

Summary
The proline catabolic enzymes proline dehydrogenase and Δ1-pyrroline-5-carboxylate dehydrogenase catalyze the 4-electron oxidation of proline to glutamate. These enzymes play important roles in cellular redox control, superoxide generation, apoptosis and cancer. In some bacteria, the two enzymes are fused into the bifunctional enzyme, proline utilization A. Here we review the three-dimensional structural information that is currently available for proline catabolic enzymes. Crystal structures have been determined for bacterial monofunctional proline dehydrogenase and Δ1-pyrroline-5-carboxylate dehydrogenase, as well as the proline dehydrogenase and DNA-binding domains of proline utilization A. Some of the functional insights provided by analyses of these structures are discussed, including substrate recognition, catalytic mechanism, biochemical basis of inherited proline catabolic disorders and DNA recognition by proline utilization A.
Keywords: Proline-Catabolism, Proline metabolism, Protein structure, X-ray crystallography, Proline dehydrogenase, P5C dehydrogenase, Proline utilization A, Ribbon-helix-helix
Introduction
Proline has a central role in metabolism and is increasingly being recognized as a critical amino acid in bioenergetics, cellular redox control, apoptosis and cancer (Phang 1985; Donald et al. 2001; Phang et al. 2001; Rivera and Maxwell 2005; Pandhare et al. 2006). Accordingly, there is great interest in understanding the biological roles and biochemical functions of the enzymes that oxidize and synthesize proline. Several articles in this issue discuss biological and medical aspects of proline metabolic enzymes. This purpose of this article is to provide a brief review of the three-dimensional structural information that is available for these enzymes, with a focus on the structural biology of proline catabolic enzymes.
The reactions catalyzed by proline oxidation and biosynthetic enzymes are shown in Fig. 1. Beginning with proline catabolism, all organisms oxidize proline to glutamate in two enzymatic steps coupled by a nonenzymatic equilibrium. In the first step, proline is oxidized to Δ1-pyrroline-5-carboxylate (P5C) by the FAD-dependent enzyme proline dehydrogenase (PRODH, EC 1.5.99.8). P5C forms a nonenzymatic equilibrium with glutamate semialdehyde (GSA). The P5C/GSA equilibrium is strongly pH-dependent with P5C favored at pH greater than approximately 6.5 (Lewis et al. 1993; Bearne and Wolfenden 1995). P5C dehydrogenase (P5CDH, EC 1.5.1.12) completes the transformation of proline to glutamate by catalyzing the oxidation of GSA, utilizing NAD+ as the electron acceptor. PRODH and P5CDH are highly conserved throughout eukaryotes and bacteria. On the other hand, archaea utilize a pair of enzymes distinct in sequence and structure from the eukaryotic/bacterial ones (Tsuge et al. 2005). Nevertheless, the essential chemistry of proline catabolism - the 4-electron oxidation of proline to glutamate via P5C/GSA - is common to all life.
Fig. 1.

Reactions catalyzed by proline catabolic and biosynthetic enzymes.
The biosynthetic pathway for proline has two routes for the formation of GSA from glutamate or ornithine. The glutamate route involves two enzymatic steps catalyzed by γ-glutamyl kinase (γ-GK, EC 2.7.2.11) and γ-glutamyl phosphate reductase (γ-GPR, EC 1.2.1.41). These enzymes are fused into the bifunctional enzyme P5C synthetase (P5CS) in plants and animals, whereas they are separate enzymes in lower organisms such as bacteria and yeast. Gamma-GK catalyzes phosphoryl transfer from ATP to glutamate to form γ-glutamyl phosphate. Gamma-GPR catalyzes the NADPH-dependent reduction of γ-glutamyl phosphate to GSA and phosphate. The route from ornithine to GSA is catalyzed by the pyridoxal-5′-phosphate-dependent enzyme ornithine aminotransferase (OAT, EC 2.6.1.13). Finally, reduction of P5C to proline is catalyzed by the NADPH-dependent enzyme P5C reductase (P5CR, EC 1.5.1.2).
Table 1 provides a list of crystal structures for the enzymes represented in Fig. 1. Although this information is not exhaustive, it does show that crystal structures are available for every enzyme. In most cases, the enzymes or domains have been crystallized in the presence of substrate analogs or products to provide insights into substrate recognition. Many of the enzymes and domains used for structure determination derive from bacterial sources, which reflects the relative ease of isolation and purification of the bacterial enzymes compared to the eukaryotic ones. Note, for example, that structures of both human proline catabolic enzymes and the human P5CS γ-GK domain have not been determined, although structures of bacterial homologs are known. Nevertheless, structures of the bacterial enzymes provide good starting places for understanding many important aspects of the human enzymes, including the overall fold, active site architecture, cofactor binding and substrate specificity. For the remainder of this review, we will focus on the proline catabolic enzymes (upper half of Table 1).
Table 1.
Crystal structures of proline catabolic and biosynthetic enzymes
| Protein or domain | PDB codes | Reference |
|---|---|---|
|
Proline catabolic enzymes | ||
| PRODH domain of E. coli PutA complexes with the proline analogs THFA, L-lactate and acetate | 1TIW, 1TJ0, 1TJ2 | (Zhang et al. 2004a) |
| PRODH domain of E. coli PutA reduced with dithionite | 2FZM | (Zhang et al. 2007) |
| PRODH from T. thermophilus | 2G37 | (White et al. 2007) |
| PRODH from T. thermophilus inactivated by N-propargylglycine | 2EKG | (White et al. 2008) |
| P5CDH from T. thermophilus complexes with NAD+, NADH, glutamate and NADP+ | 2BHP, 2BJA, 2BHQ, 2EHQ | (Inagaki et al. 2006; Inagaki et al. 2007) |
| DNA-binding domain of E. coli PutA | 2GPE | (Larson et al. 2006) |
| DNA-binding domain of E. coli PutA bound to DNA | 2RBF | (Zhou et al. 2008a) |
|
| ||
|
Proline biosynthetic enzymes | ||
| γ-GK from Campylobacter jejuni complexed with ADP | 2AKO | |
| γ-GK from E. coli complexes with glutamate and γ-glutamyl phosphate | 2J5T, 2J5V | |
| γ-GPR from Thermotoga maritima | 1O20 | (Page et al. 2004) |
| γ-GPR from Saccharomyces cerevisiae | 1VLU | |
| P5CS γ-GPR domain from human | 2H5G | |
| OAT from human | 1OAT | (Shen et al. 1998) |
| OAT from human, active site mutants | 2BYL, 2BYJ | (Markova et al. 2005) |
| OAT from human complexed with 5-fluoromethylornithine | 2OAT | (Storici et al. 1999) |
| OAT from human complexes with gabaculine and L-canaline | 1GBN, 2CAN | (Shah et al. 1997) |
| OAT from Plasmodium Yoelii | 1Z7D | |
| P5CR from Neisseria Meningitidis and Streptococcus pyogenes; apo and complexes with NADP+ and proline | 2AG8, 1YQG, 2AHR, 2AMF | (Nocek et al. 2005) |
| P5CR from human; apo and ternary complexes with NAD+/Glu and NADP+/Glu | 2GER, 2GR9, 2GRA | (Meng et al. 2006) |
| P5CR from human complexed with NAD+ | 2IZZ | |
| P5CR from Plasmodium falciparum complexed with NADP+ | 2RCY | |
Organization of proline catabolic enzymes
An interesting aspect of proline catabolism in the eukaryotic and bacterial worlds is that PRODH and P5CDH are fused together in some organisms, with the fused enzymes known as proline utilization A (PutA). The traditional view was that PRODH and P5CDH are separated in eukaryotes and fused in bacteria. Recent analysis of genome sequence data, however, reveals a more complex scheme (White et al. 2007), which is depicted in the phylogenetic tree in Fig. 2 The updated view is that PutAs are indeed restricted to bacteria (branches 1 and 2), but monofunctional PRODHs appear in both eukaryotes (branch 3A) and bacteria (branch 3B). This discovery is notable because the monofunctional bacterial enzymes are potential model systems for understanding the eukaryotic enzymes.
