces sequences telomeric of c-myc on chromosome 8. The more centromeric CH sequences are juxtaposed to c-myc, but neither CH is associated with VH sequences (not shown).
Group 2 (Fig. 3B2) includes seven HMCL in which a variety of complex chromosomal insertions result in the juxtaposition of c-myc and CH sequences. Two HMCL (KMS-12 and MM-S1) have a chromosome 8 that appears to be structurally normal but has an insertion of CH sequences near c-myc (diagram in Fig. 3A3). For each line, the juxtaposed c-myc and CH sequences are each present as more than two discrete signals, indicating a duplication of the associated CH and c-myc sequences. A second pair of HMCL has translocations involving chromosome 8 and a second chromosome (chromosome 3 in MM.1 and chromosome 1 in OPM-1), and in each instance CH sequences are inserted near c-myc at the translocation breakpoint. Note that there is no detectable chromosome 14-painting signal associated with the inserted CH for MM.1 (diagrams in Fig. 3A4), whereas there is a chromosome 14-painting signal associated with the inserted CH for OPM-1 (diagram in Fig. 3A5). A third pair of HMCL (KMS-11 and OCI-MY5) have complex insertions and duplications resulting in the juxtaposition of c-myc and CH sequences at well-separated sites on a third chromosome. In KMS-11, a segment of chromosome 8 is inserted on an unknown chromosome, with c-myc and inserted CH sequences at each junction of chromosome 8 and the unknown chromosome. For OCI-MY5, an unknown chromosome contains a segment of chromosome 14 that is sandwiched between two segments of chromosome 8, with juxtaposition of c-myc and CH sequences at each 8/14 boundary.
A seventh HMCL (KMM-1) has c-myc inserted into the IgH locus on chromosome 14 (diagram in Fig. 3A6). Previously, we had cloned for KMM-1 paired 8;14 and 14;21 breakpoints, in which IgH sequences upstream of σδ are juxtaposed to sequences upstream of c-myc on chromosome 8 (8;14 breakpoint in Fig. 2D) and to a sequence localized at 21q22 (14;21 breakpoint in Fig. 2D) (7). FISH analyses confirmed colocalization of CH, c-myc, VH (not shown), and normal chromosome 21 sequences near the 14;21 breakpoint (not shown) at 14q, but no detectable chromosome 8- or 21-painting signals (not shown). Together, the two cloned breakpoints and the FISH results demonstrate that small pieces of 8 (including c-myc) and 21 are inserted within the IgH locus (see Fig. 2D for molecular details).
Group 3 includes two HMCL that each have two different kinds of karyotypic abnormalities that juxtapose c-myc and CH (Fig. 3B3). Karpas 620 has a classic t(8;14) translocation, as described in Group 1 above. It also has a t(8;11)(q24;q13) translocation in which chromosome 11 sequences replace chromosome 8 sequences telomeric to c-myc, but with CH sequences juxtaposed to c-myc at the breakpoint. JJN3 has an apparently normal chromosome 8 with CH sequences inserted near c-myc. It also has the telomeric end of 8 with juxtaposed c-myc and CH sequences moved to the short arm of chromosome 14.
Group 4 includes two HMCL with chromosomal structural alterations that juxtapose c-myc and Cλ sequences (Fig. 3B4). For 8226, Cλ and chromosome 22q sequences telomeric to Cλ are translocated to chromosome 16, with inserted c-myc sequences juxtaposed to Cλ at the translocation breakpoint. In direct contrast, for Delta-47, c-myc and chromosome 8q24 sequences telomeric to c-myc are translocated to an unknown chromosome, with inserted Cλ sequences juxtaposed near c-myc at the breakpoint.
Group 5 includes four HMCL in which either a c-myc (Fig. 3B5a) or L-myc (Fig. 3B5b) locus is positioned in an altered chromosomal location but without any detectable association with CH, Cλ, or Cκ sequences. The H929 and L363 HMCL have translocations of chromosomes 20 and 5, respectively, to 8q24, with c-myc localized near the breakpoint. The H929 breakpoint is complex, including a deletion of c-myc 3′ untranslated sequences and downstream PVT-1 chromosome 8 sequences (28), but replacement of most sequences telomeric to c-myc by translocated sequences from chromosome 20. FLAM-76 has an insertion of c-myc on an unknown chromosome. The U266 HMCL (29), which expresses L-myc but not c-myc, has no karyotypic abnormality of c-myc. Instead, it has a translocation in which an unknown chromosome replaces sequences telomeric to 1p34, the normal site at which L-myc is located. However, L-myc is not at its normal site but is moved to 1p13, near the centromere, as a result of an inversion.
