

Abstract
Helicases are proteins that unwind double-stranded nucleic acids. Dda helicase from bacteriophage T4 has served as an excellent model for understanding the molecular mechanism of this class of enzymes. Study of the structure of Dda may reveal why some helicases translocate in a 5′-to-3′ direction on DNA, while others translocate in a 3′-to-5′ direction. Attaining a structure of Dda has proven difficult because the protein fails to readily form crystals and is too large for current NMR technologies. We have developed a homology model of the enzyme which will serve to guide studies that examine the structural and functional significance of the interaction between Dda and DNA, and how this interaction affects translocation and unwinding of DNA. We have tested the structural model by using methods for mapping protein domains and for examining protein surfaces that interact with DNA.
Keywords: Dda, helicase, homology modeling, structure, formaldehyde crosslinking, footprinting, limited proteolysis
Helicases are essential to all cellular processes that involve nucleic acids including replication, transcription, translation, recombination and repair (1-3). They function by using NTP-hydrolysis to drive catalysis of the unwinding of dsDNA and dsRNA. Helicase deficiencies can result in a number of medical conditions in humans such as Werner syndrome, xeroderma pigmentosum (XP), and Bloom’s Syndrome (4-7). Elucidation of helicase mechanism could allow for treatment of such diseases as well as an understanding and perhaps control of nucleic acid metabolism in viruses and cancer.
There are five superfamilies (SF) of helicases of which, SF1 and SF2 are the largest and most well studied. All helicases contain Walker A and Walker B motifs which are involved in NTP binding and Mg+2 binding respectively. Those helicases found in SF1 and SF2 share seven conserved motifs: I, Ia, II, III, IV, V, VI (3). Binding, translocation and unwinding can occur in either a 3′ to 5′ direction (“A” helicases) or a 5′ to 3′ direction (“B” helicases) (8). Most helicases require a single-stranded overhang to stimulate ATP hydrolysis and begin translocation along nucleic acids.
The first helicase crystal structure solved was that of PcrA, an SF1A helicase found in Bacillus stearothermophilus (9). Structures of PcrA complexed with an ATP analog and a DNA substrate have since been published (10). The combination of these structures with biochemical data provided a mechanism for helicase unwinding based on the protein/DNA/ATP interactions observed. This model, “The Mexican Wave,” shows certain amino acids acting as ratchets moving ssDNA bases along the enzyme, unwinding DNA in the process (10).
Bacteriophage T4 helicase Dda is an SF1B helicase that has recently been implicated in aiding initiation at origins of replication in T4 (11). Currently only two SF1B DNA helicase structures have been published, RecD from E. coli and RecD2 from Deinococcus radiodurans (12, 13). E. coli RecD was solved as part of the RecBCD complex from E. coli (13). The DNA that crystallized with the original RecBCD complex bound to both RecB and RecC, but was not long enough to extend to RecD. However, a recent structure of RecBCD has been crystallized with a longer oligonucleotide that extends into the RecD helicase (12). This structure has allowed the identification of some residues which may be crucial to RecD/ssDNA interaction. Many of these residues are conserved in Dda. Although an SF1B helicase structure has yet to be solved with ATP, the structure of E. coli RecD has provided much insight into SF1B helicases. It also provides a good basis for a Dda homology model.
The sequence homology between E. coli RecD and Dda is low (16% identity) but a physical investigation of amino acid sequence reveals a similarity between the two helicases with respect to the placement of their seven highly conserved helicase motifs (Figure 1A). Although little biochemical information exists for the RecD enzyme, many studies have focused on the detailed biochemical characterization of Dda (14-28). We believe that a structure of Dda in combination with the wealth of biochemical knowledge currently published about Dda will provide us an opportunity to elucidate a mechanism for SF1B helicase translocation along DNA.
Figure 1.
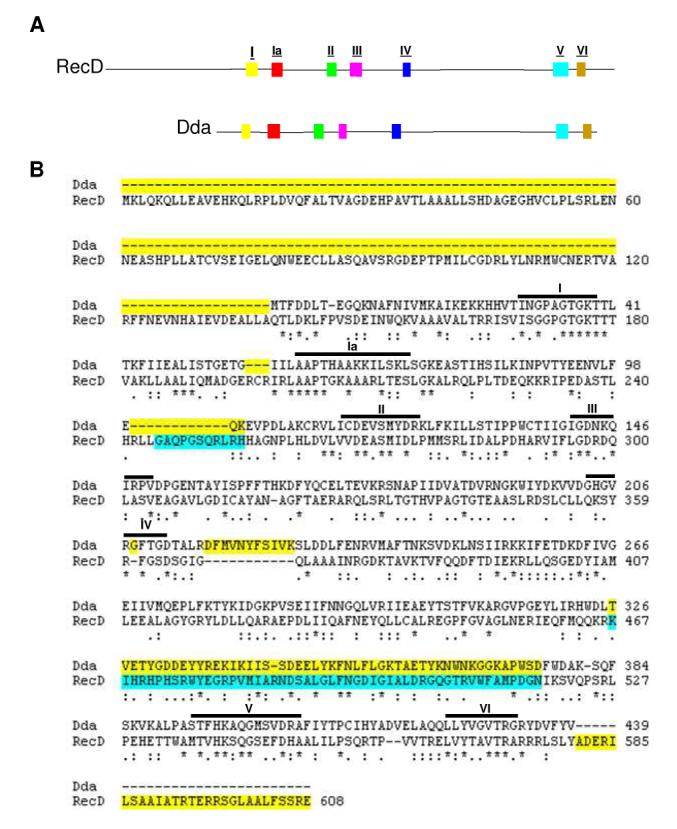
Highly conserved helicase motifs coincide between SF1B helicases Dda and RecD. (A) A side by side comparison of the amino acid sequences of Dda (bottom) and RecD (top) with conserved helicase motifs highlighted. Conserved motifs are as noted in Table 1. (B) Alignment of Dda and RecD used in homology model of Dda. The CLUSTALW program was used to attain an alignment of Dda with RecD. Identical residues are marked with an asterisk while conserved substitutions are marked with two dots and semi-conserved substitutions are marked with one dot. Regions highlighted in yellow were not included in the Dda model. Regions highlighted in blue do not exist in the original E. coli RecD crystal structure.
Dda has proven difficult to purify in quantities sufficient for crystallography (Blair and Raney unpublished results); thus we have set out to attain its structure through homology modeling using the E.coli RecD structure as a template. Also, we have used methodology for determining protein surface interactions to allow us to test our homology model, and in the process we have determined previously unreported sites of Dda/DNA interaction. We present our model as the most comprehensive representation of Dda structure available to date. We also present a model of Dda/DNA interaction based on our findings.