Fig. 2.

Unrooted phylogenetic tree representing the organization of proline catabolic enzymes in bacteria and eukaryotes. The tree has three main branches corresponding to PutAs (branches 1 and 2) and monofunctional enzymes (branch 3). PutAs are found only in bacteria. The monofunctional enzymes appear in both eukaryotes (branch 3A) and bacteria (branch 3B). Structures of proline catabolic proteins and domains are superimposed on their respective branches (clockwise from top left): E. coli PutA DNA-binding domain (branch 1), E. coli PutA PRODH domain (branch 1), T. thermophilus TtPRODH (branch 3B) and T. thermophilus TtP5CDH (branch 3B).
PutAs are peripheral membrane-associated bi-functional enzymes that contain 1000-1300 amino acid residues. The PRODH and P5CDH domains are located in the N-terminal and C-terminal halves of PutA, respectively. In some bacteria, such as E. coli (Becker and Thomas 2001; Gu et al. 2004), Salmonella typhimurium (Menzel and Roth 1981) and Pseudomonas putida (Vilchez et al. 2000a), PutA is also a transcriptional repressor of the genes putA and putP (encodes the high-affinity Na+-proline transporter). These PutAs are thus “trifunctional”. In the absence of proline, trifunctional PutA represses the expression of the put genes, which are transcribed in opposite directions, by binding to the put intergenic DNA region (Ostrovsky de Spicer et al. 1991; Brown and Wood 1992; Zhang et al. 2004b). To fulfill its roles as a transcriptional repressor and membrane-bound proline catabolic enzyme, trifunctional PutAs must undergo proline-dependent functional switching. The switching mechanism is actively studied by Prof. Donald Becker’s group using E. coli PutA as the prototype (Becker and Thomas 2001; Zhu and Becker 2003; Zhang et al. 2004b; Zhang et al. 2007). This work is also reviewed in this issue (Zhou et al. 2008b).
The PRODH fold
Crystal structures have been determined for the monofunctional PRODH from Thermus thermophilus (TtPRODH) and the PRODH domain of E. coli PutA. These structures established that PRODHs adopt a distorted (βα)8 barrel fold. The (βα)8 barrel is a common fold for enzymes and is often referred to as the TIM barrel fold because it was first observed for triosephosphate isomerase. The TIM fold is easy to recognize for TtPRODH (Fig. 3A). At 307 residues in length, TtPRODH is one of the shortest monofunctional PRODHs. It thus represents the minimalist PRODH enzyme. The classic TIM barrel fold consists of eight parallel β-strands arranged like the staves of a barrel (Fig. 3A). Each strand is followed by an α-helix, and these helices are arrayed on the outside of the β-barrel next to the strands. The strands and helices are numbered 1–8 starting at the N-terminus as shown in Fig. 3A. The active sites of TIM barrel enzymes are always formed by the loops connecting the carboxy ends of the strands with the amino ends of the helices. Accordingly, the FAD cofactor of TtPRODH binds at the C-terminal ends of the strands of the barrel (Fig. 3). The re face of the isoalloxazine packs tightly against strands 4–6, while the si face is available for hydride transfer from the substrate proline (Fig. 3A).
Fig. 3.
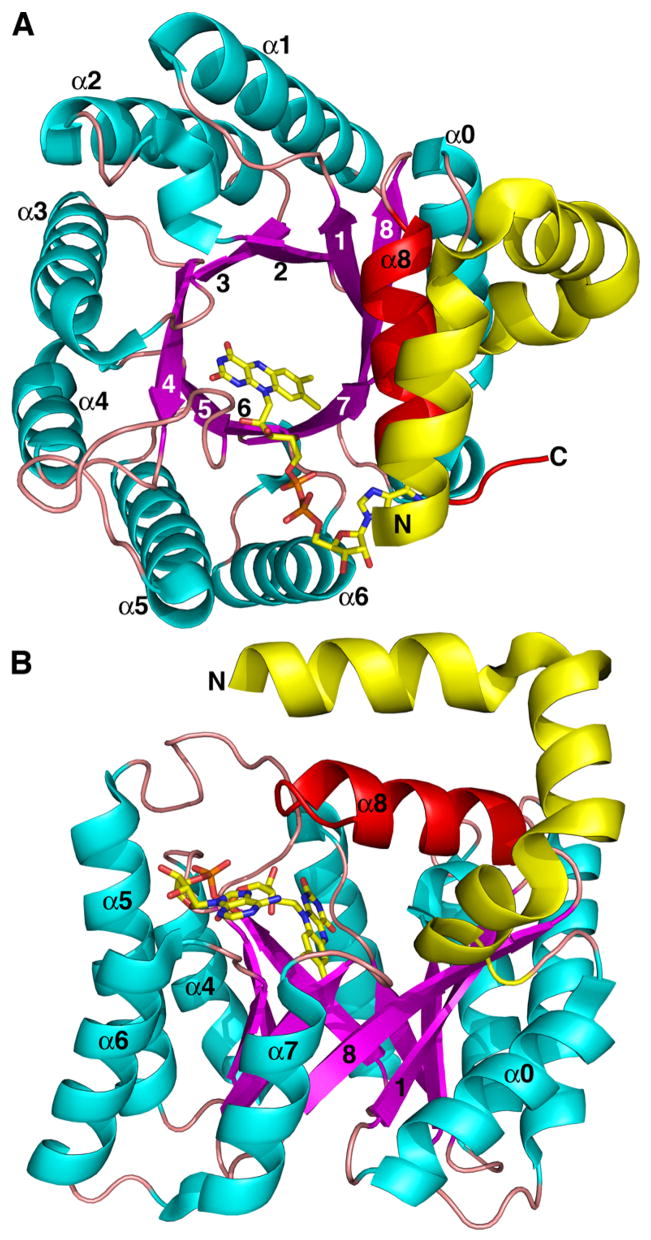
Two views of the monofunctional PRODH from T. thermophilus. β-strands and α-helices of the (βα)8 barrel are colored magenta and cyan, respectively. Helix 8 is colored red. Selected strands and helices are numbered. The FAD cofactor is shown in yellow sticks. This figure and others were prepared with PyMOL (W. L. DeLano (2002) The PyMOL Molecular Graphics System).