Chromosome-painting probes do not generate detectable signals for sequences less than about 5 Mb. Thus, insertion of c-myc without corresponding chromosome 8 painting (KMM-1, 8226, and Flam-76), CH sequences without corresponding chromosome 14 painting (LP1, KMS-12, MM-S1, MM.1, KMS-11, JJN3, and Karpas 620), or Cλ sequences without corresponding chromosome 22 painting (Delta-47) might correspond to sequences containing tens of kilobases up to about 5 Mb. For six HMCL with CH insertion but without detectable 14 painting and VH signals (KMS-12, MM-S1, MM.1, KMS-11, JJN3, and Karpas 620), we used an α-enhancer probe (A2H10.5) in FISH analyses to confirm the association of 3′ IgH enhancer sequences with c-myc. This probe hybridizes to both α1 and α2 3′ IgH enhancer sequences and was found colocalized with the c-myc probe in each of these six lines (not shown). A FISH probe containing sequences immediately centromeric to the α2 enhancer (EG3–5) also colocalized with c-myc in five of these six lines (not shown). However, in JJN3, the centromeric IgH signal was absent from both chromosomal sites at which CH and c-myc sequences are juxtaposed. Therefore, the centromeric breakpoint of the insertion occurs within the IgH locus only in JJN3.
Translocations can dysregulate an oncogene by positioning it up to approximately 500 kb from an Ig locus (9). Consistently overlapping signals on metaphase FISH analyses are compatible with sequences that are separated by as much as 1–2 Mb, whereas overlapping signals on interphase FISH analyses indicate sequences that are separated by <500 kb (31). In each case that we have shown an association of two signals (CH and c-myc, for example) by metaphase FISH, we have analyzed at least 10 corresponding interphase nuclei to confirm overlapping signals that must be separated by no more than approximately 500 kb (examples in Fig. 3B6). We conclude that an Ig locus is responsible for dysregulating c-myc in 15 HMCL (Fig. 3B1-4). It seems likely that c-myc (or L-myc) is dysregulated as a result of the karyotypic abnormalities identified in four other HMCL (Fig. 3B5), despite the apparent absence of an association with an Ig locus.
Karyotypic Abnormalities of c-myc Are Present in Primary MM Tumors.
First, it was shown previously that the H929 c-myc rearrangement also is present in primary tumor cells (28). Second, for two MM tumors, independent cell lines were generated (32, 33). Each pair of HMCL (OPM-1 and OPM-2; KMS-12BM and KMS-12PE) display many karyotypic differences (not shown) but each pair shares a distinct karyotypic abnormality that juxtaposes c-myc and CH sequences (Table 1), indicating that the karyotypic abnormality of c-myc was present in primary tumor cells. Third, seven of 14 karyotypically abnormal stage III primary MM tumors showed karyotypic abnormalities of the c-myc locus. Three tumors had juxtaposition of c-myc with Ig sequences: one tumor with a variant form of the t(8;14) translocation, as shown in Fig. 4 and the diagram in Fig. 3A2; another tumor with CH sequences inserted into the c-myc locus on chromosome 8; and a third tumor with a t(8;22) translocation. Four other tumors had karyotypic abnormalities of the c-myc locus that did not involve an IgH or IgL locus: two tumors with der(3)t(3;8) and c-myc at the breakpoint; one tumor with dic(8;9)(q24;p24) and c-myc at the breakpoint; and one tumor with a duplication of the c-myc locus. Importantly, there is heterogeneity of the c-myc abnormality in two of these tumors. For one tumor, only 20% of the tumor cells have a t(3;8) translocation with c-myc but no Ig sequences at the breakpoint. For a second tumor, 80% of the tumor cells have a t(8;22) translocation with c-myc and Cλ sequences juxtaposed at the breakpoint, and 20% of the tumor cells lack this translocation. Finally, there is a recent report in which 50 MM tumors with abnormal karyotypes were analyzed by spectral karyotyping (SKY) (34). From our analysis of this published data, there were four tumors with t(8;14), five with t(8;22), and six with other karyotypic abnormalities at or near 8q24, so that 30% of these 50 MM tumors had apparent structural abnormalities of the c-myc locus by SKY analysis.
