

Abstract

One of the leading sources of false positives in early drug discovery is the formation of organic small molecule aggregates, which inhibit enzymes nonspecifically at micromolar concentrations in aqueous solution. The molecular basis for this widespread problem remains hazy. To investigate the mechanism of inhibition at a molecular level, we determined changes in solvent accessibility that occur when an enzyme binds to an aggregate using hydrogen−deuterium exchange mass spectrometry. For AmpC β-lactamase, binding to aggregates of the small molecule rottlerin increased the deuterium exchange of all 10 reproducibly detectable peptides, which covered 41% of the sequence of β-lactamase. This suggested a global increase in proton accessibility upon aggregate binding, consistent with denaturation. We then investigated whether enzyme−aggregate complexes were more susceptible to proteolysis than uninhibited enzyme. For five aggregators, trypsin degradation of β-lactamase increased substantially when β-lactamase was inhibited by aggregates, whereas uninhibited enzyme was generally stable to digestion. Combined, these results suggest that the mechanism of action of aggregate-based inhibitors proceeds via partial protein unfolding when bound to an aggregate particle.
Introduction
Many organic small molecules form submicrometer aggregates at micromolar concentrations in aqueous solution.1,2 Such molecules are found among screening hit lists, biological reagents, and even marketed drugs.3−11 These aggregates have the unusual property of nonspecifically inhibiting enzyme targets, leading to false positive “hits” in biochemical assays, a problem that is now well-recognized, particularly in high-throughput screening.12−20 Still, exactly how aggregates cause inhibition remains poorly understood.(21) Here we revisit the specific mechanism of nonspecific inhibition by investigating the structural changes that are induced in the enzyme upon binding to the aggregate.
In 2003 McGovern et al. observed three mechanistic features of small molecule aggregates that guided our investigation.(22) First, inhibition occurs via the direct binding of enzyme to aggregate, as shown by (1) the ability to sediment protein−aggregate complexes with centrifugation, (2) the punctate fluorescence observed by microscopy in mixtures of aggregates with green fluorescent protein (GFPa), and (3) the direct observation of protein−aggregate complexes by transmission electron microscopy. Second, aggregate-based inhibition can be rapidly reversed by the addition of a nonionic detergent such as Triton X-100, indicating that enzyme can quickly (within tens of seconds) regain activity from aggregate-based inhibition. Last, several experiments appeared to be inconsistent with denaturation as a potential mechanism of action. For example, it seemed unlikely that enzyme could rapidly refold into its active state upon the addition of detergent if it were completely denatured when bound to the aggregate. It seemed equally unlikely that GFP could retain its fluorescence if it were completely denatured while bound to an aggregate. Two other experiments suggested that inhibition was not due to denaturation: (1) additional denaturants such as guanidinium or urea did not increase inhibition by aggregates (if anything, inhibition was decreased) and (2) a destabilized mutant appeared to be no more sensitive to aggregate-based inhibition than its wild type counterpart.
As a result of McGovern’s work, we considered three possible mechanisms of action that might explain aggregate-based inhibition (Figure 1). Although we did not believe that there was large scale unfolding of the enzyme, it still seemed reasonable that there might be small-scale or local unfolding, which has also been proposed by Ryan et al.(23) On the other hand, aggregate binding may have the opposite effect: instead of increasing flexibility, it may rigidify it, restricting those dynamic motions necessary for catalysis. Finally, aggregates may physically sequester enzyme away from substrate. To explore these potential mechanisms, we chose to use hydrogen−deuterium exchange mass spectrometry (HDX MS), a technique widely used to measure changes in solvent accessibility for processes such as enzyme unfolding or protein−protein interactions.24−30 HDX MS relies on the different exchange rates of the backbone amide protons with a deuterated solvent, which are measured by the change in mass as deuterium replaces hydrogen. To investigate changes in solvent accessibility, we quantified deuterium exchange of AmpC β-lactamase over 8 h in the presence or absence of an aggregating inhibitor, rottlerin. To obtain localized information, β-lactamase was digested with pepsin after exchange. We reproducibly observed 10 fragments covering 41% of the entire enzyme sequence. The differences in solvent accessibility were not localized to specific regions (given the nonspecific nature of aggregate-based inhibition, we did not expect to see peptide-specific interactions); rather, we observed a general trend across all peptides. The differences in solvent accessibility that we observed by mass spectrometry suggested that we may also see differences in protease sensitivity, which we investigated by gel electrophoresis of tryptic digests of our model enzyme in the presence or absence of several known aggregating inhibitors. Combined, these experiments suggest small scale enzyme unfolding as a molecular mechanism for aggregate-based inhibition.
Figure 1.