The PRODH barrel differs from the classic TIM barrel in that the helix following β-strand 8 in the topology (helix 8) sits atop the barrel rather than alongside β-strand 8 (Fig. 3B, red helix). We note that the location reserved for helix 8 in classic TIM barrels is occupied in PRODH by the helix that precedes β1 in the polypeptide chain (denoted α0 in Fig. 3B). This unique variation of the TIM barrel fold is crucial for catalysis because helix 8 donates side chains that interact with the substrate, as described in the next section.
Structures of the E. coli PutA PRODH domain have provided insights into how the PRODH barrel is incorporated into the PutA protein and how the substrate proline is recognized. The first structure of a PutA PRODH domain was determined from crystals of a construct containing residues 1-669 of E. coli PutA (Lee et al. 2003). Due to the susceptibility of this construct to proteolytic degradation near residues 82 and 632, subsequent structural and functional work employed the shorter constructs PutA86-669 and PutA86-630.
Crystal structures of E. coli PutA PRODH domain constructs revealed PutA-specific elaborations of the TIM barrel fold. For example, beginning at residue 87, the polypeptide chain forms three α-helices that wrap halfway around of the barrel (Fig. 4A, yellow helices), contacting helices 4–7. It was initially thought that the arms mediated dimerization of PutA PRODH domains (Lee et al. 2003), but this interpretation was later shown by Zhang et al. to be incorrect (Zhang et al. 2004a). Subsequent work showed that dimerization of PutAs is mediated, at least in part, by the DNA-binding domains (Larson et al. 2006). Currently, the role of the helical arm is unknown.
Fig. 4.

Two views of the PRODH domain of E. coli PutA complexed with the proline analog THFA. The orientations shown here are similar to those of Fig. 3. β-strands and α-helices of the (βα)8 barrel are colored magenta and cyan, respectively. Helix 8 is colored red. Selected strands and helices are numbered. The FAD cofactor and THFA inhibitor are shown in yellow and green sticks, respectively. The helical elbow that wraps around the barrel is shown in yellow. Residues following the barrel are colored slate. The dotted curve indicates poorly ordered parts of the polypeptide chain between residues 147 and 263.
The conformation of residues 141-260 is poorly defined (dotted curve in Fig. 4A), which suggests that these residues are highly flexible. However, one should use caution in interpreting structures derived from fragments of larger proteins. In this case, residues 140-260 could form a folded domain in full-length PutA that is stabilized by tertiary interactions not present in the construct used for crystallization.
Residues 261-562 of E. coli PutA form a distorted (βα)8 barrel that is very similar to that of TtPRODH (Fig. 4, magenta and cyan). The fact that this fold is observed in two different PRODHs suggests that it is the defining fold of the PRODH family. Within the barrel, there is a helix inserted between β5 and α5 (denoted α5a in Fig. 4), which is not observed in TtPRODH. This unique feature of PutA may contribute to the different FAD conformations in PutA and TtPRODH (White et al. 2007).
Following helix 8, residues 563-610 of E. coli PutA fold into 3 helices that pack against the barrel, atop the 3-helix arm (Fig. 4, slate). These residues are part of the linker that connects the PRODH and P5CDH active sites.
Structural basis of proline recognition by PRODH
Structures of the E. coli PutA PRODH domain complexed with reversible inhibitors L-tetrahydro-2-furoic acid (THFA), L-lactate and acetate have provided insights into substrate recognition (Zhang et al. 2004a). The most useful of these structures is the complex with THFA because of the high structural similarity of this compound to proline.
THFA binds at the si side of the FAD isoalloxazine such that the rings of the inhibitor and cofactor are approximately parallel (Fig. 5). FAD N5 is the hydride acceptor of the proline oxidation reaction and C5 of THFA represents the hydride donor atom of proline. These two atoms are close together (3.3 Å), as expected. THFA (or proline) is a rather small molecule, yet several residues − 9 total - contact the inhibitor. These interactions are described schematically in Fig. 6.
Fig. 5.

Stereo view of the active site the E. coli PutA PRODH domain highlighting interactions with the inhibitor THFA (green). The FAD cofactor is colored yellow. Protein side chains are colored white. Dotted lines indicate hydrogen bonds and ion pairs.
Fig. 6.
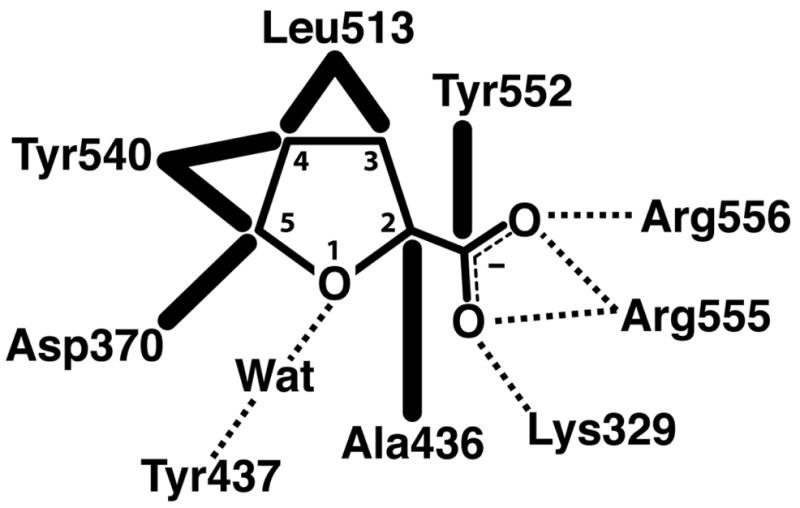
Schematic diagram of interactions between the E. coli PutA PRODH domain and the proline analog THFA. Dotted lines indicate hydrogen bonds and ion pairs. Thick solid lines indicate van der Waals interactions.
The residues that contact THFA are highly conserved. Among PutAs and bacterial monofunctional PRODHs, all but Ala436 and Tyr437 are identically conserved. Ile substitutes for Ala436 in some monofunctional PRODHs (Burkholderia, for example), and Asn substitutes for Tyr437 in branch 2A PutAs.
Conservation of these proline-contacting residues provides clues about the structural basis of the different substrate specificities of human PRODHs. Humans have two isoforms of PRODH (Adams and Frank 1980). The enzyme encoded on chromosome 22 (PRODH) is specific for proline, whereas the enzyme encoded on chromosome 19 (OH-PRODH) is specific for hydroxyproline. Analysis of sequence alignments clearly shows that seven of the nine proline-contacting residues are present in the sequences of both human enzymes: Asp370, Ala436, Tyr437, Leu513, Tyr552, Arg555 and Arg556. The sequence similarity between E. coli PutA and the human enzymes is weak near Lys329, but it appears that this residue may be Gln in the two human enzymes. Interestingly, a tyrosine residue equivalent to Tyr540 is present in human PRODH (Tyr548) but this residue is replaced by Ser in OH-PRODH (Ser485). In the PutA86-669/THFA structure, the phenol ring of Tyr540 contacts the C4 atom of THFA at a distance of 3.4 Å (Figs. 5, 6). This steric contact may prevent binding of hydroxyproline by interfering with the 4-OH group of hydroxyproline. With Ser in place of Tyr, the active site likely has enough open space to accommodate the 4-OH group. In addition, it is possible that the Ser side chain may form hydrogen bonding interactions with the 4-OH group.