Figure 4.
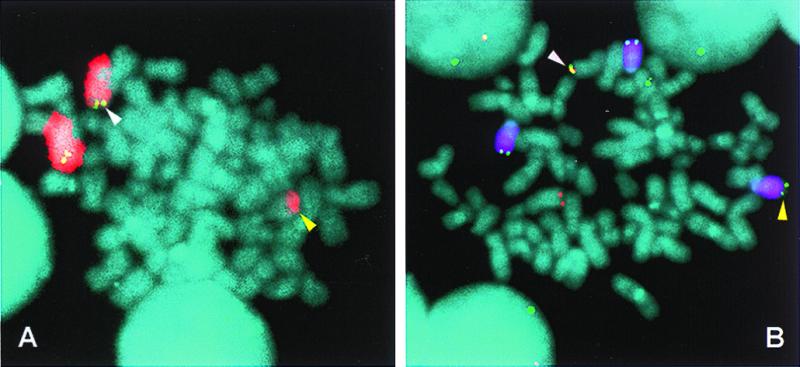
FISH detection of a variant t(8;14) translocation in an MM tumor. Metaphase chromosomes from a primary MM tumor were hybridized with chromosome 8 painting (Texas red, red) and c-myc (FITC, green) probes (A) and chromosome 14 painting (Cy5, purple), CH (FITC, green), and c-myc (rhodamine, red) probes (B) to identify a variant t(8;14) translocation (diagram in Fig. 3A2). In both A and B, white arrows indicate the der(8) chromosome with c-myc and CH signals and yellow arrows indicate the der(14) chromosome that also has a CH signal. Because CH sequences are located on both der(8) and der(14), the chromosome 14 breakpoint appears to be located between the Eα1 and Eα2 3′ IgH enhancers, but could occur within the sequences detected by the CH probe or occur as a result of a duplication of CH sequences. In separate experiments not shown, a VH probe colocalized with the CH and c-myc probes on the der(8) chromosome (white arrows) but not with the CH signal on der(14).
Discussion
Analysis of HMCL and primary tumors using the specific probes described above identifies karyotypic abnormalities near the c-myc locus that were not and could not have been detected by conventional cytogenetics or SKY analyses. For the 18 HMCL with karyotypic abnormalities of the c-myc locus, the original conventional cytogenetic analyses identified 8q24 abnormalities in only five lines. Even SKY analyses would not have detected a karyotypic abnormality at 8q24 in the seven HMCL in which c-myc, CH, or Cλ sequences inserted into an altered chromosomal position without a gross karyotypic change. Our results emphasize the possibility that the chromosomal structural changes that occur in many kinds of tumors may include recurrent oncogenic abnormalities that cannot be appreciated unless one does FISH analyses with specific probes.
The c-myc and L-myc genes are not expressed in resting germinal center B cells or terminally differentiated plasma cells, but either c-myc or L-myc is expressed in all HMCL. In addition, we show that there is expression of only one c-myc allele in eight informative HMCL that express c-myc. As summarized above, specific FISH analyses have identified structural abnormalities of the c-myc (or L-myc) locus in all but one of 20 HMCL. The selective expression of L-myc or one c-myc allele in all nine informative HMCL indicates that the chromosomal structural abnormalities are associated with tumor-specific cis-dysregulation of c- or L-myc. However, our FISH analyses of 14 primary MM tumors, together with a recent report of SKY analyses of 50 MM tumors (34), have identified karyotypic abnormalities of the c-myc locus less frequently (about 30–50%) in advanced primary MM tumors.