EXPERIMENTAL PROCEDURES
Molecular Modeling
Amino acid alignments of Dda (P32270.2 GI:20141288) and E. coli RecD (BAB37099.1 GI:13363148) were produced using the CLUSTAL W program (29). Our Dda homology model was created by replacing each amino acid in the RecD structure (1W36d) with its corresponding Dda amino acid from an alignment of the two proteins. Modeling was done using the COMPOSER portion of SYBYL software (Tripos). The energy of the structure was periodically minimized as the residues were replaced. The model was then modified using results from the PredictProtein website which determines the secondary structure of a molecule (30). WebLab Viewer Pro was used to create the figures (Molecular Simulations Inc.).
Materials
HEPES, glycerol, NaCl, glycine, ammonium bicarbonate and lysine-HCl were from Fisher. Iodoacetamide was from Sigma. Sulfo-SS-NHS-Biotin and TECP-HCl were from Pierce. Trypsin, GluC and AspN were from Roche. MALDI matrices 2,5-dihydroxybenzoic acid (DHB) and α-cyano-4-hydroxycinnamic acid (HCCA) were purchased from Sigma and recrystallized in house. C-18 ziptips were from Millipore. Dynabeads M280 streptavidin was from Invitrogen. All oligonucleotides were purchased from Integrated DNA Technologies and purified as described previously (25).
Protein Overexpression
An ATPase deficient version of Dda (DdaK38A) was used for all structural studies in this work due to its relatively high yield when overexpressed in E. coli cells. DdaK38A was purified as described previously for wtDda (24) with slight modifications, namely the use of a microfluidizer rather than the freeze thaw method for cell lysis. 54 mg of purified DdaK38A was acquired from 63 g of cells.
NHS-Biotin Footprinting
Protein footprinting experiments were adapted from Kvaratskhelia, et al. (31). Buffer conditions consisted of 50 mM HEPES pH 7.5 and 50 mM NaCl. Reactions containing 5 μM DdaK38A alone or with 5 μM DNA consisting of ten thymine residues (10T) were exposed to 1 mM Sulfo-SS-NHS-Biotin for labeling. All samples were incubated at room temperature for 0, 1, 5 or 10 minutes. Reactions were quenched with 100 mM lysine-HCl and divided into two aliquots. One aliquot was separated by SDS-PAGE and stained with colloidal Coomassie. Before digestion, samples were reduced using TCEP-HCl and alkylated using iodoacetamide. Bands containing Dda were excised from the gels and destained using 50 mM ammonium bicarbonate/50% methanol before being subjected to 100 ng GluC or 100 ng AspN digestion for mass spectrometric analysis. The second aliquot was digested in solution also using 100 ng GluC or 100 ng AspN. Samples were desalted using a C18 Ziptip and eluted in DHB MALDI matrix for mass spectrometric analysis. The final monoisotopic mass of the label was determined to be 145.18 Da.
Mass Spectrometry
Mass spectra of peptides were collected with a MALDI-prOTOF mass spectrometer (PerkinElmerSciex) (32, 33). Spectra were viewed using MoverZ software from Genomic Solutions. Monoisotopic peak lists were searched using the PeptideMap feature on the PROWL website (http://prowl.rockefeller.edu/prowl/prowl.html) which was set to identify peptides with lysines modified by the Sulfo-SS-NHS-Biotin reagent (+145.18 Da). Any peptides identified were then verified using tandem mass spectrometry on a vMALDI-LTQ ion trap mass spectrometer (Thermo). Theoretical tandem fragment ions were produced using the ProteinInfo function of the PROWL website and manually matched to MS/MS spectra.
Formaldehyde Crosslinking
Formaldehyde crosslinking of DdaK38A was performed using a method adapted from Kim, et al. (34). Buffer conditions consisted of 20 mM HEPES pH 7.5, 4 mM MgOAc, 1 mM DTT, 2 μM DdaK38A and 4 μM biotinylated 10mer (5′bio-ACCGACGCCA3′) DNA oligonucleotide. Dda and DNA were incubated for two minutes at room temperature to allow the protein/DNA interaction to occur. Formaldehyde was then added to the reaction and was quenched at 0.5, 1, 5 or 10 minutes using 200 mM glycine. The reactions were then digested using 75 ng trypsin at 37 °C overnight. Peptides covalently linked to biotinylated DNA were isolated using Dynabead M280 streptavidin beads. Crosslinking was reversed by incubation at 70°C for an hour. Elutant was collected by centrifugation for 3 minutes at 3,000 X g and concentrated using a C18 Ziptip. Samples were eluted in HCCA MALDI matrix and subjected to analysis by mass spectrometry.
Limited Proteolysis
A sample containing 0.7 mg/mL DdaK38A was incubated with GluC protease at concentrations varying from 0 U to 26.3 U for fifteen minutes at room temperature (35). Sample buffer (62.5 mM Tris HCl pH 6.8, 10% glycerol, 375 mM β-mercaptoethanol, 1% SDS and 0.008% bromophenol blue) was added to quench the reaction and samples were separated by SDS-PAGE using a 5% stacking gel and 10% resolving gel, which was stained using Coomassie Brilliant Blue. Bands containing Dda proteolysis products were excised, reduced with TCEP-HCl, alkylated with iodoacetamide and digested with 75 ng trypsin at 37 °C overnight. Tryptic peptides were analyzed by mass spectrometry and verified by tandem mass spectrometry as described above.
RESULTS
Homology modeling of Dda
In the absence of an x-ray crystallographic or NMR structure, homology modeling is often used to predict a protein’s tertiary structure. Although Dda does not have any obvious homologs, the structures of several superfamily 1 helicases are known. The SF1B DNA helicase, RecD from the RecBCD complex in E. coli, provides a good basis for Dda homology modeling due to its close alignment with Dda amino acid sequence. Figure 1A shows a side by side comparison of E. coli RecD and Dda amino acid sequences highlighting the seven highly conserved motifs of SF1 helicases. This comparison indicates a similarity in spacing of motifs between the two proteins. One exception is motif IV which has proven difficult to assign due to its variable amino acid sequence in different helicases (36).
The structure of the E. coli RecD protein (pdb: 1w36d) was imported into the SYBYL program and the COMPOSER function was used to mutate RecD amino acids one at a time to their corresponding Dda amino acids from the ClustalW alignment (Figure 1B). The published crystal structure of RecD lacks some regions (245-255 and 467-518) thus some portions of our final model of Dda (242-243 and 326-376) are missing. Figure 1B highlights these particular regions in the RecD/Dda alignment. Sequences shown in turquoise were not reported in the published RecD structure and are thus not represented in our Dda model. Some portions of RecD were not included in the final Dda model because they did not correspond to any amino acid in the Dda alignment. These are shown in Figure 1B highlighted in yellow. The amino acid sequence of RecD2 did not align well with Dda so, although it is a more complete structure, it was not chosen for homology modeling (Supplemental Figure 1).