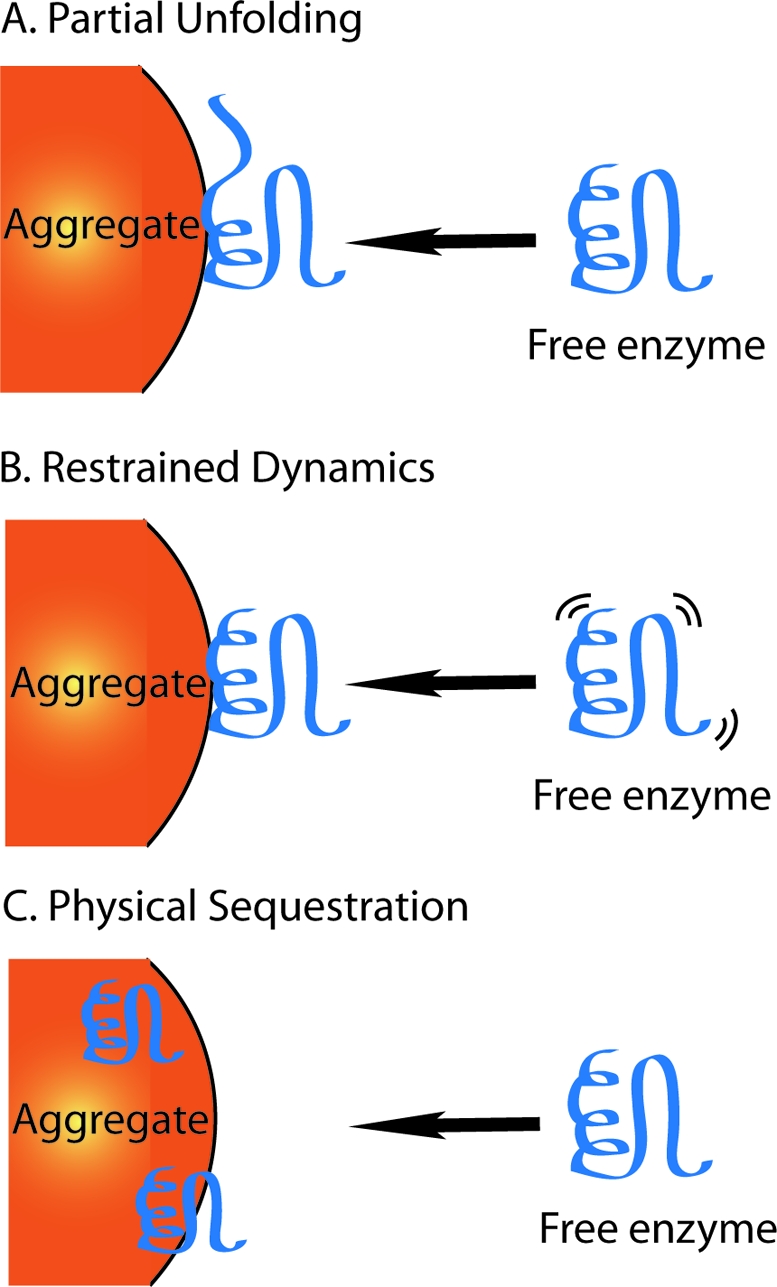
Three models for the mechanism of action of promiscuous small-molecule aggregators. (A) Binding to the aggregate promotes a partial unfolding event. (B) Binding to the aggregate constrains protein dynamics and restricts catalytic motions. (C) The aggregate physically blocks the active site and sequesters enzyme away from substrate.
Results
To examine the structural changes that occur in an enzyme when bound to a small molecule aggregate, we began by measuring changes in solvent accessibility using HDX MS. The experiments were conducted with AmpC β-lactamase, which is perhaps the enzyme best characterized for aggregate-based inhibition. Rottlerin was chosen as a model aggregator because of the large concentration range between its critical aggregation concentration (CAC) and when it begins to precipitate and because of its relatively high “potency” as an aggregator (low micromolar IC50 vs AmpC). β-Lactamase was incubated in deuterated MOPS buffer between 10 min and 8 h, in either the presence or absence of rottlerin. After exchange, the aggregates were disrupted with detergent and the exchange reaction was quenched by the addition of cold acid. β-Lactamase was subsequently digested with immobilized pepsin to obtain regional exchange information. These peptides were then separated and analyzed by electrospray ionization liquid chromatography−mass spectrometry (ESI LCMS).
Two modifications were necessary to accommodate the presence of aggregates in the HDX MS experiment. First, aggregate−enzyme complexes had to be exchanged in the deuterated buffer at a lower concentration and then concentrated before further sample preparation. Aggregates are a phase between soluble, free small molecule and precipitant, the latter of which does not typically inhibit enzymes, so there is an upper limit to the concentration that we can take an aggregating inhibitor: the concentration when it ceases to form more aggregates and instead begins to precipitate. Typically, micromolar concentrations of an aggregating molecule will inhibit nanomolar concentrations of enzyme. These HDX MS experiments required micromolar concentrations of protein, and if a corresponding concentration of inhibitor were used, we would need an inhibitor that was soluble at millimolar concentrations. As aggregation is a form of insolubility, there are very few examples of molecules that have such high solubility and these molecules are often weak aggregators and unsuitable for this analysis.
Since we did not have an aggregator that could be used at millimolar concentrations, we instead reduced the concentration of the enzyme so that it would be mostly inhibited by a preprecipitant colloidal form of the aggregator (100 μM for rottlerin). We relied on the fact that aggregates and aggregate-bound enzyme can be pelleted and therefore concentrated by centrifugation. Although incubation was performed at a lower concentration of enzyme, we could concentrate the enzyme−aggregate complexes prior to analysis by collecting the pellet and removing the supernatant. This has the additional benefit of guaranteeing that predominantly aggregate-bound complexes would be analyzed, as free enzyme would not be pulled down by centrifugation and would be many-fold lower in concentration.
The second necessary modification was the addition of detergent. Although we tried several detergents, none were nearly as effective as Triton X-100. It was necessary to add Triton mainly to release bound enzyme from the aggregates so that the complexes were not pulled down again when centrifugation was used to remove the immobilized pepsin. This resulted in a delicate balance between using enough detergent to release the enzyme for analysis but not so much that the detergent signals overwhelmed the peptide signals in the mass spectra, where Triton signals were both numerous and strong. To increase separation between the peptide and detergent signals, we used ESI LCMS rather than matrix-assisted laser desorption/ionization (MALDI, which does not require peptide separation).31,32 As a result of the presence of aggregates, the inhibited samples generally showed much weaker signal intensities compared to uninhibited samples. Often, the peptide signals were so weak that they could not be analyzed. Those that were observed were noisy, probably because of the weak intensities.