The incidence of c-myc abnormalities and increased c-myc expression is reported to be more frequent in extramedullary and more proliferative intramedullary tumors compared with less aggressive intramedullary tumors (35). Moreover, cell lines rarely can be made even from advanced intramedullary MM tumors but can be made from most extramedullary MM tumors (36). Therefore, we hypothesize that c-myc dysregulation is usually a late event of tumor progression in MM, consistent with the higher proliferative capacity as manifested by an increased DNA-labeling index late in the disease (37, 38). Direct evidence that c-myc dysregulation is a late event is provided by the heterogeneity of two advanced intramedullary MM tumors in which not all of the tumor cells have the karyotypic abnormality of c-myc. Thus, MM differs from murine plasmacytoma and Burkitt's lymphoma tumors, in which dysregulation of c-myc appears to be one of the earliest oncogenic events (1, 2, 39). We note, however, that dysregulation of c-myc sometimes may occur as a late oncogenic event in other B lymphocyte tumors (40, 41).
Translocations involving c-myc and an Ig locus in Burkitt's lymphoma and murine plasmacytoma appear to be early events that are mediated by errors in V(D)J or IgH switch recombination or somatic hypermutation (1, 2, 39). Similarly, translocations involving IgH switch regions are thought to occur as one of the earliest steps in molecular pathogenesis of MM (7, 10). However, from two MM tumors and 13 HMCL, we have cloned and characterized 19 IgH switch translocation breakpoints and found no example in which the c-myc locus is joined to a switch region (7, 9, 42). Moreover, additional FISH analyses and Southern blot assays (not shown) demonstrate that the telomeric IgH breakpoints are located between γ4 and ɛ in JJN3 and within the α2 locus in MM.1, but not within or near a switch region in either MM cell line. We predict that many—perhaps all—of the c-myc translocations and insertions described here are not caused by errors in V(D)J or IgH switch recombination or in somatic hypermutation. Instead, they result from other, yet to be defined, mechanisms that are responsible for chromosomal translocations that occur during progression of many kinds of tumors. However, despite an apparent lack of involvement of B lymphocyte-specific recombination mechanisms in the c-myc translocations in MM, there seems to be a strong association with Ig loci, consistent with previous suggestions that translocations seem to target regions that are active and/or “accessible” in specific cell lineages (43).
Acknowledgments
We thank H. Nash for SKY analyses on OPM-2 and H929, W. A. Wyatt for technical assistance, and M. Chesi, M. Gellert, S. Lipkowitz, and L. Staudt for reviewing the manuscript. P.L.B. is supported in part by The Howard Temin Award (CA74265).
Abbreviations
- MM
multiple myeloma
- HMCL
human myeloma cell lines
- FISH
fluorescence in situ hybridization
- DAPI
4′,6-diamidino-2-phenylindole
- SKY
spectral karyotyping
- BAC
bacterial artificial chromosome
References
- 1.Wiener F, Potter M. In: The Causes And Consequences Of Chromosomal Aberrations. Kirsch I R, editor. New York: CRC; 1993. pp. 91–124. [Google Scholar]
- 2.Dalla-Favera R. In: The Causes And Consequences Of Chromosomal Aberrations. Kirsch I R, editor. New York: CRC; 1993. pp. 313–332. [Google Scholar]