The initial homology model had some discrepancies in secondary structure that could be discerned by simple observation. There were portions of the model that were classified in the COMPOSER program as alpha helices, but could easily be inferred to be beta sheets and vice versa. The PredictProtein program was used to predict the secondary structure of Dda based on its amino acid sequence (Supplemental Figure 2) and appropriate modifications were made to the model using WebLab viewer. The structure was then imported back into COMPOSER and energetically minimized.
RecD has N-terminal and C-terminal tails that do not align with Dda (Figure 1B), but otherwise the two proteins align well despite their low sequence similarities (16% identity based on ClustalW alignment). Also, we submitted the Dda amino acid sequence to the FUGUE database which provided a homology model based on the protein of known crystal structure most similar to Dda. The database returned RecD as most similar to Dda and produced a model almost identical to our own (Supplemental Figure 3). All of these factors contributed to our decision to use RecD as a basis for our Dda homology model.
Our current working homology model is shown in Figure 2. Dda is oriented with the C-terminus on the left because this is the orientation of the RecD structure in the RecBCD complex. All seven helicase motifs are highlighted and correspond in color to those in the side by side comparison of Dda and RecD primary structures in Figure 1A and in Table 1. Five of the motifs (I, II, III, IV, and IV) are located in a central cleft of the model where ATP binding and hydrolysis are most likely to occur based on other known helicase structures (37). Motifs Ia and V are located along the bottom portion of the protein near the putative DNA binding site. All motifs are placed similarly in our Dda model relative to their positioning in the RecD. The majority of helicase studies to date indicate that motif IV is involved in ssDNA binding although in our model it is positioned in proximity to the ATP hydrolysis pocket. Motif IV is of special interest because it appears to function differently in SF1 and SF2 helicases. In SF2 helicases it has a different conserved sequence and has been shown to interact with DNA (38). In SF1 helicases, however, it has been shown to interact with ATP (38, 39). The difficulty in assigning motif IV is illustrated by the fact that Satapathy et al have re-evaluated the position of this motif in RecD compared to earlier assignments (36, 37). Our model shows motif IV in the “flexible linker” between the two domains of Dda, which corresponds to the newer definition of the RecD motif IV placement described by Satapathy et al.
Figure 2.
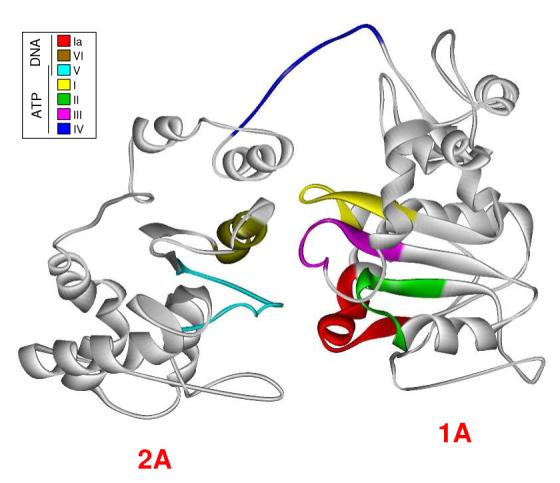
Homology model of Dda. The crystal structure of RecD was replaced residue for residue with its corresponding amino acid residue from its alignment with Dda. The energy of the structure was minimized periodically during the process. Helicase motifs are shown in the inset box and are grouped by their conserved functions. Motifs known to interact with ATP/ADP are located in the cleft between RecA-like domains of the protein while motifs associated with ssDNA interaction are located on the bottom face of the protein. The exception is Motif IV which has been reported to be involved in ssDNA interaction but is located near the ATP binding pocket in our model.
Table 1.
Conserved helicase motif function
| Motif | Function |
|---|---|
| I (yellow) | amino group of lysine interacts with phosphates of MgATP/MgADP; hydroxyl of serine or threonine coordinates Mg2+ ion. |
| Ia (red) | interacts with ssDNA, |
| II (green) | aspartic acid in position 1 coordinates Mg2+ |
| III (purple) | interacts with ATP |
| IV (blue) | interacts with ATP |
| V (turquoise) | interacts with ssDNA and/or ATP |
| VI (brown) | interacts with ATP |
Caruthers and McKay, (2002) Curr. Opin. Struct. Biol. 12, 123-133., Korolev et al. (1998) Protein Science 7, 605-610., Phillips et al. (1997) Mol. Gen. Genet. 254, 319-329.
The domains connected by this linker correspond to RecD domains 1A and 2A. The N-terminal domain of RecD, which is involved in protein/protein interaction, does not align to any portion of Dda and so it is not included in our model. Domains 1B and 2B as noted in the RecD2 structure are not included in our model because they did not crystallize in the E. coli RecD structure. Interestingly, in the RecD2 structure domain 2B was found to have an SH3 fold which is most commonly seen in signal transduction pathways and is known to bind peptides and occasionally DNA (40-42).
Because the seven helicase motifs are highly conserved between SF1 helicases, we can use information from other helicases to determine what portions of our Dda model should bind ssDNA, ATP and Mg2+. For example: the amino group of lysine 38 in motif I (yellow) is part of the P-loop in the Walker A motif which is known to interact with phosphates of MgATP/MgADP complexes (37). We have previously reported a variant of Dda that has a lysine to alanine point mutation at position 38 which does not exhibit ATPase activity (24). Wild type Dda appears to be toxic to E. coli cells which makes it difficult to purify in amounts sufficient for structural studies, therefore, we have used the ATPase deficient variant (DdaK38A) in all of our structural studies in this report. The lack of ATPase activity results in about a tenfold increase in purified protein. DdaK38A has been shown to bind DNA with an affinity similar to wtDda suggesting that its structure is similar to that of wtDda (24).
The motifs that are represented as being in the “middle” of the molecule are all conserved for ATP and Mg2+ interaction, strongly suggesting that this portion of Dda is involved in ATP binding and hydrolysis. We know from other SF1 helicase studies that the hydroxyl group of the threonine in motif I (yellow) coordinates with the Mg2+ ion. Motif II (green) has an aspartic acid (D115) which has been shown to coordinate Mg2+ in other SF1 helicases while motif III (purple) and motif VI (brown) have been reported to interact with ATP.
The motifs shown to interact with ssDNA reside at the “bottom” of the molecule. Motif V (turquoise) has been shown to interact with ssDNA and/or ATP, which coincides with its placement between the putative ssDNA binding site and the ATP hydrolysis pocket of our model (37). Motif IA (red) has a proline that is highly conserved in SF1B helicases and is proposed to be involved in ssDNA interactions (37).
The placement of motif IV in our model of Dda is of particular interest because it sits atop the ATP hydrolysis pocket. The RecD structure also shows motif IV located in the hinge region of the protein between domains 1A and 2A. We believe that this placement, combined with published information on the function of this motif in other SF1 helicases supports the finding that Motif IV is involved in ATP hydrolysis. Based on the side by side comparison with RecD, the amino acid alignment of RecD and Dda, and the location of the highly conserved helicase motifs, this structural model should be useful for conducting structure-function studies.