The peptides that we reproducibly observed covered 41% of the β-lactamase sequence (Figure 2), representing several regions of the enzyme spanning both buried and exposed regions. Although our results are not strong enough to determine whether specific areas experienced more exchange than others, it was also not our goal to do so. Given that aggregates are nonspecific inhibitors, we were searching for a global effect, a mechanism that could explain inhibition of many enzymes and that was not restricted to specific residues or peptide sequences. The results suggested such a trend. Across all of the peptides that we measured, enzyme−aggregate samples showed deuterium incorporation greater than or equal to the deuterium incorporation of the enzyme alone (Table 1). Levels of deuteration were very low in both samples with and without inhibitor; however, the trend of higher deuterium content in the aggregate-containing samples is consistent across all of the peptides. We suspect the low levels of incorporation are due to increased back exchange occurring during the protein concentration and chromatography steps. Many peptides incorporated levels of deuterium that were not, in themselves, entirely convincing; however, even here, each showed more deuterium exchange in the presence of aggregates than for the free enzyme. We never observed a peptide that had reduced deuterium exchange in the presence of aggregates. Two peptides that did show significant exchange are shown in Figure 3 (a complete list of peptides is available in the Supporting Information). Again, we observed very low deuterium incorporation, but the time points repeatedly indicated a significant difference between samples with and without aggregates. The higher deuterium incorporation of aggregate-bound enzyme across all 10 peptides suggested that the enzyme may be unfolded when bound to the aggregate.
Figure 2.

Amino acid sequence and structure of AmpC β-lactamase: (A) enzyme sequence showing the peptic fragments that could be quantified reproducibly in the mass spectrum both with and without the aggregator rottlerin; (B) two orientations of the structure of AmpC β-lactamase indicating the location of the peptic fragments that were observed in the mass spectrum (fragments shown in purple, active site residues shown in yellow).(39)
Table 1. Summary of H/D Exchange Data for AmpC β-Lactamase in the Presence or Absence of the Aggregating Inhibitor Rottlerin.
| fold increase in deuteration (Dinhibited/Duninhibited) |
deuterium incorporation after 8 hd |
|||||||
|---|---|---|---|---|---|---|---|---|
| peptide | no. of amide bonds in peptidea | deuterium incorporation after 24 hb | 10 minc | 1 hc | 8 hc | no inhibitor | + rottlerin | % solvent accessibilitye |
| 8−18 | 8 | 1.8 | 1.3 | 2.3 | 2.5 | 0.2 ± 0.2 | 0.5 ± 0.2 | 35.7 |
| 42−56 | 12 | 6 | 1.9 | 2.7 | 1.8 | 0.8 ± 0.2 | 1.4 ± 0.2 | 44.7 |
| 105−128 | 20 | 6.6 | 1.6 | 2.6 | 1.8 | 1.5 ± 0.6 | 2.7 ± 0.5 | 31.3 |
| 109−128 | 16 | 5.4 | 1.5 | 2.5 | 1.6 | 1.4 ± 0.4 | 2.2 ± 0.3 | 36.9 |
| 132−146 | 11 | 4.2 | 2.6 | 4.2 | 3.4 | 0.3 ± 0.1 | 1.1 ± 0.1 | 34.8 |
| 243−250 | 6 | 1.8 | 1.2 | NMf | 1.7 | 0.4 ± 0.2 | 0.7 ± 0.2 | 20.7 |
| 271−289 | 15 | 6.6 | 1.4 | 2.6 | 1.4 | 2.3 ± 0.5 | 3.1 ± 0.6 | 40.7 |
| 291−322 | 26 | 8 | 1.6 | 2.5 | 1.8 | 1.7 ± 0.6 | 3.1 ± 0.6 | 22.3 |
| 323−332 | 7 | 1 | 1.3 | 3.0 | 2.7 | 0.2 ± 0.0 | 0.5 ± 0.1 | 11.8 |
| 336−350 | 11 | 2.7 | 1.7 | 3.7 | 2.0 | 0.5 ± 0.2 | 1.0 ± 0.2 | 21.6 |
Equal to number of amide bonds + 1, excluding prolines.
Deuterium incorporation is calculated by subtracting the mass of the control undeuterated centroid from the centroid of the deuterated sample. Deuterium incorporation is not corrected for back exchange.
Indicates the amount of time the samples were exchanged in the deuterated solvent.
Deuterium incorporation is calculated as the (deuterated mass × charge – charge) minus (undeuterated mass × charge – charge) with the standard deviation of three replicate measurements.
Average ratio of side chain surface area to random coil values per residue calculated using Getarea.(38) Greater than 50% is considered exposed, and less that 20% is considered buried.
Not measured.
Figure 3.

Mass envelopes and corresponding deuterium incorporation plots for two fragments from the mass spectrum of β-lactamase in the presence (dotted line) or absence (solid line) of the aggregator rottlerin. The spectra have been expanded to show the isotopic distribution of the ions of interest: (A) the peptide containing residues 132−146 (monoisotopic m/z = 908.4, +2 charge state) and (B) the peptide containing residues 291−322 (monoisotopic m/z = 813.7, +4 charge state). For each peptide, the isotopic distributions are shown for (i) the undeuterated sample, (ii) the sample that was deuterated for 4 h in the absence of rottlerin, and (iii) the sample that was deuterated for 4 h in the presence of rottlerin.