- 3.Lazo P A, Lee J S, Tsichlis P N. Proc Natl Acad Sci USA. 1990;87:170–173. doi: 10.1073/pnas.87.1.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Korsmeyer S J. Annu Rev Immunol. 1992;10:785–807. doi: 10.1146/annurev.iy.10.040192.004033. [DOI] [PubMed] [Google Scholar]
- 5.Goossens T, Klein U, Kuppers R. Proc Natl Acad Sci USA. 1998;95:2463–2468. doi: 10.1073/pnas.95.5.2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sale J E, Neuberger M S. Immunity. 1999;9:1–15. [Google Scholar]
- 7.Bergsagel P L, Chesi M, Nardini E, Brents L A, Kirby S L, Kuehl W M. Proc Natl Acad Sci USA. 1996;93:13931–13936. doi: 10.1073/pnas.93.24.13931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chesi M, Nardini E, Brents L A, Schrock E, Ried T, Kuehl W M, Bergsagel P L. Nat Genet. 1997;16:260–264. doi: 10.1038/ng0797-260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chesi M, Bergsagel P L, Shonukan O O, Martelli M L, Brents L A, Chen T, Schrock E, Ried T, Kuehl W M. Blood. 1998;91:4457–4463. [PubMed] [Google Scholar]
- 10.Nishida K, Tamura A, Nakazawa N, Ueda Y, Abe T, Matsuda F, Kashima K, Taniwaki M. Blood. 1997;90:526–534. [PubMed] [Google Scholar]
- 11.Avet-Loiseau H, Brigaudeau C, Morineau N, Talmant P, Lai J Y, Daviet A, Li J-Y, Praloran V, Rapp M-J, Harousseau J L, et al. Genes Chromosomes Cancer. 1999;24:9–15. doi: 10.1002/(sici)1098-2264(199901)24:1<9::aid-gcc2>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- 12.Dewald G W, Kyle R A, Hicks G A, Greipp P R. Blood. 1985;66:380–390. [PubMed] [Google Scholar]
- 13.Sawyer J R, Waldron J A, Jagannath S, Barlogie B. Cancer Genet Cytogenet. 1995;82:41–49. doi: 10.1016/0165-4608(94)00284-i. [DOI] [PubMed] [Google Scholar]
- 14.Lai J L, Zandecki M, Mary J Y, Bernardi F, Izydorczyk V, Flactif M, Morel P, Jouet J P, Bauters F, Facon T. Blood. 1995;85:2490–2497. [PubMed] [Google Scholar]
- 15.Calasanz M J, Cigudosa J C, Odero M D, Ferreira C, Ardanaz M T, Fraile A, Carrasco J L, Sole F, Cuesta B, Gullon A. Genes Chromosomes Cancer. 1997;18:84–93. [PubMed] [Google Scholar]
- 16.Bakkus M H C, Brakel-van Peer K M J, Michiels J J, van't Veer M B, Benner R. Oncogene. 1990;5:1359–1364. [PubMed] [Google Scholar]
- 17.Gabrea A, Bergsagel P L, Chesi M, Shou Y, Kuehl W M. Mol Cell. 1999;3:119–123. doi: 10.1016/s1097-2765(00)80180-x. [DOI] [PubMed] [Google Scholar]
- 18.Cook G P, Tomlinson I M, Walter G, Riethman H, Carter N P, Buluwela L, Winter G, Rabbitts T H. Nat Genet. 1994;7:162–168. doi: 10.1038/ng0694-162. [DOI] [PubMed] [Google Scholar]
- 19.Mills F C, Harindranath N, Mitchell M, Max E E. J Exp Med. 1997;186:845–858. doi: 10.1084/jem.186.6.845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kaye F, Battey J, Nau M, Brooks B, Seifter E, DeGreve J, Birrer M, Sausville E, Minna J. Mol Cell Biol. 1988;8:186–195. doi: 10.1128/mcb.8.1.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nau M M, Borrks B J, Carney D N, Gazdar A F, Battey J F, Sausville E A, Minna J D. Proc Natl Acad Sci USA. 1986;83:1092–1096. doi: 10.1073/pnas.83.4.1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kuehl W M, Brents L A, Chesi M, Bergsagel P L. Cancer Res. 1996;56:4370–4373. [PubMed] [Google Scholar]
- 23.Durie B G M, Grogan T M, Spier C, Vela E, Baum V, Rodriquez M A, Fruktiger Y. Blood. 1989;73:770–776. [PubMed] [Google Scholar]
