

Summary
Intracellular signaling is often mediated by a family of functionally overlapping signal mediators that contain multiple sites interacting with other proteins or ligands with weak affinity (Kd > μM). Conjugation of multiple low-affinity ligands into a high-affinity multivalent molecule provides a means to control the entire protein family within a single intracellular pathway. The N-end rule pathway is a ubiquitin (Ub)-dependent proteolytic system where at least four Ub ligases, called N-recognins, have a common domain critical for binding to type-1 (basic) and type-2 (bulky hydrophobic) destabilizing N-terminal residues of substrates as degrons. The recent development of a heterodivalent inhibitor targeting type-1 and type-2 substrate binding sites of the N-recognin family provides new opportunities to manipulate this proteolytic pathway in biochemical and pathophysiological conditions. We overview the N-end rule pathway as an intracellular target for heterodivalent molecules and discuss the basis of thermodynamics and kinetics related to heterodivalent interactions.
Running Title: N-end rule, UBR box, multivalent interaction, polyvalency, heterodivalent, divalent inhibitors, ubiquitin, ubiquitin-proteasome system
Introduction
Nature employs multivalent interactions to increase selectivity and avidity of protein/protein or protein/ligand interactions in various processes, such as antigen-antibody, virus-cell and bacterial toxin-cell interactions (Choi, 2004; Kiessling et al., 2000; 2006; Huskens, 2006; Basha et al., 2006). Examples of natural multivalent molecules include the trimeric hemagglutinin complex of the influenza virus that recognizes host cells through multivalent binding to N-acetyl neuraminic acid (Spaltenstein and Whitesides, 1991). The enhancement, often dramatic, in selectivity and avidity of multivalent interaction is manifested by synthetic multivalent sialic acid molecules capable of binding to the hemagglutinin receptor on the viral surface with a multivalent enhancement factor of greater than 107 (Choi et al., 1996). As such, natural and synthetic multivalent interactions have been extensively investigated to explain the basis of multivalency and in an attempt to inhibit undesired ligand-receptor interactions or to induce desired biological responses. Various synthetic multivalent compounds were proven to be able to efficiently control physiological processes in different contexts, including receptor clustering (Gestwicki and Kiessling, 2002; Alarcón et al., 2006; Dam and Brewer, 2008), receptor selectivity (Lee and Lee, 2000), bacterial toxins (Rai et al., 2006; Kitov et al., 2000), pathogen-cell adhesion (Matrosovich, 1989), and protein-protein interactions (Gestwicki and Marinec, 2007). Most of the multivalent molecules synthesized to date are interhomovalent (Figure 1A) in that two identical ligands target the same binding site of two identical proteins on the surface of viruses, bacteria, or cells (reviewed in Choi, 2004). In contrast, rapamycin, an immunosuppressant drug produced from the bacterium Streptomyces hygroscopicus, is an interheterodivalent compound (Figure 1B) that can simultaneously bind two cytoplasmic proteins, FKBP12 (FK506 binding protein) and FRB (FKBP-rapamycin binding domain), to form the FKBP-rapamycin-FRB ternary complex (Sabatini et al., 1994). Some synthetic rapamycin derivatives were demonstrated to alter various intracellular pathways, including protein relocalization (de Graffenried et al., 2004; Haruki et al., 2008), conditional induction of apoptosis (Mallet et al., 2002), protein degradation (Janse et al., 2004), and conditional protein splicing (Schwartz et al., 2007).
Figure 1. Different types of multivalent ligands and a model showing the influence of the linker on effective concentrations (Ceff) of divalent molecules.

(A-C) Shown are interhomovalent (A), interheterovalent (B), and intraheterovalent (C) molecules.
(D-F) The bound ligand in a divalent molecule confines the other ligand to the hemispherical proximity, influencing the effective concentration (Ceff) as a function of its linker length. Shown are RF-Cn-type molecules (see below), in which the linker is longer (D), optimal (E), or shorter (F) compared to the distance between two binding sites of the target.