If aggregates were unfolding bound enzyme, we might expect aggregate-bound enzyme to be more susceptible to proteolytic degradation than free, uninhibited enzyme. If an enzyme is even slightly unfolded, it should be measurably more sensitive to digestion by proteases (Figure 4). Given recent results showing the very slow off rate of enzyme from the aggregate and that the aggregate can be saturated with enzyme and so maintain a large excess of uninhibited protease, this experiment seemed feasible.33−35 We chose five known aggregators: rottlerin, Congo Red, Eriochrome Blue Black B, nicardipine, and L-755,507. We again used β-lactamase as our model enzyme and chose trypsin as the protease. β-Lactamase was preincubated with or without aggregates before the addition of trypsin to avoid competition with trypsin for binding space on the aggregate. Free or aggregate-bound enzyme was then incubated with trypsin between zero min and 4 h, and the digestion was monitored by gel electrophoresis.
Figure 4.

Schematic representation of the tryptic digest experiment and interpretation. If the enzyme is unfolded on the aggregate, it should be more susceptible to proteolysis compared to enzyme in the absence of aggregates (A). If the enzyme is in its native state when bound to the aggregate, identical to unbound enzyme, it should be degraded at a comparable rate (B). Although some trypsin may be bound to the aggregate, trypsin is used in excess to ensure that there is free, active trypsin in solution: (+) indicates that aggregates are present; (−) indicates free enzyme.
For all of the aggregators, the presence of aggregates had no effect on the band representing β-lactamase in the absence of trypsin (Figure 5, lanes 2 and 3 or 1 and 3). In contrast, when we added trypsin, we observed significant digestion in the presence of aggregates but not in the absence (Figure 5). For example, in Figure 5A, the β-lactamase band is present in both the aggregate-inhibited and free enzyme samples at zero minutes (lanes 6 and 7), but as time progresses, the β-lactamase band in the presence of rottlerin becomes weaker and weaker until it is completely gone by 4 h (lane 15). Conversely, β-lactamase in the absence of rottlerin appears almost as strong after 4 h with trypsin as it did after zero min (lane 14). For Congo Red and Eriochrome, the β-lactamase band is present in both samples at zero min of trypsin incubation (Figure 5B,C, lanes 6 and 7), but after only 15 min the β-lactamase band is gone in the presence of aggregates (lane 9). Again, β-lactamase in the absence of aggregates remains undigested by trypsin even after 4 h (lane 14). Although the effect was less pronounced, two other aggregators, nicardipine and L-755,507, also increased sensitivity to trypsin (Figure 5D,E). In many of these samples, it is also possible to observe the formation of a large degradation product of β-lactamase running below the trypsin band (Figure 5). Intriguingly, trypsin also appears to experience more self-degradation in the presence of aggregators (Figure 5A,C, lanes 4 and 5).
Figure 5.

SDS−PAGE and silver stain of tryptic digests of β-lactamase in the presence (+) or absence (−) of aggregating inhibitors. The inhibitors are (A) 100 μM rottlerin, the same inhibitor used in the HDX MS experiments, (B) 250 μM Congo Red, (C) 250 μM Eriochrome Blue Black B, (D) 100 μM L-755,507, and (E) 250 μM nicardipine. For gels B, C, and E, lane 1 is a molecular weight ladder. For gels A and D, lane 2 is a molecular weight ladder. Lanes 2 and 3 for gels B, C, and E or lanes 1 and 3 for gels A and D show β-lactamase without trypsin, in the absence (−) or presence (+) of aggregator. Lanes 4 and 5 contain trypsin (no β-lactamase) in the absence and presence of aggregator, respectively. Lanes 6−15 contain tryptic digests of β-lactamase in the absence or presence of aggregator for digestion times of 0 min, 15 min, 30 min, 1 h, and 4 h (from left to right). β-Lactamase was present at 0.5 μM in all experiments. Trypsin was used at a concentration of 0.01 mg/mL for (A), 0.025 mg/mL for (B), (C), and (D), and 0.05 mg/mL for (E). A suspected degradation product of AmpC is indicated with an arrow and labeled “dp.”
Discussion
The key result to emerge from this study is the mechanism by which association with colloidal aggregates leads to enzyme inhibition: partial protein denaturation. Two observations support this. The increased hydrogen−deuterium exchange of β-lactamase when bound to rottlerin aggregates suggests that the enzyme’s backbone amides are more exposed to solvent when bound to an aggregate, indicating at least partial enzyme denaturation (Figure 3). Consistent with this view is the increased susceptibility of the enzyme to trypsin degradation when bound to the characteristic aggregators rottlerin, Congo Red, Eriochrome Blue Black B, nicardipine, and L-755,507 (Figure 5).
That proteins directly associate with aggregates has been established for some time and is supported by the precipitation of aggregate-bound protein on centrifugation, by the detection of aggregate binding with surface plasmon resonance,(20) and by imaging of aggregate−protein complexes by transmission electron microscopy and fluorescence microscopy.(22) What remained perplexing was why such association should lead to inhibition. Proteins can be attached to solid supports, as in chromatography, without compromising their function, and so association, per se, seemed unlikely to lead to inhibition. An early hypothesis was that the aggregates were acting as denaturants, but several lines of evidence seemed to tilt against this. First, GFP retained fluorescence while bound to aggregate particles, which should not be true if GFP was substantially denatured on aggregate association. Second, destabilized and up-stabilized mutants of the same enzyme were inhibited to the same extent by colloidal inhibitors, indicating that liability to denaturation did not increase susceptibility to aggregate-based inhibition. Third, classical denaturants such as urea or guanidinium failed to potentiate aggregate-based inhibition; rather, inhibition was actually reduced in the presence of these denaturants. Finally, disruption of the aggregates by detergent returned enzyme activity within the dead time of the experiment, less than 15 s, suggesting that no substantial refolding was occurring. Whereas none of these experiments were in themselves conclusive, taken together they alluded to another, unknown mechanism behind aggregate-based inhibition, and our understanding has remained at this point for the past 5 years.