Dda is protected by DNA at lysines 123, 177, and 243
DNA binding studies have shown that Dda has a binding site size of 6 nt and has little or no sequence specificity (14, 24, 26). A specific DNA binding site on Dda, however, has not yet been reported. Chemical footprinting is a technique for determining protein-protein and protein-DNA interactions, whereby the chemical reactivity of specific amino acids is evaluated in the presence or absence of a ligand (31). We used Sulfo-SS-NHS-biotin to label surface lysines of Dda in the presence and absence of DNA. Dda contains 42 lysine residues (9.7% of the molecule), so it is a good candidate for chemical footprinting.
Dda was subjected to Sulfo-SS-NHS-biotin labeling for various times after which the reaction was quenched using lysine. This served to determine which lysines are accessible and reactive towards the labeling agent. Lysines in peptides identified by mass spectrometry are listed in Table 2 with their reactivity to the label noted. Dda was then incubated with an oligonucleotide, dT10, for two minutes to allow binding to occur and was then subjected to the same labeling reaction. Using mass spectrometry, we determined which peptides were labeled in the presence or absence of DNA (Table 2). Any reduction in reactivity indicates sites on Dda that interact with the DNA and are thereby protected from the labeling reaction.
Table 2.
Lysines identified by mass spectrometry
| Peptide* | Lysine | Modified Dda Alone |
Modified Dda with DNA (dT10) |
|---|---|---|---|
| 9 to 23 | 11 | no | no |
| 19 | no | no | |
| 24 to 47 | 22 | no | yes |
| 24 | yes | no | |
| 25 | yes | no | |
| 38 | no | no | |
| 43 | yes | yes | |
| 55 to 77 | 67 | no | no |
| 68 | no | no | |
| 72 | no | no | |
| 76 | yes | yes | |
| 78 to 93 | 86 | yes | yes |
| 121 to 142 | 123 | yes | no |
| 126 | yes | no | |
| 143 to 166 | 145 | no | no |
| 166 | no | no | |
| 167 to 184 | 177 | yes | no |
| 189 to 197 | 194 | no | no |
| 217 to 229 | 227 | yes | no |
| 230 to 245 | 243 | yes | no |
| 246 to 261 | 247 | no | no |
| 254 | no | no | |
| 255 | no | no | |
| 261 | yes | yes | |
| 268 to 288 | 277 | no | no |
| 280 | yes | yes | |
| 284 | no | no | |
| 349 to 361 | 351 | no | no |
| 358 | yes | yes | |
| 379 to 403 | 381 | yes | yes |
| 386 | yes | yes | |
| 388 | yes | yes | |
| 397 | yes | yes |
All peptides showed only one lysine modification.
The homology model shows lysine 123 positioned at the base of Dda in motif II, consistent with ssDNA binding in other helicases. According to our footprinting results, this residue is protected from chemical labeling when it is allowed to interact with ssDNA. Figure 3A shows mass spectrometry results for the 121-142 peptide. The unlabeled mass (2540.4 Da) can be observed in both samples due to the slight inefficiency of the labeling reaction. The labeled mass (2685.4 Da), however, can only be observed in the samples containing no DNA. This suggests that ssDNA is protecting Dda at K123 indicating a protein/DNA interaction at this site. Tandem mass spectrometry was used to show that the 2685.4 Da peak observed in the absence of DNA was the 121-142 peptide while the background peaks observed in the presence of DNA did not correspond to that peptide. Tandem mass spectrometry data (Supplemental Figure 4A) shows the sequence of the labeled peptide in the sample containing only Dda. Samples containing DNA had no peptide fragments matching the labeled peptide (Supplemental Figure 5). The MS/MS data verifies that the specific residue being labeled is K123.
Figure 3.

Identification of amino acid residues that interact with ssDNA by protein footprinting. ssDNA protects Dda from lysine-reactive labeling at three positions: K123, K177 and K243. Dda was exposed to NHS-biotin in the presence or absence of a ssDNA oligonucleotide, (dT)10. (A) Mass spectrometry results indicate an unlabeled 121-142 peptide peak in samples with and without ssDNA, but the labeled 121-142 peptide mass only exists in the samples without ssDNA. This peptide contains two lysines that could be labeled by NHS-Biotin, K123 or K126. Tandem mass spectrometry fragmentation ruled out K126 (Supplementary Figure 4). (B) Mass spectrometry results indicate an unlabeled 167-184 peptide peak in both samples, but the labeled peptide only exists in the samples with no ssDNA. This peptide has one lysine residue, K177. (C) Mass spectrometry results indicate an unlabeled 230-245 peptide peak in both samples, but the labeled peptide only exists in the samples including ssDNA. This peptide has one lysine residue, K243. (D) Representation of protected amino acids in the model. K123 (blue) is located in motif II which has been shown to interact with ssDNA in other helicases. K177 (red) is not located in a conserved helicase motif. K243 (green) is also not located in a conserved helicase motif, but it is located near Motif IV which has been reported to bind ssDNA in other helicases and is in proximity to the ATP hydrolysis pocket in our model. All spectra were matched within an error of 10 ppm.
Lysine 177, which is not located in a conserved helicase motif, also exhibits differential reactivity in the presence of DNA. In Figure 3B the unlabeled mass of the 167-184 peptide (2182.1 Da) can be seen in both samples, while samples containing no DNA show both the unlabeled and the labeled masses (2327.1 Da). This indicates that ssDNA is protecting Dda at K177 and suggests another protein/DNA interaction. K177 is located on the “top” surface of our Dda model, which is removed from the predicted DNA binding site at the “bottom”. This may be an indication that DNA is wrapping around Dda after it is unwound, or that the path of ssDNA is somewhat different on Dda than other SF1 helicases. Tandem mass spectrometry data (Supplemental Figure 4B) shows the sequence of the labeled peptide in the Dda alone sample. No fragment ions matching the labeled peptide were identified in the samples containing DNA (Supplemental Figure 5). The MS/MS data verifies that the specific residue being labeled is K177.
Lysine 243 is also not located in a conserved helicase motif but it is protected by ssDNA. Mass spectrometry results seen in Figure 3C indicate the unlabeled mass of peptide 230-245 (1884.9 Da) occurs in both Dda alone and Dda with DNA samples but the labeled peptide mass (2029.9 Da) only occurs in samples containing no DNA. Protection at K243 indicates a protein/DNA interaction at this site. Tandem mass spectrometry data (Supplemental Figure 4C) shows the sequence of the labeled peptide in the Dda alone sample. Once again, no fragments of the labeled peptide were identified in the tandem mass spectra of the samples with DNA (Supplemental Figure 5). The MS/MS allows determination that labeling is specifically occurring at K243 in samples without DNA. A comprehensive representation of DNA binding sites determined by footprinting as represented in our model is shown in Figure 3D.