Because so many residues cluster around THFA (and presumably proline), the inhibitor is fully buried by the protein and inaccessible to solvent. Based on the high sequence conservation of active site residues throughout the PutA/PRODH family, it is likely that all PutAs and PRODHs (including the human enzymes) bind the substrate in this closed, buried active site. Thus, the inhibitor-bound PutA PRODH structures raised the question of how the substrate enters the active site and how the product exits. Clearly protein motion must occur during substrate binding and product release.
Although crystals of the inhibitor-free PutA PRODH domain remain elusive, the structure of inhibitor-free monofunctional PRODH from T. thermophilus provides clues about protein motion involved in substrate binding and product release. As discussed above, TtPRODH adopts the distorted (βα)8 barrel fold in which helix 8 is poised atop the barrel. However, this critical helix is shifted 4 Å away from the FAD and toward the rim of the β-barrel, compared to the PutA86-669/THFA structure (compare Figs. 3A and 4A). As a result, the active site of TtPRODH is open and highly solvent exposed (White et al. 2007). Comparison of the TtPRODH and PutA86-669/THFA structures suggests that α8 is crucial for protecting the flavin from solvent and that this helix may be a flexible element of the active site that moves in response to substrate binding and product release. Flexibility of α8 may be relevant to understanding the oxygen reactivity of some PRODHs. Most notably, production of proline-dependent superoxide is central to the role of human PRODH in apoptosis (Donald et al. 2001; Liu et al. 2005). Also, it has been shown that TtPRODH generates superoxide in vitro (White et al. 2007). For PRODH to generate superoxide, molecular oxygen must have access to the reduced flavin. It is thus tempting to speculate that movement of helix 8 plays a role in the mechanism of superoxide production by modulating accessibility of the FAD cofactor.
Mechanism-based inactivation of PRODH
Reversible inhibition of PRODHs and PutAs by proline analogs THFA, L-lactate, and acetate has been studied in detail using kinetic and structural methods (Zhu et al. 2002; Zhang et al. 2004a). These compounds are classic competitive inhibitors with inhibition constant values (Ki) ranging from sub-millimolar for THFA to 30 mM for acetate. Irreversible inhibition of PRODH by mechanism-based inactivators is less well studied. In one study, for example, Tritsch, et al., reported that 4-methylene-L-proline is a mechanism-based inactivator of PRODH activity in rat liver mitochondrial suspensions (Tritsch et al. 1993).
A new development in this area is the discovery that N-propargylglycine ((1) in Scheme 1) is a mechanism-based inactivator of PRODH (White et al. 2008). Measurements of the kinetics of inhibition have been studied for TtPRODH, and a 1.9 Å resolution structure of the inactivated enzyme has been determined (White et al. 2008). The structure shows that the N5 atom of the FAD cofactor is covalently connected to the ε-amino group of Lys99 via a 3-carbon linkage (3). This residue is equivalent to Lys329 of E. coli PutA, which forms ion pair interactions with the carboxylate group of the substrate (Figs. 5, 6). Furthermore, the isoalloxazine ring has a butterfly angle of 25°, which suggests that the flavin cofactor is reduced. Although the mechanism of inactivation is unknown at this time, the likely first step is the oxidation of N-propargylglycine to N-propargyliminoglycine (2) with concomitant reduction of FAD. This step is analogous to the oxidation of proline to P5C (Fig. 1). N-propargyliminoglycine is an α, β-unsaturated iminium compound and two possible mechanisms can be envisioned that lead to the observed FAD-Lys adduct (White et al. 2008). In the first mechanism, the flavin N5 atom serves as a nucleophile that attacks the acetylene group of (2) to produce a 1,4-addition product. Schiff base formation between lysine and the imine of the 1,4-addition product releases glycine and links the enzyme to the modified flavin. In the second mechanism, hydrolysis of (2) yields propynal and glycine. This step is analogous to hydrolysis of P5C to GSA (Fig. 1). A 1,4-addition reaction with propynal coupled with Schiff base formation between Lys and the carbonyl group tethers the enzyme to the flavin.
Scheme 1.

Preliminary studies show that N-propargylglycine inactivates PutAs from B. japonicum and E. coli, as well as PutA86-630. It will be interesting to see whether this compound inactivates eukaryotic PRODHs. We note that the active site Lys that forms the adduct with reduced FAD is replaced by Gln in the human enzymes, although as noted above, the sequence alignments are not robust in this region of the polypeptide chain. Whether the active site Lys is necessary for inactivation remains to be seen.
The discovery that N-propargylglycine is a mechanism-based inactivator of PRODHs and PutAs provides a new avenue for developing specific inhibitors by elaboration of the N-propargylglycine framework to make it more structurally analogous to proline. Such inactivators could have potential applications. One possible use is insect control because proline, via proline catabolism, is a major fuel source for insects during flight (Saktor 1976; Custer 2005). Another possibility is the development of inactivators that target PRODH from procyclic forms of trypanosomatids, which utilize proline catabolism for energy (Bringaud et al. 2006). Specific, irreversible inactivators of PRODH also offer a new way to modulate proline catabolism in eukaryotic cells. These compounds may be useful in studies of the role of PRODH in cancer and apoptosis.
Structural basis of PutA-DNA recognition
Trifunctional PutAs are transcriptional repressors of the put regulon in addition to serving as membrane-associated bifunctional proline catabolic enzymes (Menzel and Roth 1981; Ostrovsky de Spicer et al. 1991; Vilchez et al. 2000b; Gu et al. 2004). The put regulon consists of the genes that encode PutA and the Na+- proline transporter PutP, which are transcribed divergently. PutA represses transcription of the PutA and PutP genes by binding to sites in a control region that separates the two genes. In E. coli, for example, the control region consists of 419 base pairs and there are PutA five binding sites, each containing the consensus sequence GTTGCA (Zhou et al. 2008a).
Molecular dissection studies showed that the DNA-binding domain of E. coli PutA resides within the N-terminal 47 residues of the polypeptide chain, and analysis of amino acid sequence data further suggested that the DNA-binding domain is a ribbon-helix-helix (RHH) domain (Gu et al. 2004). Crystal structures of a polypeptide corresponding to the first 52 residues of E. coli PutA (PutA52) confirmed that PutA is a member of the RHH family of DNA-binding proteins (Larson et al. 2006). Well-known members of this large family include the Arc repressor, which controls the bacteriophage lytic cycle, MetJ, (regulation of methionine biosynthesis genes) and NikR (Ni(II) response regulator).
PutA52 is typical of RHH domains in terms of overall fold and dimeric structure (Fig. 7A). The RHH fold consists of a β-strand (β1) followed by two α-helices (αA, αB). Two RHH subunits assemble into a dimer featuring an intermolecular two-stranded antiparallel β-sheet. We note that all trifunctional PutAs studied to date form apparent dimers in solution. It is likely that RHH domains of the full-length protein dimerize as shown in Fig. 7A because the intermolecular β-sheet is necessary for DNA recognition, as described next.