This study compels us to reevaluate our previous results. We can reconcile the earlier and present observations by noting that the previous study only seemed to exclude large scale protein denaturation. More local denaturation would not be expected to extinguish GFP fluorescence(36) nor would substantial refolding be necessary for the enzyme to regain activity upon aggregate disruption by detergent. As to the reduced inhibition observed in the presence of classical denaturants, we have since discovered that urea and guanidinium themselves disrupt colloidal aggregates, preventing any additive denaturant effect. Finally, our ability to interpret the inhibitory effects of colloidal aggregates on destabilized and up-stabilized mutant enzymes was confounded by the CAC of the colloidal particles and their unusually tight binding to proteins. Neither of these phenomena were understood in 2003, but recent studies have shown that aggregates undergo a CAC phase transition, above which their affinities for proteins are in the picomolar dissociation range or better.20,34,35 Given micromolar CAC values and picomolar Kd values for the protein−aggregate interaction, one would not expect to see differential inhibition of the mutant enzymes that were more or less stabilized by no more than 5 kcal/mol.
Returning to our three models (Figure 1), we have ruled out reduced dynamics and physical sequestration, as both would have resulted in reduced solvent accessibility (less deuterium exchange) and protection from proteolysis; rather, the reverse is observed in both cases. Taken together, our previous studies and current experiments suggest that on association with colloidal, promiscuous inhibitors, proteins undergo partial denaturation, which manifests itself as enzyme inhibition. In such a mechanism, hydrogen−deuterium exchange would increase, as would proteolysis, but the enzyme would not be so denatured that it could not rapidly return to its active conformation or that GFP would not retain fluorescence.
Two important caveats to these conclusions are the weak peptide signals in the mass spectra and the low overall deuterium incorporation. The presence of the aggregates interfered with the HDX MS, reducing signal-to-noise and experimental reproducibility. We suspect that we were only partially successful at disrupting aggregate−enzyme complexes, and so lost peptide during sample preparation. Typically this problem would be addressed by the addition of further Triton X-100, but this posed its own hazards for mass spectrometry because the detergent peaks easily overwhelmed our peptide signals. Detergents better tolerated by mass spectrometry, such as β-octaglucoside, were ineffective at disrupting the aggregates to free the peptides for analysis. The levels of deuterium incorporation in both the aggregate-inhibited and uninhibited samples were disconcertingly low, which we attribute to high back exchange. Combined with the complicating factors of low peptide signal strength and overwhelming detergent signals, the chromatographic run time had to be increased to allow separation of the peptide and detergent peaks, thus leading to more back exchange. We suspect that the somewhat erratic incorporation of deuterium as a function of time is due to the low deuterium content (which is more subject to signal-to-noise interference).
Because of the low signal-to-noise and low deuterium incorporation, we draw no conclusions about which parts of the enzyme are more exposed to solvent upon aggregate binding, as would ordinarily be tempting to do with this technique. That admitted, every single peptide that we observed had greater hydrogen−deuterium exchange in the aggregate-bound protein compared to the free protein. This suggests that whereas we cannot resolve the specific parts of β-lactamase that are more denatured, the enzyme is being partially denatured. The protease digestion experiments support this conclusion. Inhibition by each of five different aggregators increased susceptibility of β-lactamase to proteolysis to the point where degradation was apparent within 15 min of trypsin addition, whereas digestion of uninhibited enzyme took place over several hours (Figure 5).
Partial protein denaturation on association with colloidal aggregates offers the first molecular mechanism for the inhibitory effects of these particles. This mechanism is attractive in that it is general, relying on no particular feature of enzyme or aggregate structure. All that would be required are the nonpolar, buried residues that all enzymes possess, which might be expected to associate with the nonpolar surfaces that the colloidal aggregates inevitably feature. This explains the lack of specificity of the colloidal aggregates with soluble proteins. It also suggests the opportunity for more detailed, biophysical studies of the sort that have long been used to characterize protein stability and hydrophobic binding. As an aside, we note that as denaturants, colloidal aggregates are peculiar. Their denaturation-by-sequestration mechanism on large isolatable particles may be unique and may lend itself to pragmatic uses. These might include the ability to sequester proteins in a relatively concentrated, inactive form and from a medicinal chemistry perspective to affect the adsorption and distribution of drugs and reagents.(4) This would be an interesting twist for a species that until now, owing to its pervasiveness in early drug and reagent discovery, has been only a great and recurring problem.
At this time, several key characteristics of aggregate behavior have come into focus. Particle formation appears to be a common feature of organic molecules in aqueous buffer: it is ubiquitous in early drug discovery and particularly problematic in high-throughput screening.1,2,9,21 As has been known for some time, small molecule colloidal aggregates inhibit protein via direct binding, an association that we now believe is surprisingly tight, picomolar or better.20,22,34 The low dissociation constant results in little to no measurable exchange between aggregate-bound and free protein.(33) Conversely, the aggregates themselves are in dynamic equilibrium with their monomer small molecule components and will rapidly dissociate when diluted below their threshold of formation, the CAC.(35) As an aside, we note that the CAC of an organic molecule in a particular buffer will be relatively invariant. Whether this molecule, above its CAC, will observably inhibit depends on other variables, including the stoichiometric ratio of the protein it is sequestering; if unrecognized, this can lead to confusion in reconciling the behavior of a particular molecule from assay to assay. The aggregates appear to be solid, densely packed particles, the larger of which can bind on the order of 10 000 protein molecules. Although we have yet to conclusively determine that protein is not absorbed within the aggregate, adsorption to the surface has been observed by microscopy(22) and our calculations indicate that the particle surface has more than sufficient binding capacity to accommodate all bound protein.(35) The final insight to this picture is the conclusion that we have drawn here: that the aggregate−protein interaction results in partial denaturation and subsequent inhibition. This model appears to be true for a number of aggregating molecules, suggesting it may be general, but it remains unclear whether there may be different subtypes of aggregates. Evidence of differing detergent sensitivity, size, and binding strength suggest that there are aggregates that behave uncharacteristically and may operate by additional mechanisms.20,23 Regardless, we now have a far clearer understanding of these so often perplexing particles.