The NHS-biotin footprinting experiment was repeated with a longer oligonucleotide (15T) which verified the three sites identified here as being protected as well as a novel site in the 349-361 peptide (Supplemental Figure 6). The 349-361 peptide is located in a portion of Dda that is not represented in our model but correlates to the SH3 domain found in RecD2.
Dda crosslinks to DNA in the 167-199 peptide
Another way to determine protein-DNA interactions is by using formaldehyde crosslinking. This method is commonly applied in chromatin immunoprecipitation (ChIP) assays when trying to determine the specific DNA binding sites of a protein. Here we used this technology to identify the DNA binding site of Dda using a biotinylated oligonucleotide. Formaldehyde covalently crosslinks lysine residues to adenine, cytosine and guanine DNA bases but not thymines. Therefore, we used a 10mer DNA oligonucleotide with a 5′ biotin tag that contained no thymines for this analysis. A 10 base ssDNA strand will allow one Dda molecule to bind and we used a saturating concentration of DNA to ensure all Dda molecules were bound (18). The efficiency of formaldehyde crosslinking is low (∼1%, Blair and Raney, unpublished observation) so the peptides that are crosslinked to DNA are in low abundance. In this analysis we used the biotin tag on the DNA to enrich for peptides that crosslinked to the ssDNA oligomer, thereby increasing the sensitivity of the method.
We identified one peptide of interest using the PeptideMap database to search mass spectrometry data. The mass of this peptide (3886.99 Da) corresponds to the 167-199 amino acid portion of Dda (Figure 4A) with four missed tryptic cut sites. We expected missed cuts due to the covalent crosslinking of the DNA to Dda occupying lysines and protecting some arginines, thus blocking typical trypsin cleavage sites. Tandem mass spectrometry was performed to determine the validity of this identification. The low abundance of the crosslinked peptide translated to minimal MS2 peptide fragment ions but two highly abundant ions could be identified in the spectra that correspond to theoretical fragment ions for the 167-199 peptide (Figure 4B). The peptide uncovered using this analysis is highlighted in Figure 4C. It encompasses K177 which was shown to be protected by DNA in the protein footprinting experiments described previously (Figure 3B). This further validates the interaction between Dda and DNA within this region of the protein.
Figure 4.
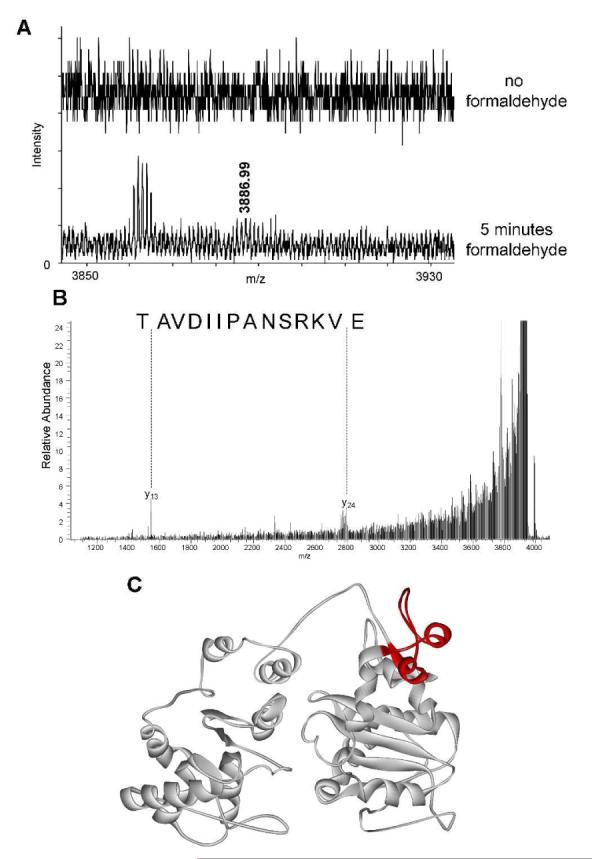
Crosslinking of Dda to DNA occurs in the 167-199 peptide. Dda was exposed to formaldehyde for 5 minutes in the presence of ssDNA to allow a covalent crosslink to occur. (A) Mass spectrometry results indicate a peptide of mass 3886.99 Da that can be found only in samples containing formaldehyde. (B) MS/MS results confirm that the 3886.99 Da peak mass correlates to Dda peptide 167-199. (C) This peptide, shown in red, contains K177 which was shown to be protected by ssDNA in the footprinting experiments shown in Figure 3B. All spectra were matched within an error of 10 ppm.
Dda residue E175 is solvent accessible
Limited proteolysis is another tool commonly used to characterize protein structure (43). Protease dependant digestion of a protein can help determine the solvent accessibility of certain residues. Buried amino acid residues are less accessible to the protease, while residues located on the surface of a protein will be much better candidates for proteolysis. These proteolysis products can be visualized using SDS-PAGE and their sequences determined by mass spectrometry.
We performed limited proteolysis on DdaK38A using increasing concentrations of GluC protease, which cleaves C-terminally to glutamate residues. We observed three distinct products, which were identified using SDS-PAGE followed by mass spectrometry (Figure 5A & 5B). One major product (Product 1) was identified to be a C-terminal truncation of Dda. Two glutamate residues exist near the N-terminal cut site of Product 1: E172 and E175 (Figure 5B and 5C, red). Both residues are located in the alpha helix containing K177 which was shown to bind DNA in Figures 3B and 4. Based on Product 2, an N-terminal truncation of Dda, and Product 3 , a mixture of N-and C-terminal truncations of Dda, we determined the cut site to be glutamine 175 (Figure 5B and 5C, red). This result indicates that this particular alpha helix is very accessible to GluC protease and so it is most likely to be a flexible region of the protein. Upon addition of dT15 ssDNA to the proteolysis reaction, the E175 product (Figure 5A, product 1) is reduced suggesting protection of the peptide containing E175 by ssDNA (Figure 5A).
Figure 5.
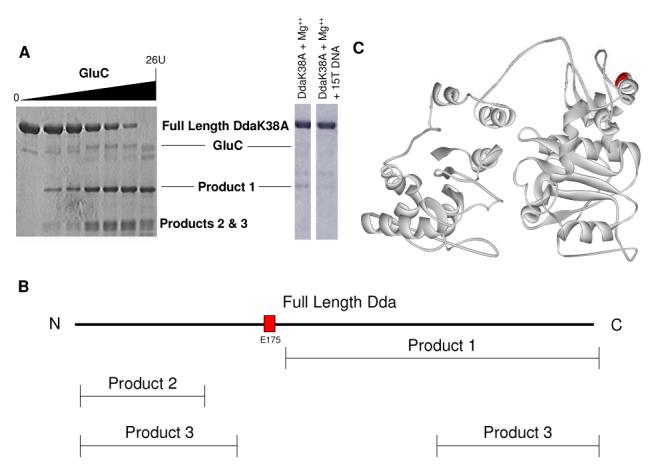
Limited proteolysis indicates E175 is solvent exposed. (A) Proteolysis was performed using varying concentrations of GluC. Three products were identified by SDS-PAGE. Proteolysis including dT15 DNA was performed under the following conditions: 10 μM DdaK38A, 2 mM Mg(OAc)2, 1 U GluC, 20 mM HEPES pH 7.5, 10 mM KOAc, 10% glycerol, 0.1 mM EDTA and 5 μM dT15 for 15 minutes. (B) Mass spectrometric analysis of the three proteolysis products indicates that proteolysis occurred at E175 (red). (C) E175 is located in a portion of the Dda model that has been shown to interact with ssDNA (red). All spectra were matched within an error of 10 ppm.