Fig. 7.

Structure of the E. coli PutA DNA-binding domain. A, PutA52 dimer highlighting residues important for binding DNA. The two chains are colored green and red. Positions of residues of the β-sheet that form hydrogen bonds with DNA bases are indicated in yellow. Side chains of residues at the N-terminus of αB that interact with the DNA backbone are drawn as cyan sticks. B, Structure of PutA52 complexed with DNA, highlighting interaction of the protein β-sheet with the DNA major groove. Coloring of the protein is the same as in panel A. Selected protein side chains are drawn as spheres.
The structure of PutA52 complexed with a 21-bp DNA fragment corresponding to one of the consensus binding sites was recently determined (Zhou et al. 2008a). The β-sheet of PutA52 inserts into the DNA major groove (Fig. 7B), which is typical for RHH proteins (Schreiter and Drennan 2007). With this arrangement, residues near the N-terminus of αB interact with the DNA backbone, and residues of the sheet interact with DNA bases. Residues forming interactions with the DNA backbone include Thr28, Pro29 and His30 (Fig. 7, cyan). Three residues of the β-sheet form base-specific hydrogen bonds with the consensus DNA motif: Thr5, Gly7 and Lys9 (Fig. 7, yellow). All six of these critical residues appear to be identically conserved in trifunctional PutAs (Larson et al. 2006).
The roles of Gly7 and Pro29 in DNA recognition are particularly interesting because these residues are universally conserved among trifunctional PutAs, yet are rarely found in other RHH domains. Typically, RHH domains have a polar side chain, such as Thr or Asn, at the position of the β-sheet equivalent to PutA Gly7. This polar residue in other RHH domains forms hydrogen bonds with DNA bases. The PutA52/DNA structure shows that Gly7 contributes to base-specific recognition, despite lacking a polar side chain (Zhou et al. 2008a). Gly7 of one protein chain donates a hydrogen bond to a guanine base, while Gly7 of the other protein chain forms van der Waals interactions with the C5 methyl group of a thymine base. Pro29 is the first residue of αB (Fig. 7A). This position is fairly variable in RHH domains but it is rarely proline. The structure shows that the Cδ atom of Pro29 is located 3.4 Å from oxygen atoms of the DNA backbone (Zhou et al. 2008a). This observation suggests the intriguing possibility that Pro29 may be donating unconventional C-H…O hydrogen bonds to the DNA backbone. The unique interactions formed by PutA-specific residues Gly7 and Pro29 are likely important for proper recognition of operator sites in the put control region.
Structural studies of P5CDH
P5CDH catalyzes the oxidation of GSA to glutamate using NAD+ as the electron acceptor (Fig. 1). This enzyme belongs to the aldehyde dehydrogenase (ALDH) superfamily, which reminds us that GSA - not P5C - is the substrate. ALDHs have been classified into families and subfamilies based on amino acid sequence analysis (Sophos and Vasiliou 2003). Human P5CDH belongs to eukaryotic ALDH family 4 and is referred to in the literature as ALDH4 and ALDH4A1.
Several structures of P5CDH from T. thermophilus (TtP5CDH) have been determined (Inagaki et al. 2006; Inagaki et al. 2007), including complexes with NAD+, NADH, NADP+, and glutamate (Table 1). TtP5CDH displays the ALDH fold, which consists of three domains: NAD+-binding domain, catalytic domain and dimerization domain (Fig. 8A). The NAD+-binding domain adopts a distorted Rossmann dinucleotide-binding fold featuring a five-stranded parallel β-sheet. This particular variation of the classic Rossmann dinucleotide-binding fold has been described (Liu et al. 1997). As in other Rossmann-fold enzymes, the NAD+ cofactor binds in an extended conformation along the carboxy-terminal edge of the β-sheet (Fig. 8A). The catalytic domain adopts an α/β fold featuring a central 7-stranded β-sheet with all but one strand in parallel. This domain provides the catalytic Cys residue (Cys322 in TtP5CDH). The active site is located between the dinucleotide-binding and catalytic domains. The dimerization domain is a three-pronged extension with two of the prongs forming a β-structure (Fig. 8A, pink).
Fig. 8.
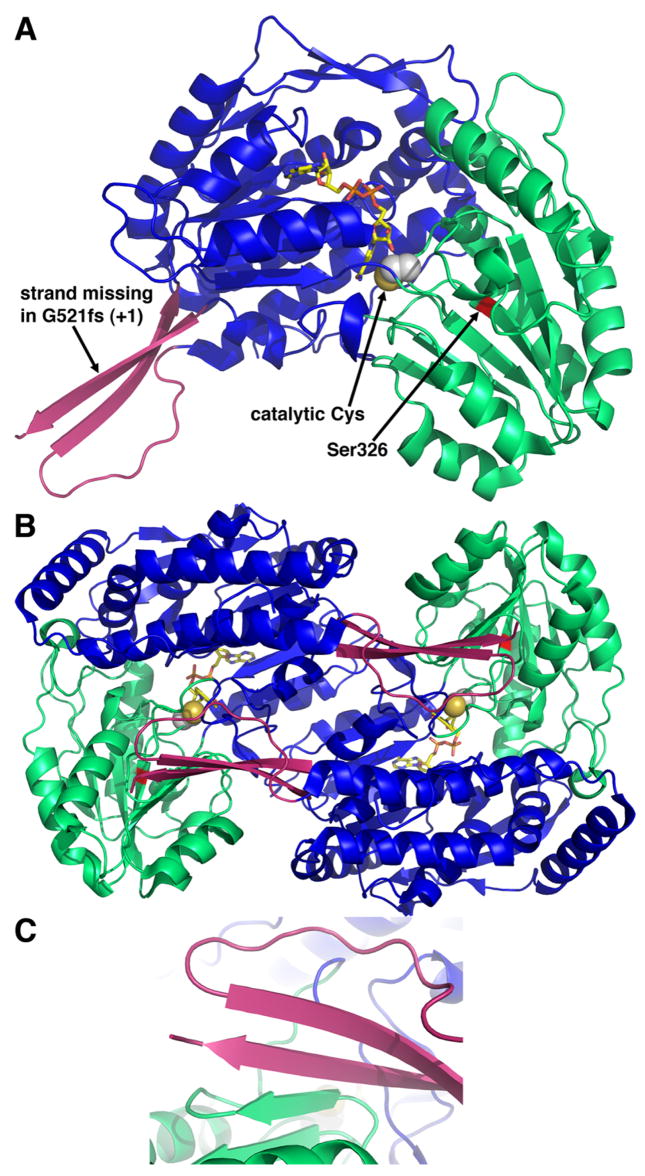
Structure P5CDH from T. thermophilus (Inagaki et al. 2006). A, ribbon drawing of a P5CDH subunit. The three domains are colored blue (NAD+-binding), green (catalytic) and pink (dimerization). The NAD+ cofactor is shown in yellow sticks. Catalytic Cys322 is represented in spheres. B, ribbon drawing of a P5CDH dimer. The two subunits of the dimer are colored as in panel A. C, close-up view of the intermolecular β-sheet in the dimer interface.