Experimental Section
Materials
AmpC β-lactamase was expressed and purified as previously described.(37) Rottlerin, Congo Red, Eriochrome Blue Black B, nicardipine, trypsin from porcine pancreas, and trifluoroacetic acid were purchased from Sigma-Aldrich. L-755,507 was purchased from Tocris. Immobilized pepsin was purchased from Pierce.
HDX MS Sample Preparation
AmpC β-lactamase (215 μM stock in 50 mM KPi, pH 7.0) was delivered to the deuterated solvent (50 mM MOPS buffer in D2O, pH 7.0) to a final concentration of 16 μM for uninhibited enzyme or 0.16 μM for samples containing the inhibitor rottlerin. The total volume of samples without rottlerin (including the undeuterated and 100% deuterated controls) was 10 μL, whereas the total volume for each sample with rottlerin was 1 mL. The concentration of rottlerin in these samples was 100 μM (0.5% DMSO) delivered from a 20 mM stock in DMSO. The difference in β-lactamase concentration and total volume was to accommodate for the limited solubility of rottlerin. Deuterium exchange was not increased in samples containing 0.5% DMSO (no aggregates). After incubation in the deuterated solvent for 5 min or 1, 2, 4, or 8 h, the samples containing rottlerin were centrifuged for 5 min at 16000g to pull down and concentrate aggregate−enzyme complexes and the top 990 μL amount was discarded. The bottom 10 μL volume was considered the pellet and were treated identically to the 10 μL samples without rottlerin from this point on.
To disrupt the aggregates, 1 μL of 3% Triton X-100 in 50 mM deuterated MOPS buffer was added to every sample and the samples were mixed vigorously by pipetting. Immediately after, the samples were quenched with 90 μL of ice cold 0.1% trifluoroacetic acid in water, pH 2.5. The samples were mixed and transferred to 100 μL of immobilized pepsin beads. Samples were kept on ice with occasional mixing for 5 min, and then the pepsin beads were sedimented by centrifugation for 1 min at 16000g at 4 °C. The supernatant was collected and frozen with liquid nitrogen. Samples were kept at −80 °C until analysis.
Mass Spectrometry
The mass spectrometry data were acquired on an Applied Biosystems QSTAR Pulsar hybrid LC/MS/MS system. The mass spectrometer was equipped with the optional Applied Biosystems MicroIonSpray source. Chromatography was performed using an Eldex MicroPro pump equipped with an in-house splitter capable of producing column flow rates of 0.3−1.0 μL/min at pump flow rates of 50−80 μL/min. A Phenomenex Onyx monolithic column (0.100 mm × 150 mm) was used for all experiments. Samples were injected manually using a Valco C2-1006 injection valve with a 1.0 μL loop. Solvent A was 0.1% formic acid in water, and solvent B was 0.1% formic acid in acetonitrile.
Peptide Identification
The identities of the peptides formed by digestion with pepsin were determined by LC/MS/MS analysis using the following conditions: column flow rate was 0.4 μL/min, and a gradient was run from 3% to 50% solvent B in 35 min. The mass spectrometer was run using the standard information dependent acquisition method. The data obtained were searched against a database consisting only of the AmpC sequence using Protein Prospector.
Analysis of Deuterium Incorporation
For analysis of deuterated peptide samples, the same chromatography system was used. To minimize back exchange, the column flow rate was raised to 1.0 μL/min and the gradient used was from 0% to 70% solvent B in 5 min. Total analysis time was 10−12 min per sample. Furthermore, the injector, injection syringe, sample loop, column, and all transfer lines were maintained in an ice bath. Data were collected in MS mode only. All data analysis was performed with the standard Analyst 1.1 software native to the QSTAR. The average number of deuterons incorporated for each peptide was determined from the mass spectra. The centroid of the isotopic peak cluster for the undeuterated control was subtracted from the centroid of the isotopic peak cluster for each deuterated sample. The experimental conditions were the same in each analysis, and only the difference in deuterium incorporation between identical peptides was measured, so it was not necessary to correct for back exchange. Solvent accessible surface area was calculated using Getarea (version 1.0 beta) available at http://curie.utmb.edu/getarea.html, using a radius of 1.4 Å and default atomic radii and atomic solvent parameters.(38) The average percent accessibility was calculated as the average of the ratios of each side chain surface area to the random coil value.
Tryptic Digests and Gel Electrophoresis
A sample of AmpC β-lactamase (0.5 μM) was incubated in 50 mM KPi, pH 7.0, for 1 h with or without each aggregating inhibitor: 100 μM rottlerin, 250 μM Congo Red, 250 μM Eriochrome Blue Black B, 250 μM nicardipine, or 100 μM L-755,507 (with a final concentration of 1% DMSO in all samples, with or without inhibitor). Samples of trypsin alone (0.01 or 0.025 mg/mL) were prepared with or without each of the inhibitors and incubated for 1 h at room temperature in 50 mM KPi, pH 7.0. Tryptic digests of β-lactamase were prepared by first adding 0.5 μM of the enzyme to a solution with or without each inhibitor. After 5 min, trypsin was added to the solution and the digests were incubated at room temperature for 0 min, 15 min, 30 min, 1 h, or 4 h. The total volume of all samples was 25 μL. To prepare the samples for SDS−PAGE, 5 μL of loading buffer (containing 0.25% bromophenol blue, 0.25% xylene cyanol FF, 30% glycerol by volume, and 5 mM dithiothreitol) was added to each sample and the samples were boiled for 2 min. Samples were mixed by inversion, and 13 μL was added to each well. After SDS−PAGE, protein bands were detected by silver staining.