- 24.Tagawa S, Doi S, Taniwaki M, Abe T, Kanayama Y, Nojima J, Matsubara K, Kitani T. Leukemia. 1990;4:600–605. [PubMed] [Google Scholar]
- 25.Kirchner H H, Fonatsch C, Schaadt M, Gunzel M, Hellriegel K P, Diehl V. Blut. 1981;43:93–97. doi: 10.1007/BF00320467. [DOI] [PubMed] [Google Scholar]
- 26.Nacheva E, Fischer P E, Sherrington P D, Labastide W, Lawlor E, Conneally E, Blaney C, Hayhoe F G J, Karpas A. Br J Haematol. 1990;74:70–76. doi: 10.1111/j.1365-2141.1990.tb02540.x. [DOI] [PubMed] [Google Scholar]
- 27.Pegoraro L, Malavasi F, Bellone G, Massaia M, Boccadoro M, Saglio G, Guerrasio A, Benetton G, Lombardi L, Coda R, Avanzi G C. Blood. 1989;73:1020–1027. [PubMed] [Google Scholar]
- 28.Hollis G F, Gazdar A F, Bertness V, Kirsch I R. Mol Cell Biol. 1988;8:124–129. doi: 10.1128/mcb.8.1.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jernberg-Wiklund H, Pettersson M, Larsson L-G, Anton R, Nilsson K. Int J Cancer. 1992;51:116–123. doi: 10.1002/ijc.2910510121. [DOI] [PubMed] [Google Scholar]
- 30.Kuehl W M, Brents L A, Chesi M, Huppi K, Bergsagel P L. Curr Topics Microbiol Immunol. 1997;224:277–282. doi: 10.1007/978-3-642-60801-8_29. [DOI] [PubMed] [Google Scholar]
- 31.Popescu N C, Zimonjic D B. Cancer Genet Cytogenet. 1997;93:10–21. doi: 10.1016/s0165-4608(96)00262-2. [DOI] [PubMed] [Google Scholar]
- 32.Katagiri S, Yonezawa T, Kuyama J, Kanayama Y, Nishida K, Abe T, Tamaki T, Ohnishi M, Tarui S. Int J Cancer. 1985;36:241–246. doi: 10.1002/ijc.2910360217. [DOI] [PubMed] [Google Scholar]
- 33.Namba M K, Ohtsuki T, Mori M, Togawa A, Wada H, Sugihara T, Yawata Y, Kimoto T. In Vitro Cell Dev Biol. 1989;25:723–729. doi: 10.1007/BF02623725. [DOI] [PubMed] [Google Scholar]
- 34.Sawyer J R, Lukacs J L, Munshi N, Desikan K R, Singhal S, Mehta J, Siegel D, Shaughnessy J, Barlogie B. Blood. 1998;92:4269–4278. [PubMed] [Google Scholar]
- 35.Nobuyoshi M, Kawano M, Tanaka H, Ishikawa H, Tanabe O, Iwato K, Asaoku H, Sakai A, Kuramoto A. Br J Haematol. 1991;77:523–528. doi: 10.1111/j.1365-2141.1991.tb08620.x. [DOI] [PubMed] [Google Scholar]
- 36.Zhang X G, Gaillard J P, Robillard N, Lu Z Y, Gu Z J, Jourdan M, Boiron J M, Bataille R, Klein B. Blood. 1994;83:3654–3663. [PubMed] [Google Scholar]
- 37.Lokhorst H M, Boom S E, Terpstra W, Rohol L P, Gerdes J, Bast B J E G. Br J Haematol. 1988;69:477–481. doi: 10.1111/j.1365-2141.1988.tb02402.x. [DOI] [PubMed] [Google Scholar]
- 38.Greipp P R, Katzmann J A, O'Fallan W M, Kyle R A. Blood. 1988;72:219–223. [PubMed] [Google Scholar]
- 39.Muller J R, Potter M, Janz S. Proc Natl Acad Sci USA. 1994;91:12066–12070. doi: 10.1073/pnas.91.25.12066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McDonnell T J, Korsmeyer S J. Nature (London) 1991;349:254–256. doi: 10.1038/349254a0. [DOI] [PubMed] [Google Scholar]
- 41.Thangavelu M, Olopade O, Beckman E, Vardiman J W, Larson R A, McKeithan T W, LeBeau M M, Rowley J D. Genes Chromosomes Cancer. 1990;2:147–158. doi: 10.1002/gcc.2870020211. [DOI] [PubMed] [Google Scholar]
- 42.Bergsagel P L, Nardini E, Brents L, Chesi M, Kuehl W M. Curr Top Microbiol Immunol. 1997;224:283–287. doi: 10.1007/978-3-642-60801-8_30. [DOI] [PubMed] [Google Scholar]
- 43.Kirsch I R, Brown J A, Lawrence J, Korsmeyer S, Morton C C. Cancer Genet Cytogenet. 1985;18:159–171. doi: 10.1016/0165-4608(85)90066-4. [DOI] [PubMed] [Google Scholar]