Two P5CDH subunits associate into a domain-swapped dimer via intermolecular β-sheet formation involving the β-sheet of the catalytic domain and the dimerization domain of the adjacent subunit (Fig. 8B–C). The available structural information suggests that all ALDHs form this type of dimer. In some ALDHs, such as retinal dehydrogenase (Lamb and Newcomer 1999), these dimers associate to form a tetramer (“dimer of dimers”). In TtP5CDH, however, three dimers assemble into a hexamer, which is unique for ALDHs.
Structures of TtP5CDH complexed with NAD+ and glutamate have provided insights into the catalytic mechanism (Inagaki et al. 2006). The generally accepted mechanism of ALDHs begins with nucleophilic attack by Cys on the C atom of the substrate aldehyde group. Interactions in the active site help increase the electrophilicity of this C atom. In TtP5CDH, these interactions are hydrogen bonds between the O atom of the aldehyde group and Asn 184 and the backbone N-H group of the catalytic Cys. This initial nucleophilic attack results in formation of a hemithioacetal tetrahedral intermediate. Hydride transfer from the hemithioacetal intermediate to NAD+ produces the thioacylenzyme intermediate and NADH. Upon dissociation of NADH from the enzyme, residues 288-290 are predicted to change conformation. The most important of these dynamic residues is Glu288, which is proposed to swing into the active site to activate a water molecule for hydrolysis of the thioacylenzyme to generate the product glutamate.
Finally, the TtP5CDH structures have been used to help understand the structural and functional consequences of mutations associated with type II hyperprolinemia, including the missense mutation S352L and the frameshift mutation G521fs (+1) (Inagaki et al. 2006). Expression of these alleles in a P5CDH-deficient strain of yeast has shown that the mutant enzymes are nonfunctional (Geraghty et al. 1998; Phang et al. 2001). Ser352 of the human enzyme corresponds to Ser326 of TtP5CDH (red patch in Fig. 8A). This residue is located on the β-strand that follows catalytic Cys322, so it is close to the active site. The Ser-to-Leu mutation is predicted to introduce a hydrophobic side chain into a hydrophilic environment (Inagaki et al. 2006). This change is expected to disrupt hydrogen bonding interactions that are important for substrate binding and proper positioning of the catalytic Cys. The frameshift mutation is predicted to cause premature termination of translation nine codons downstream (Geraghty et al. 1998). Based on the structure, the truncated protein would lack the β-strand of the dimerization domain (Fig. 8A), which mediates intermolecular β-sheet formation in the dimer (Figs. 8B, 8C). Thus, the dimer interface of the truncated protein would be severely compromised, resulting in impaired foldability, decreased stability and lower activity.
Future challenges
The first crystal structure of a proline catabolic enzyme (PRODH) was reported in 2003 (Lee et al. 2003). The list of known structures has grown to include both of the bacterial monofunctional enzymes and the DNA-binding domain of PutA. These structures have informed us about catalytic mechanisms (Lee et al. 2003; Baban et al. 2004; Zhang et al. 2004a; Inagaki et al. 2006; White et al. 2007), biochemical basis of proline metabolic disorders (Zhang et al. 2004a; Bender et al. 2005; Inagaki et al. 2006), redox-linked conformational changes in PutA (Zhang et al. 2007) and DNA recognition by PutA (Larson et al. 2006; Zhou et al. 2008a). Although our knowledge of the structural basis of proline catabolism has matured, several outstanding challenges remain.
Structures of the eukaryotic enzymes are needed to answer basic biochemical questions and to further understand the biological roles of these enzymes. For example, whereas the C-terminal halves of human PRODH and OH-PRODH are homologous to bacterial PRODHs, the function of the N-terminal parts of the enzymes are unknown. Also, structures of human PRODHs will provide insights into the different substrate specificities of PRODH (proline) and OH-PRODH (hydroxyproline). Additionally, several missense mutations in human PRODH that are linked to diseases map to regions outside of the active site, where sequence identity with the bacterial homologs is low. In these regions, interpretations based on structures of the bacterial enzymes can be problematic. Finally, structures of human PRODH would aid in understanding the mechanism of superoxide generation.
Several structure-based questions about PutAs remain unanswered at this time. The major one is how the functional domains of PutA are spatially arranged. This information is essential for understanding how PutAs coordinate sequential oxidation reactions and the mechanism by which trifunctional PutAs switch between regulatory and enzymatic functions. The fusion of PRODH and P5CDH activities in PutA may provide a kinetic advantage because P5C/GSA can be channeled though the protein interior from the PRODH active site to the P5CDH active site rather than diffusing into bulk solvent, which is inefficient (Miles et al. 1999; Huang et al. 2001). Maloy’s group reported kinetic data indicating substrate channeling for S. typhimurium PutA (Surber and Maloy 1998), but structural and mechanistic descriptions of channeling in PutAs are lacking.
Channeling is potentially possible for the monofunctional enzymes as well. Eisenberg and co-workers refer to fused proteins, such as PutA, as Rosetta Stone proteins because they decipher interactions between protein pairs (Marcotte et al. 1999). Thus, the Rosetta Stone hypothesis of protein evolution predicts that monofunctional PRODH and P5CDH form physical and functional interactions. Substrate channeling could be particularly important in eukaryotic cells because of the role of P5C as a signalling molecule (Phang 1985). Whether monofunctional PRODH and P5CDH interact and engage in substrate channeling is a fundamental unanswered question in proline catabolism.
Acknowledgments
I would like to express my sincere gratitude to my former and current students and postdoctoral associates who have contributed to the work reviewed here: Dr. Tommi White, Dr. John Larson, Min Zhang, Dr. Jermaine Jenkins, Dr. Jonathan Schuermann, Dr. Yong-Huan Lee and Shorena Nadaraia. I also thank Prof. Donald Becker and his students for their crucial contributions to this research. Finally, I thank Dr. David Valle for insightful discussions about substrate specificities of human PRODHs. This research was supported by NIH grant GM065546. Part of the research reviewed here was performed at Advanced Light Source beamline 4.2.2. The Advanced Light Source is supported by the Director, Office of Science, Office of Basic Energy Sciences, Materials Sciences Division, of the U.S. Department of Energy under Contract No. DE-AC03-76SF00098 at Lawrence Berkeley National Laboratory.