Acknowledgments
This work was supported by NIH Grant GM71630 (to B.K.S.) and NIH Grant NCRR BRTP 01614 from the Biomedical Research Technology Program of the National Center for Research Resources (to A.L.B., Director of the Bio-Organic Biomedical Mass Spectrometry Resource at UCSF). K.E.D.C. was partly supported by a fellowship in the field of Pharmaceutics awarded by the PhRMA Foundation. We thank Dr. A. Falick at the University of California, Berkeley, for helpful discussions. We also thank A. McReynolds and D. Teotico for β-lactamase purification, and M. Mysinger and P. Kolb for solvent accessibility calculations. We also thank D. Teotico, M. Keiser, and R. Ferreira for reading this manuscript.
Supporting Information Available
Kinetic plots of the eight additional peptides reproducibly observed by HDX MS. This material is available free of charge via the Internet at http://pubs.acs.org.
Funding Statement
National Institutes of Health, United States
Footnotes
Abbreviations: GFP, green fluorescent protein; HDX MS, hydrogen−deuterium exchange mass spectrometry; ESI LCMS, electrospray ionization liquid chromatography−mass spectrometry; MALDI, matrix-assisted laser desorption/ionization; CAC, critical aggregation concentration.
Supplementary Material
References
- Feng B. Y.; Shelat A.; Doman T. N.; Guy R. K.; Shoichet B. K. High-throughput assays for promiscuous inhibitors. Nat. Chem. Biol. 2005, 1, 146–148. [DOI] [PubMed] [Google Scholar]
- McGovern S. L.; Caselli E.; Grigorieff N.; Shoichet B. K. A common mechanism underlying promiscuous inhibitors from virtual and high-throughput screening. J. Med. Chem. 2002, 45, 1712–22. [DOI] [PubMed] [Google Scholar]
- Davies S.; Reddy H.; Caviano M.; Cohen P. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem. J. 2000, 351, 95–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frenkel Y. V.; Clark A. D. J.; Das K.; Wang Y.-H.; Lewi P. J.; Janssen P. A. J.; Arnold E. Concentration and pH dependent aggregation of hydrophobic drug molecules and relevance to oral bioavailability. J. Med. Chem. 2005, 48, 1974–1983. [DOI] [PubMed] [Google Scholar]
- Lingameneni R.; Vysotskaya T.; Duch D.; Hemmings H. Inhibition of voltage-dependent sodium channels by Ro 31-8220, a ”specific” protein kinase inhibitor. FEBS Lett. 2000, 473, 265–268. [DOI] [PubMed] [Google Scholar]
- Liu H.; Wang Z.; Regni C.; Zou X.; Tipton P. A. Detailed kinetic studies of an aggregating inhibitor. Inhibition of phosphomannomutase/phosphoglucomutase by Disperse Blue 56. Biochemistry 2004, 27, 8662–8669. [DOI] [PubMed] [Google Scholar]
- McGovern S. L.; Shoichet B. K. Kinase inhibitors: not just for kinases anymore. J. Med. Chem. 2003, 46, 1478–1483. [DOI] [PubMed] [Google Scholar]
- Reddie K.; Roberts D.; Dore T. Inhibition of kinesin motor proteins by adociasulfate-2. J. Med. Chem. 2006, 49, 4857–4860. [DOI] [PubMed] [Google Scholar]
- Seidler J.; McGovern S. L.; Doman T.; Shoichet B. K. Identification and prediction of promiscuous aggregating inhibitors among known drugs. J. Med. Chem. 2003, 46, 4477–4486. [DOI] [PubMed] [Google Scholar]
- Turk B. Targeting proteases: successes, failures and future prospects. Nat. Rev. Drug Discovery 2006, 5, 785–799. [DOI] [PubMed] [Google Scholar]
- Feng B. Y.; Shoichet B. K. Small-molecule aggregates inhibit amyloid polymerization. Nat. Chem. Biol. 2008, 4, 197–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeWitte R. S. Avoiding physiochemical artifacts in early ADME-Tox experiments. Drug Discovery Today 2006, 11, 855–859. [DOI] [PubMed] [Google Scholar]
- Hann M. M.; Oprea T. I. Pursuing the leadlikeness concept in pharmaceutical research. Curr. Opin. Chem. Biol. 2004, 8, 255–263. [DOI] [PubMed] [Google Scholar]
- Keseru G.; Makara G. Hit discovery and hit-to-lead approaches. Drug Discovery Today 2006, 11, 741–748. [DOI] [PubMed] [Google Scholar]
- Lipinski C. A.; Lombardo F.; Dominy B. W.; Feeney P. J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Delivery Rev. 1997, 23, 3–25. [DOI] [PubMed] [Google Scholar]
- Rishton G. M. Nonleadlikeness and leadlikeness in biochemical screening. Drug Discovery Today 2003, 8, 86–96. [DOI] [PubMed] [Google Scholar]
- Roche O.; Schneider P.; Zuegge J.; Guba W.; Kansy M.; Alanine A.; Bleicher K.; Danel F.; Gutknecht E. M.; Rogers-Evans M.; Neidhart W.; Stalder H.; Dillon M.; Sjogren E.; Fotouhi N.; Gillespie P.; Goodnow R.; Harris W.; Jones P.; Taniguchi M.; Tsujii S.; Vvon Der Saal W.; Zimmermann G.; Schneider G. Development of a virtual screening method for identification of “frequent hitters” in compound libraries. J. Med. Chem. 2002, 45, 137–142. [DOI] [PubMed] [Google Scholar]