DISCUSSION
Elucidating the mechanism for unwinding of dsDNA by SF1 helicases will allow for important advances in the understanding of replication, recombination and repair; and may provide avenues for the development of treatments for cancer, viral infection, and diseases related to helicase activitiess (4-7). A wealth of biochemical knowledge has been reported on the SF1B helicase Dda, but no structure has been obtained. In this work, we present a homology model of Dda that will allow detailed structure/function studies of this enzyme, which is the best characterized SF1B helicase to date. We use this model along with novel Dda/ssDNA interaction data and previously published biochemical data to provide an explanation for how Dda interacts with DNA.
The homology model resembles the structure of RecD in that it has two domains that correspond to RecD domains 1A and 2A. However, after the modifications that we made to the RecD structure to attain our model, there are many subtle differences between the two structures that allow us to infer that the homology model accurately represents Dda structure. The secondary structure of some portions of RecD does not correspond to the same secondary structure in Dda. This was apparent in our model and led us to use PredictProtein to make the appropriate changes. Also, the helicase motifs are placed in a central portion of both RecD and our Dda model but they do not overlay perfectly. When the amino acid sequence of Dda was submitted to the FUGUE database a model based on RecD was produced (Supplemental Figure 3). FUGUE selects the proteins of known structure most similar to the sequence submitted and makes a homology model based on them. The RecD-based FUGUE model is very similar to the model in Figure 2 which was produced by molecular replacement. There are slight differences between the two structures but their similarities indicate that RecD is an ideal basis for Dda homology modeling.
X-ray crystal structure(s) of the PcrA helicase led to a proposal of an enzymatic mechanism for coupling of ATP binding and hydrolysis and DNA unwinding. The model, referred to as the “Mexican Wave”, indicates specific amino acid residues involved in transfer of one base of DNA through a series of “base flipping” steps that lead to one base translocated per ATP hydrolyzed (9, 44). SF1 helicases share similar helicase motifs, however SF1A helicases translocate in the 3′-to-5′ direction whereas SF1B helicases translocate in the 5′-to-3′ direction. The difference in directionality between SF1A and SF1B helicases may result from the placement of particular amino acids that are responsible for “base flipping” in the two classes of enzymes. Although the work presented here does not explain the directional bias of Dda, it does provide us with information pertinent to the amino acids crucial in Dda binding and translocation, which will be valuable for future structure-function studies.
Dda has been shown to function as a monomer in vitro and the enzyme has a six nucleotide DNA binding site (14, 24, 45). One of the most widely accepted models for monomeric helicase unwinding is called the inchworm model which indicates that two domains of a helicase can “walk” along ssDNA and unwind dsDNA in the process (10, 43). We have previously suggested that the displaced strand of DNA can interact with regions of Dda that may not be part of the conserved helicase motifs (14). We have also shown that Dda monomers act with functional cooperativity, unwinding DNA with greater processivity when multiple molecules bind a DNA substrate (18). Using this information we propose a model for the Dda/ssDNA interaction sites identified in this work (Figure 6). We suggest that monomeric Dda (gray) binds ssDNA and uses the inchworm mechanism to unwind and translocate using ATPase activity to drive the reaction. This schematic shows the displaced DNA strand wrapping around the protein and interacting at K177 and K243. Dda is known to interact with the single stranded binding protein, gp32, and the model shows the path of the displaced strand continuing through a putative site for binding of gp32 (shown in yellow in Figure 6; Blair et al, in preparation), (19). The model allows Dda to translocate along the tracking strand in a 5′ to 3′ direction allowing single-stranded-binding proteins to bind to the displaced strand, thereby preventing reannealing. After unwinding a short region of dsDNA, the newly available ssDNA could bind more than one Dda molecule to increase the processivity of unwinding as shown in vitro (15, 18). The schematic presented in Figure 6 takes into account the ssDNA binding patterns, crosslinking data and proteolysis data that we report here. The model also takes into account the interaction with the displaced strand and binding to gp32 that have been reported previously (14, 15, 18, 19).
Figure 6.
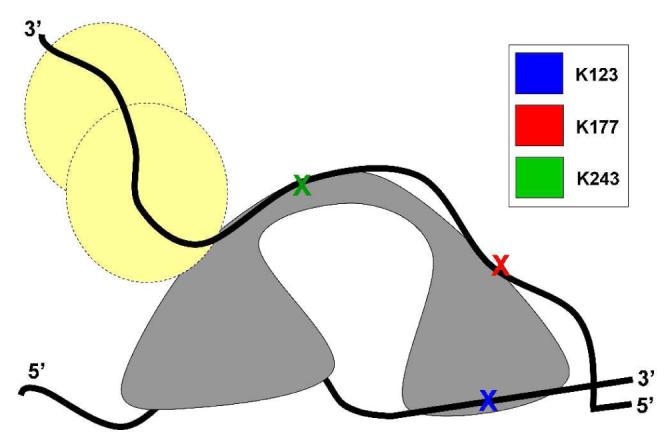
Representation of Dda interaction with DNA during unwinding. Dda (gray) is shown bound to a partially unwound DNA substrate. Dda/DNA interaction sites described in this work are colored as shown in Figure 3D. In this schematic, the tracking strand of DNA is unwound and interacts with K123 whereas the displaced strand wraps around Dda and interacts with K177 and K243, which are removed from the site of interaction with the tracking strand. The displaced strand, which corresponds to the leading strand template in a DNA replication fork, then proceeds to interact with T4 SSB gp32 (yellow circles; Blair, et al., in preparation) or replication machinery.
In conclusion, we have produced and characterized a homology model of the SF1B helicase Dda based on the structure of SF1B helicase RecD from the RecBCD complex in E. coli. This model is the most comprehensive structural representation of Dda reported to date. We have also reported three regions of Dda that are protected by ssDNA binding, one region of Dda that crosslinks DNA in the presence of formaldehyde and a region of Dda that is readily accessible to GluC digestion. This data has allowed us to propose a model for Dda/DNA interactions that can now be tested to further develop a mechano-chemical mechanism for DNA unwinding.