References
- Adams E, Frank L. Metabolism of proline and the hydroxyprolines. Annu Rev Biochem. 1980;49:1005–1061. doi: 10.1146/annurev.bi.49.070180.005041. [DOI] [PubMed] [Google Scholar]
- Baban BA, Vinod MP, Tanner JJ, Becker DF. Probing a hydrogen bond pair and the FAD redox properties in the proline dehydrogenase domain of Escherichia coli PutA. Biochim Biophys Acta. 2004;1701:49–59. doi: 10.1016/j.bbapap.2004.06.001. [DOI] [PubMed] [Google Scholar]
- Bearne SL, Wolfenden R. Glutamate gamma-semialdehyde as a natural transition state analogue inhibitor of Escherichia coli glucosamine-6-phosphate synthase. Biochemistry. 1995;34:11515–11520. doi: 10.1021/bi00036a026. [DOI] [PubMed] [Google Scholar]
- Becker DF, Thomas EA. Redox properties of the PutA protein from Escherichia coli and the influence of the flavin redox state on PutA-DNA interactions. Biochemistry. 2001;40:4714–4721. doi: 10.1021/bi0019491. [DOI] [PubMed] [Google Scholar]
- Bender HU, Almashanu S, Steel G, Hu CA, Lin WW, Willis A, Pulver A, Valle D. Functional consequences of PRODH missense mutations. Am J Hum Genet. 2005;76:409–420. doi: 10.1086/428142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bringaud F, Riviere L, Coustou V. Energy metabolism of trypanosomatids: adaptation to available carbon sources. Mol Biochem Parasitol. 2006;149:1–9. doi: 10.1016/j.molbiopara.2006.03.017. [DOI] [PubMed] [Google Scholar]
- Brown ED, Wood JM. Redesigned purification yields a fully functional PutA protein dimer from Escherichia coli. J Biol Chem. 1992;267:13086–13092. [PubMed] [Google Scholar]
- Custer AV. Stoichiometric estimates of the biochemical conversion efficiencies in tsetse metabolism. BMC Ecol. 2005;5:6. doi: 10.1186/1472-6785-5-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donald SP, Sun XY, Hu CA, Yu J, Mei JM, Valle D, Phang JM. Proline oxidase, encoded by p53-induced gene-6, catalyzes the generation of proline-dependent reactive oxygen species. Cancer Res. 2001;61:1810–1815. [PubMed] [Google Scholar]
- Geraghty MT, Vaughn D, Nicholson AJ, Lin WW, Jimenez-Sanchez G, Obie C, Flynn MP, Valle D, Hu CA. Mutations in the Delta1-pyrroline 5-carboxylate dehydrogenase gene cause type II hyperprolinemia. Hum Mol Genet. 1998;7:1411–1415. doi: 10.1093/hmg/7.9.1411. [DOI] [PubMed] [Google Scholar]
- Gu D, Zhou Y, Kallhoff V, Baban B, Tanner JJ, Becker DF. Identification and characterization of the DNA-binding domain of the multifunctional PutA flavoenzyme. J Biol Chem. 2004;279:31171–31176. doi: 10.1074/jbc.M403701200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X, Holden HM, Raushel FM. Channeling of substrates and intermediates in enzyme-catalyzed reactions. Annu Rev Biochem. 2001;70:149–180. doi: 10.1146/annurev.biochem.70.1.149. [DOI] [PubMed] [Google Scholar]
- Inagaki E, Ohshima N, Sakamoto K, Babayeva ND, Kato H, Yokoyama S, Tahirov TH. New insights into the binding mode of coenzymes: structure of Thermus thermophilus [Delta]1-pyrroline-5-carboxylate dehydrogenase complexed with NADP+ Acta Crystallogr. 2007;F63:462–465. doi: 10.1107/S1744309107021422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inagaki E, Ohshima N, Takahashi H, Kuroishi C, Yokoyama S, Tahirov TH. Crystal structure of Thermus thermophilus Delta1-pyrroline-5-carboxylate dehydrogenase. J Mol Biol. 2006;362:490–501. doi: 10.1016/j.jmb.2006.07.048. [DOI] [PubMed] [Google Scholar]
- Lamb AL, Newcomer ME. The structure of retinal dehydrogenase type II at 2.7 A resolution: implications for retinal specificity. Biochemistry. 1999;38:6003–6011. doi: 10.1021/bi9900471. [DOI] [PubMed] [Google Scholar]
- Larson JD, Jenkins JL, Schuermann JP, Zhou Y, Becker DF, Tanner JJ. Crystal structures of the DNA-binding domain of Escherichia coli proline utilization A flavoprotein and analysis of the role of Lys9 in DNA recognition. Protein Sci. 2006;15:1–12. doi: 10.1110/ps.062425706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YH, Nadaraia S, Gu D, Becker DF, Tanner JJ. Structure of the proline dehydrogenase domain of the multifunctional PutA flavoprotein. Nat Struct Biol. 2003;10:109–114. doi: 10.1038/nsb885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis ML, Rowe CJ, Sewald N, Sutherland JD, Wilson EJ, Wright MC. The effect of pH on the solution structure of delta-1-pyrroline-2-carboxylic acid as revealed by NMR and electrospray mass spectroscopy. Bioorg Med Chem Lett. 1993;3:1193–1196. [Google Scholar]
- Liu Y, Borchert GL, Donald SP, Surazynski A, Hu CA, Weydert CJ, Oberley LW, Phang JM. MnSOD inhibits proline oxidase-induced apoptosis in colorectal cancer cells. Carcinogenesis. 2005;26:1335–1342. doi: 10.1093/carcin/bgi083. [DOI] [PubMed] [Google Scholar]
- Liu ZJ, Sun YJ, Rose J, Chung YJ, Hsiao CD, Chang WR, Kuo I, Perozich J, Lindahl R, Hempel J, et al. The first structure of an aldehyde dehydrogenase reveals novel interactions between NAD and the Rossmann fold. Nat Struct Biol. 1997;4:317–326. doi: 10.1038/nsb0497-317. [DOI] [PubMed] [Google Scholar]
- Marcotte EM, Pellegrini M, Ng HL, Rice DW, Yeates TO, Eisenberg D. Detecting protein function and protein-protein interactions from genome sequences. Science. 1999;285:751–753. doi: 10.1126/science.285.5428.751. [DOI] [PubMed] [Google Scholar]
- Markova M, Peneff C, Hewlins MJ, Schirmer T, John RA. Determinants of substrate specificity in omega-aminotransferases. J Biol Chem. 2005;280:36409–36416. doi: 10.1074/jbc.M506977200. [DOI] [PubMed] [Google Scholar]
- Meng Z, Lou Z, Liu Z, Li M, Zhao X, Bartlam M, Rao Z. Crystal structure of human pyrroline-5-carboxylate reductase. J Mol Biol. 2006;359:1364–1377. doi: 10.1016/j.jmb.2006.04.053. [DOI] [PubMed] [Google Scholar]
- Menzel R, Roth J. Regulation of genes for Proline Utilization in Salmonella typhimurium: Autogenous Repression by the putA gene Product. J Mol Biol. 1981;148:21–44. doi: 10.1016/0022-2836(81)90233-3. [DOI] [PubMed] [Google Scholar]
- Miles EW, Rhee S, Davies DR. The molecular basis of substrate channeling. J Biol Chem. 1999;274:12193–12196. doi: 10.1074/jbc.274.18.12193. [DOI] [PubMed] [Google Scholar]