- Veber D. F.; Johnson S. R.; Cheng H. Y.; Smith B. R.; Ward K. W.; Kopple K. D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [DOI] [PubMed] [Google Scholar]
- Walters W. P.; Ajay Murcko M. A. Recognizing molecules with drug-like properties. Curr. Opin. Chem. Biol. 1999, 3, 384–387. [DOI] [PubMed] [Google Scholar]
- Giannetti A. M.; Koch B. D.; Browner M. F. Surface plasmon resonance based assay for the detection and characterization of promiscuous inhibitors. J. Med. Chem. 2008, 51, 574–580. [DOI] [PubMed] [Google Scholar]
- Feng B.; Simeonov A.; Jadhav A.; Babaoglu K.; Inglese J.; Shoichet B.; Austin C. A high-throughput screen for aggregation-based inhibition in a large compound library. J. Med. Chem. 2007, 50, 2385–2390. [DOI] [PubMed] [Google Scholar]
- McGovern S. L.; Helfand B. T.; Feng B. Y.; Shoichet B. K. A specific mechanism of nonspecific inhibition. J. Med. Chem. 2003, 46, 4265–4272. [DOI] [PubMed] [Google Scholar]
- Ryan A. J.; Gray N. M.; Lowe P. N.; Chung C. W. Effect of detergent on “promiscuous” inhibitors. J. Med. Chem. 2003, 3448, 3451. [DOI] [PubMed] [Google Scholar]
- Smith D.; Deng Y.; Zhang Z. Probing the non-covalent structure of proteins by amide hydrogen exchange and mass spectrometry. J. Mass Spectrom. 1997, 32, 135–146. [DOI] [PubMed] [Google Scholar]
- Zhang Z.; Smith D. L. Determination of amide hydrogen exchange by mass spectrometry: a new tool for protein structure elucidation. Protein Sci. 1993, 2, 522–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Englander S. W.; Sosnick T. R.; Englander J. J.; Mayne L. Mechanisms and uses of hydrogen exchange. Curr. Opin. Chem. Biol. 1996, 6, 18–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandell J. M.; Falick A. M.; Komives E. A. Identification of protein−protein interfaces by decreased amide proton solvent accessibility. Proc. Natl. Acad. Sci. U.S.A. 1998, 95, 14705–14710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoofnagle A. N.; Resing K. A.; Goldsmith E. J.; Ahn N. G. Changes in protein conformational mobility upon activation of extracellular regulated protein kinase-2 as detected by hydrogen exchange. Proc. Natl. Acad. Sci. U.S.A. 2001, 98, 956–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan X.; Watson J.; Ho P. S.; Deinzer M. L. Mass spectrometric approaches using electrospray ionization charge states and hydrogen−deuterium exchange for determining protein structures and their conformational changes. Mol. Cell. Proteomics 2004, 3, 10–23. [DOI] [PubMed] [Google Scholar]
- Wales T. E.; Engen J. R. Hydrogen exchange mass spectrometry for the analysis of protein dynamics. Mass Spectrom. Rev. 2006, 25, 158–170. [DOI] [PubMed] [Google Scholar]
- Burlingame A. L.; Boyd R. K.; Gaskell S. J. Mass Spectrometry. Anal. Chem. 1996, 68, 634R–683R. [DOI] [PubMed] [Google Scholar]
- Mandell J. M.; Falick A. M.; Komives E. A. Measurement of amide hydrogen exchange by MALDI-TOF mass spectrometry. Anal. Chem. 1998, 70, 3987–3995. [DOI] [PubMed] [Google Scholar]
- Coan K. E. D.; Shoichet B. K. Stability and equilibria of promiscuous aggregates in high protein milieus. Mol. BioSyst. 2007, 3, 208–213. [DOI] [PubMed] [Google Scholar]
- Shoichet B. K. Interpreting steep dose-response curves in early inhibitor discovery. J. Med. Chem. 2006, 49, 7274–7277. [DOI] [PubMed] [Google Scholar]
- Coan K. E. D.; Shoichet B. K. Stoichiometry and physical chemistry of promiscuous aggregate-based inhibitors. J. Am. Chem. Soc. 2008, 130, 9606–9612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alkaabi K. M.; Yafea A.; Ashraf S. S. Effect of pH on thermal- and chemical-induced denaturation of GFP. Appl. Biochem. Biotechnol. 2005, 126, 149–156. [DOI] [PubMed] [Google Scholar]
- Weston G. S.; Blazquez J.; Baquero F.; Shoichet B. K. Structure-based enhancement of boronic acid-based inhibitors of AmpC beta-lactamase. J. Med. Chem. 1998, 41, 4577–4586. [DOI] [PubMed] [Google Scholar]
- Fraczkiewicz R.; Braun W. Exact and efficient analytical calculation of the accessible surface areas and their gradients for macromolecules. J. Comput. Chem. 1998, 19, 319–333. [Google Scholar]
- Powers R. A.; Caselli E.; Focia P. J.; Prati F.; Shoichet B. K. Structures of ceftazidine and its transition-state analogue in complex with AmpC beta-lactamase: implications for resistance mutations and inhibitor design. Biochemistry 2001, 40, 9207–9214. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.