Supplementary Material
ACKNOWLEDGEMENT
We thank Dr. Cesar Compadre for general assistance with molecular modeling, Dr. Yaoqi Zhou for initial Dda models based on SF1A helicases and the UAMS Proteomics Core Facility.
† This research was funded by the NIH COBRE Grant P20RR15569 (KDR and AJT) and NIH Grant R01GM054900 (KDR), NIH INBRE grant P20RR016460 (AJT) and the Arkansas Biosciences Institute (AJT).
Abbreviations
- ABC
ammonium bicarbonate
- Dda
DNA-dependent ATPase
- DNA
deoxyribonucleic acid
- dsDNA
double stranded DNA
- NA
nucleic acids
- NMR
nuclear magnetic resonance
- SF
superfamily
- ssDNA
single-stranded DNA
Footnotes
SUPPORTING INFORMATION AVAILABLE Alignment of RecD2 with Dda, secondary structure of Dda as predicted by the PredictProtein database, homology model of Dda based on RecD from the FUGUE program, tandem mass spectrometry data verifying the location of protection in protein footprinting experiments. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- (1).Delagoutte E, von Hippel PH. Helicase mechanisms and the coupling of helicases within macromolecular machines. Part I: Structures and properties of isolated helicases. Q.Rev.Biophys. 2002;35:431–478. doi: 10.1017/s0033583502003852. [DOI] [PubMed] [Google Scholar]
- (2).Delagoutte E, von Hippel PH. Helicase mechanisms and the coupling of helicases within macromolecular machines. Part II: Integration of helicases into cellular processes. Q.Rev.Biophys. 2003;36:1–69. doi: 10.1017/s0033583502003864. [DOI] [PubMed] [Google Scholar]
- (3).Gorbalenya AE, Koonin EV. Helicases - Amino-Acid-Sequence Comparisons and Structure-Function-Relationships. Current Opinion in Structural Biology. 1993;3:419–429. [Google Scholar]
- (4).Brosh RM, Jr., Li JL, Kenny MK, Karow JK, Cooper MP, Kureekattil RP, Hickson ID, Bohr VA. Replication protein A physically interacts with the Bloom’s syndrome protein and stimulates its helicase activity. J.Biol.Chem. 2000;275:23500–23508. doi: 10.1074/jbc.M001557200. [DOI] [PubMed] [Google Scholar]
- (5).Ellis NA. DNA helicases in inherited human disorders. Curr.Opin.Genet.Dev. 1997;7:354–363. doi: 10.1016/s0959-437x(97)80149-9. [DOI] [PubMed] [Google Scholar]
- (6).Wu L, Davies SL, North PS, Goulaouic H, Riou JF, Turley H, Gatter KC, Hickson ID. The Bloom’s syndrome gene product interacts with topoisomerase III. J.Biol.Chem. 2000;275:9636–9644. doi: 10.1074/jbc.275.13.9636. [DOI] [PubMed] [Google Scholar]
- (7).German J. Bloom’s syndrome. Dermatol.Clin. 1995;13:7–18. [PubMed] [Google Scholar]
- (8).Singleton MR, Dillingham MS, Wigley DB. Structure and mechanism of helicases and nucleic acid translocases. Annu Rev Biochem. 2007;76:23–50. doi: 10.1146/annurev.biochem.76.052305.115300. [DOI] [PubMed] [Google Scholar]
- (9).Subramanya HS, Bird LE, Brannigan JA, Wigley DB. Crystal structure of a DExx box DNA helicase. Nature. 1996;384:379–383. doi: 10.1038/384379a0. [DOI] [PubMed] [Google Scholar]
- (10).Velankar SS, Soultanas P, Dillingham MS, Subramanya HS, Wigley DB. Crystal structures of complexes of PcrA DNA helicase with a DNA substrate indicate an inchworm mechanism. Cell. 1999;97:75–84. doi: 10.1016/s0092-8674(00)80716-3. [DOI] [PubMed] [Google Scholar]
- (11).Brister JR. Origin activation requires both replicative and accessory helicases during T4 infection. J Mol Biol. 2008;377:1304–1313. doi: 10.1016/j.jmb.2008.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Saikrishnan K, Griffiths SP, Cook N, Court R, Wigley DB. DNA binding to RecD: role of the 1B domain in SF1B helicase activity. Embo J. 2008;27:2222–2229. doi: 10.1038/emboj.2008.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Singleton MR, Dillingham MS, Gaudier M, Kowalczykowski SC, Wigley DB. Crystal structure of RecBCD enzyme reveals a machine for processing DNA breaks. Nature. 2004;432:187–193. doi: 10.1038/nature02988. [DOI] [PubMed] [Google Scholar]
- (14).Eoff RL, Raney KD. Intermediates revealed in the kinetic mechanism for DNA unwinding by a monomeric helicase. Nat Struct Mol Biol. 2006;13:242–249. doi: 10.1038/nsmb1055. [DOI] [PubMed] [Google Scholar]
- (15).Byrd AK, Raney KD. Displacement of a DNA binding protein by Dda helicase. Nucleic Acids Res. 2006;34:3020–3029. doi: 10.1093/nar/gkl369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Eoff RL, Spurling TL, Raney KD. Chemically modified DNA substrates implicate the importance of electrostatic interactions for DNA unwinding by Dda helicase. Biochemistry. 2005;44:666–674. doi: 10.1021/bi0484926. [DOI] [PubMed] [Google Scholar]
- (17).Eoff RL, Raney KD. Helicase-catalysed translocation and strand separation. Biochem Soc Trans. 2005;33:1474–1478. doi: 10.1042/BST0331474. [DOI] [PubMed] [Google Scholar]
- (18).Byrd AK, Raney KD. Increasing the length of the single-stranded overhang enhances unwinding of duplex DNA by bacteriophage T4 Dda helicase. Biochemistry. 2005;44:12990–12997. doi: 10.1021/bi050703z. [DOI] [PubMed] [Google Scholar]
- (19).Ma Y, Wang T, Villemain JL, Giedroc DP, Morrical SW. Dual functions of single-stranded DNA-binding protein in helicase loading at the bacteriophage T4 DNA replication fork. J.Biol.Chem. 2004;279:19035–19045. doi: 10.1074/jbc.M311738200. [DOI] [PubMed] [Google Scholar]
- (20).Byrd AK, Raney KD. Protein displacement by an assembly of helicase molecules aligned along single-stranded DNA. Nat Struct Mol Biol. 2004;11:531–538. doi: 10.1038/nsmb774. [DOI] [PubMed] [Google Scholar]