- Nocek B, Chang C, Li H, Lezondra L, Holzle D, Collart F, Joachimiak A. Crystal structures of delta1-pyrroline-5-carboxylate reductase from human pathogens Neisseria meningitides and Streptococcus pyogenes. J Mol Biol. 2005;354:91–106. doi: 10.1016/j.jmb.2005.08.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostrovsky de Spicer P, O’Brien K, Maloy S. Regulation of proline utilization in Salmonella typhimurium: a membrane-associated dehydrogenase binds DNA in vitro. J Bacteriol. 1991;173:211–219. doi: 10.1128/jb.173.1.211-219.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page R, Nelson MS, von Delft F, Elsliger MA, Canaves JM, Brinen LS, Dai X, Deacon AM, Floyd R, Godzik A, et al. Crystal structure of gamma-glutamyl phosphate reductase (TM0293) from Thermotoga maritima at 2.0 A resolution. Proteins. 2004;54:157–161. doi: 10.1002/prot.10562. [DOI] [PubMed] [Google Scholar]
- Pandhare J, Cooper SK, Phang JM. Proline oxidase, a proapoptotic gene, is induced by troglitazone: evidence for both peroxisome proliferator-activated receptor gamma-dependent and -independent mechanisms. J Biol Chem. 2006;281:2044–2052. doi: 10.1074/jbc.M507867200. [DOI] [PubMed] [Google Scholar]
- Phang JM. The regulatory functions of proline and pyrroline-5-carboxylic acid. Curr Top Cell Reg. 1985;25:92–132. doi: 10.1016/b978-0-12-152825-6.50008-4. [DOI] [PubMed] [Google Scholar]
- Phang JM, Hu CA, Valle D. Disorders of proline and hydroxyproline metabolism. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. Metabolic and molecular basis of inherited disease. McGraw Hill; New York: 2001. pp. 1821–1838. [Google Scholar]
- Rivera A, Maxwell SA. The p53-induced gene-6 (proline oxidase) mediates apoptosis through a calcineurin-dependent pathway. J Biol Chem. 2005;280:29346–29354. doi: 10.1074/jbc.M504852200. [DOI] [PubMed] [Google Scholar]
- Saktor B. Biochemical adaptations for flight in the insect. Biochem Soc Symp. 1976;41:111–131. [PubMed] [Google Scholar]
- Schreiter ER, Drennan CL. Ribbon-helix-helix transcription factors: variations on a theme. Nat Rev Microbiol. 2007;5:710–720. doi: 10.1038/nrmicro1717. [DOI] [PubMed] [Google Scholar]
- Shah SA, Shen BW, Brunger AT. Human ornithine aminotransferase complexed with L-canaline and gabaculine: structural basis for substrate recognition. Structure. 1997;5:1067–1075. doi: 10.1016/s0969-2126(97)00258-x. [DOI] [PubMed] [Google Scholar]
- Shen BW, Hennig M, Hohenester E, Jansonius JN, Schirmer T. Crystal structure of human recombinant ornithine aminotransferase. J Mol Biol. 1998;277:81–102. doi: 10.1006/jmbi.1997.1583. [DOI] [PubMed] [Google Scholar]
- Sophos NA, Vasiliou V. Aldehyde dehydrogenase gene superfamily: the 2002 update. Chem Biol Interact. 2003;143–144:5–22. doi: 10.1016/s0009-2797(02)00163-1. [DOI] [PubMed] [Google Scholar]
- Storici P, Capitani G, Muller R, Schirmer T, Jansonius JN. Crystal structure of human ornithine aminotransferase complexed with the highly specific and potent inhibitor 5-fluoromethylornithine. J Mol Biol. 1999;285:297–309. doi: 10.1006/jmbi.1998.2289. [DOI] [PubMed] [Google Scholar]
- Surber MW, Maloy S. The PutA protein of Salmonella typhimurium catalyzes the two steps of proline degradation via a leaky channel. Arch Biochem Biophys. 1998;354:281–287. doi: 10.1006/abbi.1998.0697. [DOI] [PubMed] [Google Scholar]
- Tritsch D, Mawlawi H, Biellmann JF. Mechanism-based inhibition of proline dehydrogenase by proline analogues. Biochim Biophys Acta. 1993;1202:77–81. doi: 10.1016/0167-4838(93)90065-y. [DOI] [PubMed] [Google Scholar]
- Tsuge H, Kawakami R, Sakuraba H, Ago H, Miyano M, Aki K, Katunuma N, Ohshima T. Crystal structure of a novel FAD-, FMN-, and ATP-containing L-proline dehydrogenase complex from Pyrococcus horikoshii. J Biol Chem. 2005;280:31045–31049. doi: 10.1074/jbc.C500234200. [DOI] [PubMed] [Google Scholar]
- Vilchez S, Manzanera M, Ramos JL. Control of expression of divergent Pseudomonas putida put promoters for proline catabolism. Appl Environ Microbiol. 2000a;66:5221–5225. doi: 10.1128/aem.66.12.5221-5225.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilchez S, Molina L, Ramos C, Ramos JL. Proline catabolism by Pseudomonas putida: cloning, characterization, and expression of the put genes in the presence of root exudates. J Bacteriol. 2000b;182:91–99. doi: 10.1128/jb.182.1.91-99.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White TA, Krishnan N, Becker DF, Tanner JJ. Structure and kinetics of monofunctional proline dehydrogenase from Thermus thermophilus. J Biol Chem. 2007;282:14316–14327. doi: 10.1074/jbc.M700912200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White TAWH, Johnson J, Whitman CP, Tanner JJ. Structural basis for the inactivation of Thermus thermophilus proline dehydrogenase by N-propargylglycine. 2008 doi: 10.1021/bi800055w. Submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, White TA, Schuermann JP, Baban BA, Becker DF, Tanner JJ. Structures of the Escherichia coli PutA proline dehydrogenase domain in complex with competitive inhibitors. Biochemistry. 2004a;43:12539–12548. doi: 10.1021/bi048737e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Zhang M, Zhu W, Zhou Y, Wanduragala S, Rewinkel D, Tanner JJ, Becker DF. Redox-induced changes in flavin structure and roles of flavin N(5) and the ribityl 2′-OH group in regulating PutA--membrane binding. Biochemistry. 2007;46:483–491. doi: 10.1021/bi061935g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Zhou Y, Becker DF. Regulation of PutA-membrane associations by flavin adenine dinucleotide reduction. Biochemistry. 2004b;43:13165–13174. doi: 10.1021/bi048596g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Larson JD, Bottoms CA, Arturo E, Henzl MT, Jenkins JL, Nix JC, Becker DF, Tanner JJ. Molecular and structural basis of transcriptional regulation of the put regulon in Escherichia coli. 2008a doi: 10.1016/j.jmb.2008.05.084. Submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Zhu W, Bellur PS, Rewinkel D, Becker DF. Direct linking of metabolism and gene expression in the proline utilization A protein from Escherichia coli. Amino Acids. 2008b doi: 10.1007/s00726-008-0053-6. to be published. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu W, Becker DF. Flavin redox state triggers conformational changes in the PutA protein from Escherichia coli. Biochemistry. 2003;42:5469–5477. doi: 10.1021/bi0272196. [DOI] [PubMed] [Google Scholar]
- Zhu W, Gincherman Y, Docherty P, Spilling CD, Becker DF. Effects of proline analog binding on the spectroscopic and redox properties of PutA. Arch Biochem Biophys. 2002;408:131–136. doi: 10.1016/s0003-9861(02)00535-0. [DOI] [PubMed] [Google Scholar]