Intracellular signaling is often mediated by a family of functionally overlapping signal mediators that contain one or more structurally conserved domain(s) interacting with other ligands or proteins. Protein-protein and ligand-protein interactions are the combined effect of multiple microscopic interactions, such as electrostatic interactions between amino acids and van der Waals interactions between atoms. The communication between many signaling molecules is governed by weak, transient interactions (Kd > μM), as opposed to high-affinity drug-receptor interactions estimated to have mean Kd of 10-7.3 M (Houk et al., 2003). Not surprisingly, the paradigm in drug discovery has been focused on screening or synthesizing the highest-affinity ligand (Kd, sub-μM or nM) on the hopes that the resulting ligand will lead to a druggable compound with maximal therapeutic and minimal side effects. Under this paradigm, weak-affinity molecules are neglected based on a general notion that a weak-affinity molecule binds to a target with low selectivity and, thus, is pharmacologically useless. It is increasingly clear, however, that many weak-affinity biological interactions can become a useful target when multiple low-affinity ligands are combined into a multivalent molecule. For instance, various compounds with tethered ligands have been designed to enhance affinity for target enzymes (reviewed in Erlanson et al., 2004), such as carbonic anhydrase I (Banerjee et al., 2005), glutathione S-transferase (Maeda et al., 2006), and thrombin (Tolkatchev et al., 2005). The concept of multivalency is also successfully used in fragment-based drug discovery (FBDD), where a functional drug with high affinity and selectivity is synthesized or screened in smaller pieces that have low affinity and selectivity (Congreve et al., 2008). In this approach, initial high throughput screening identifies simple molecular fragments, which usually are small (120-250 Da) and of weak affinity (Kd, 10 μM-mM). However, some of the resulting fragment hits may have high unit affinity per atom, and the combination of these monovalent molecules may yield a drug-like compound with high selectivity and affinity to the target, thermodynamically (enhanced binding affinity) and kinetically (reduced dissociation rate). One noteworthy technique based on the concept of FBDD is ‘SAR by NMR’ (Structure-Activity Relationships by Nuclear Magnetic Resonance), in which multiple small fragments that bind to proximal sites on a protein are screened and linked together using NMR-assisted structural analysis (Shuker et al., 1996; Hajduk, 2006; Weigelt et al., 2002). Bearing in mind the demonstrated effectiveness of multivalency in various interactions, one would speculate that a multivalent molecule targeting multiple sites within a single domain or of multiple domains conserved in signaling molecules would enable the control of the entire protein family within a specific intracellular signaling pathway.
The purpose of this review is to overview the N-end rule pathway as an intracellular target for heterodivalent molecules, introduce the design and characterization of model heterodivalent compounds, and discuss the basis of thermodynamics and kinetics related to multivalent molecules, in particular those with long, flexible linkers. The N-end rule pathway is a ubiquitin (Ub)-dependent proteolytic system that plays a critical role in a variety of physiological processes, including cardiovascular signaling, oxygen/nitric oxide sensing, and viral and bacterial life cycles (Tasaki and Kwon, 2007). There are at least four recognition E3 components, called N-recognins, that contain a common domain critical for binding to type-1 (basic) and type-2 (bulky hydrophobic) destabilizing N-terminal residues of substrates as degrons. Recent development of a heterodivalent inhibitor targeting type-1 and type-2 substrate binding sites of the N-recognin family provides new opportunities to manipulate this proteolytic pathway in biochemical and pathophysiological conditions (Lee et al., 2008).
Multivalent Interaction: Thermodynamics and Kinetics
We discuss thermodynamics and kinetics related to heterodivalent molecules (Figure 1) that have a long, flexible linker to simultaneously target type-1 and type-2 binding sites of N-recognins. Whereas the binding of a monovalent molecule is mainly determined by the ligand's binding affinity, the overall avidity of a multivalent molecule to the target is affected not only by the affinity of individual ligands but also by other parameters such as the characteristics of the linkers connecting the individual ligands (Mammen et al., 1998; Krishnamurthy et al., 2006; Kitov and Bundle, 2003; Kiessling et al., 2000). As noted by Kitov and Bundle (2003), the free energy of binding for a multivalent interaction (ΔG0multi) can be described by the equation: ΔG0multi = nΔG0mono + ΔG0interaction (1), where ΔG0mono is the free energy of binding for the corresponding monovalent interaction, n represents the number of ligands that are bound to receptors, and ΔG0interaction contains contributions from the favorable and unfavorable effects of tethering. The various factors that contribute to ΔG0interaction are illustrated in the expression for ΔG0multi proposed by Krishnamurthy et al. (2006):
ΔG0multi = nΔG0mono + (n-1)(TΔS0mono,trans+rot +ΔH0linker −TΔS0conf +ΔG0coop) − RTln(Ωn/Ω0) (2). The term [(n-1) TΔS0mono,trans+rot] is based on the assumption that the unfavorable translational and rotational entropy of binding is approximately the same for a multivalent interaction as for a monovalent one. The term [(n-1) ΔH0linker] represents the change in enthalpy due to interactions between the linker and the target. The term [-(n-1) TΔS0conf] represents the loss of conformational entropy of the linkers following binding of the multivalent ligand. The term [(n-1) ΔG0coop] represents contributions from cooperativity – the influence of one binding event on subsequent events. The final term is a statistical factor based on the degeneracy (Ωn) for the multivalent ligand-receptor complex (Kitov and Bundle, 2003).