- (21).Tackett AJ, Corey DR, Raney KD. Non-Watson-Crick interactions between PNA and DNA inhibit the ATPase activity of bacteriophage T4 Dda helicase. Nucleic Acids Res. 2002;30:950–957. doi: 10.1093/nar/30.4.950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Nanduri B, Eoff RL, Tackett AJ, Raney KD. Measurement of steady-state kinetic parameters for DNA unwinding by the bacteriophage T4 Dda helicase: use of peptide nucleic acids to trap single-stranded DNA products of helicase reactions. Nucleic Acids Res. 2001;29:2829–2835. doi: 10.1093/nar/29.13.2829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Morris PD, Tackett AJ, Raney KD. Biotin-streptavidin-labeled oligonucleotides as probes of helicase mechanisms. Methods. 2001;23:149–159. doi: 10.1006/meth.2000.1116. [DOI] [PubMed] [Google Scholar]
- (24).Morris PD, Tackett AJ, Babb K, Nanduri B, Chick C, Scott J, Raney KD. Evidence for a functional monomeric form of the bacteriophage T4 DdA helicase. Dda does not form stable oligomeric structures. J Biol Chem. 2001;276:19691–19698. doi: 10.1074/jbc.M010928200. [DOI] [PubMed] [Google Scholar]
- (25).Morris PD, Raney KD. DNA helicases displace streptavidin from biotin-labeled oligonucleotides. Biochemistry. 1999;38:5164–5171. doi: 10.1021/bi9822269. [DOI] [PubMed] [Google Scholar]
- (26).Raney KD, Benkovic SJ. Bacteriophage T4 Dda helicase translocates in a unidirectional fashion on single-stranded DNA. J.Biol.Chem. 1995;270:22236–22242. doi: 10.1074/jbc.270.38.22236. [DOI] [PubMed] [Google Scholar]
- (27).Hacker KJ, Alberts BM. Overexpression, purification, sequence analysis, and characterization of the T4 bacteriophage dda DNA helicase. J.Biol.Chem. 1992;267:20674–20681. [PubMed] [Google Scholar]
- (28).Jongeneel CV, Formosa T, Alberts BM. Purification and characterization of the bacteriophage T4 dda protein. A DNA helicase that associates with the viral helix-destabilizing protein. J.Biol.Chem. 1984;259:12925–12932. [PubMed] [Google Scholar]
- (29).Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Rost B, Yachdav G, Liu J. The PredictProtein server. Nucleic Acids Res. 2004;32:W321–326. doi: 10.1093/nar/gkh377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Kvaratskhelia M, Miller JT, Budihas SR, Pannell LK, Le Grice SF. Identification of specific HIV-1 reverse transcriptase contacts to the viral RNA:tRNA complex by mass spectrometry and a primary amine selective reagent. Proc Natl Acad Sci U S A. 2002;99:15988–15993. doi: 10.1073/pnas.252550199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Collom SL, Jamakhandi AP, Tackett AJ, Radominska-Pandya A, Miller GP. CYP2E1 active site residues in substrate recognition sequence 5 identified by photoaffinity labeling and homology modeling. Arch Biochem Biophys. 2007;459:59–69. doi: 10.1016/j.abb.2006.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Gradolatto A, Rogers RS, Lavender H, Taverna SD, Allis CD, Aitchison JD, Tackett AJ. Saccharomyces cerevisiae Yta7 Regulates Histone Gene Expression. Genetics. 2008;179:291–304. doi: 10.1534/genetics.107.086520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Kim YC, Russell WK, Ranjith-Kumar CT, Thomson M, Russell DH, Kao CC. Functional analysis of RNA binding by the hepatitis C virus RNA-dependent RNA polymerase. J.Biol.Chem. 2005;280:38011–38019. doi: 10.1074/jbc.M508145200. [DOI] [PubMed] [Google Scholar]
- (35).Bird LE, Hakansson K, Pan H, Wigley DB. Characterization and crystallization of the helicase domain of bacteriophage T7 gene 4 protein. Nucleic Acids Res. 1997;25:2620–2626. doi: 10.1093/nar/25.13.2620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Satapathy AK, Pavankumar TL, Bhattacharjya S, Sankaranarayanan R, Ray MK. ATPase activity of RecD is essential for growth of the Antarctic Pseudomonas syringae Lz4W at low temperature. Febs J. 2008;275:1835–1851. doi: 10.1111/j.1742-4658.2008.06342.x. [DOI] [PubMed] [Google Scholar]
- (37).Caruthers JM, McKay DB. Helicase structure and mechanism. Curr.Opin.Struct.Biol. 2002;12:123–133. doi: 10.1016/s0959-440x(02)00298-1. [DOI] [PubMed] [Google Scholar]
- (38).Korolev S, Yao N, Lohman TM, Weber PC, Waksman G. Comparisons between the structures of HCV and Rep helicases reveal structural similarities between SF1 and SF2 super-families of helicases. Protein Sci. 1998;7:605–610. doi: 10.1002/pro.5560070309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Phillips RJ, Hickleton DC, Boehmer PE, Emmerson PT. The RecB protein of Escherichia coli translocates along single-stranded DNA in the 3′ to 5′ direction: a proposed ratchet mechanism. Mol Gen Genet. 1997;254:319–329. doi: 10.1007/pl00008605. [DOI] [PubMed] [Google Scholar]
- (40).Lodi PJ, Ernst JA, Kuszewski J, Hickman AB, Engelman A, Craigie R, Clore GM, Gronenborn AM. Solution structure of the DNA binding domain of HIV-1 integrase. Biochemistry. 1995;34:9826–9833. doi: 10.1021/bi00031a002. [DOI] [PubMed] [Google Scholar]
- (41).Eijkelenboom AP, Lutzke RA, Boelens R, Plasterk RH, Kaptein R, Hard K. The DNA-binding domain of HIV-1 integrase has an SH3-like fold. Nat Struct Biol. 1995;2:807–810. doi: 10.1038/nsb0995-807. [DOI] [PubMed] [Google Scholar]
- (42).Vink C, Groeneger A. M. Oude, Plasterk RH. Identification of the catalytic and DNA-binding region of the human immunodeficiency virus type I integrase protein. Nucleic Acids Res. 1993;21:1419–1425. doi: 10.1093/nar/21.6.1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Bird LE, Brannigan JA, Subramanya HS, Wigley DB. Characterisation of Bacillus stearothermophilus PcrA helicase: evidence against an active rolling mechanism. Nucleic Acids Res. 1998;26:2686–2693. doi: 10.1093/nar/26.11.2686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Soultanas P, Wigley DB. DNA helicases: ‘inching forward’. Curr.Opin.Struct.Biol. 2000;10:124–128. doi: 10.1016/s0959-440x(99)00059-7. [DOI] [PubMed] [Google Scholar]
- (45).Nanduri B, Byrd AK, Eoff RL, Tackett AJ, Raney KD. Pre-steady-state DNA unwinding by bacteriophage T4 Dda helicase reveals a monomeric molecular motor. Proc Natl Acad Sci U S A. 2002;99:14722–14727. doi: 10.1073/pnas.232401899. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.