The above discussion can be used to guide the design of high avidity multivalent or divalent ligands by focusing on the various contributions to ΔG0interaction. For instance, as noted by Krishnamurthy et al. (2006), the magnitude of the contribution due to “entropic enhancement”, [(n-1) TΔS0mono,trans+rot], may be reduced by enthalpy/entropy compensation (EEC), since binding events with more favorable enthalpies of binding are associated with more unfavorable translational and rotational entropies of binding. They related TΔS0mono,trans+rot to ΔH0mono by the expression TΔS0mono,trans+rot =c ΔH0mono (3), where c is a constant (0<c< 1). Collectively, equations 2 and 3 suggest that for a constant ΔG0mono, the highest avidity multivalent ligands will be generated from monovalent ligands that bind with the most favorable enthalpy, ΔH0mono.
The avidity of a multivalent ligand is influenced not only by the choice of monovalent ligand, but also by the choice of linker. Equation 2 suggests that the use of a rigid linker might be optimal, as it would lower the conformational entropy penalty [-(n-1) TΔS0conf]; however, a rigid linker might also result in unfavorable interactions between the linkers or ligands and the receptor. On the other hand, a flexible linker would facilitate multivalent binding without steric obstruction, but might result in a significant loss in conformational entropy on binding. Models that assume that bonds are free rotors predict severe losses in conformational entropy for flexible linkers (TΔS0conf ∼ 0.7 kcal/mol per freely rotating bond of a linker when it is bound at both ends) (Krishamurthy et al., 2007). Flexible linkers have, however, been used successfully to design potent multivalent ligands (Kramers and Karpen, 1998), and models based on effective concentration (Ceff) predict a much smaller loss in conformational entropy on binding for long and flexible linkers than models based on the assumption that bonds are free rotors which become completely restricted following multivalent binding (Gargano et al., 2001; Diestler and Knapp, 2008; Krishnamurthy et al., 2007). An effective strategy for the design of a multivalent ligand might therefore be to connect the individual ligands by a flexible linker that is significantly longer than the spacing between the binding sites (Figure 1D-1F). We note that the principles described above should be applicable for the design of not only multivalent ligands but also homodivalent and heterodivalent ligands, including heterodivalent molecules that simultaneously target the type-1 and type-2 binding sites of N-recognins.
While the above discussion focused primarily on thermodynamics, the kinetics of interaction of multivalent ligands with their targets are also of interest. Studies on the kinetics of multivalent interaction suggest that enhancements in avidity are primarily due to decreases in the rates of dissociation (koff) of the multivalent entities than due to increases in the rates of association (kon) (Mammen et al., 1998). There are also fundamental differences between the dissociation of high avidity multivalent complexes and the dissociation of high affinity monovalent complexes. For instance, the dissociation of multivalent complexes occurs in stages, enabling the rate of dissociation to be enhanced by the addition of sufficiently high concentrations of competing monovalent ligand (Rao et al., 1998; 2000). As discussed above, these principles are generally applicable for multivalent ligands as well as for homodivalent and heterodivalent ligands.
The N-End Rule Pathway as an Intracellular Model for Heterodivalent Inhibitors
The N-end rule pathway is a subset of the ubiquitin-proteasome system (UPS), where recognition E3 components called N-recognins recognize type-1 and type-2 N-terminal residues of substrates as part of degrons (N-degrons). Recent proteomic studies identified four N-recognins containing the 70-residue UBR box that functions as a substrate recognition domain for type-1 and type-2 N-termini. We introduce the N-end rule pathway to those who are interested in designing multivalent inhibitors for intracellular pathways.
The ubiquitin-proteasome system
Ub is a 76-residue protein whose conjugation to other proteins regulates a variety of biological processes (Varshavsky, 1997). Ub-dependent proteolysis involves the marking of a target protein through covalent conjugation of Ub to an internal Lys residue of a substrate, which is mediated by the E1-E2-E3 enzymatic cascade (Figure 2). E1 is the ATP-dependent Ub-activating enzyme, which forms a high-energy thioester bond between the C-terminal Gly of Ub and a specific Cys of E1. The activated Ub is transesterified to a Cys residue of an E2 enzyme. E3 recognizes a substrate's degradation signal (degron) and conjugates, as a complex with E2, Ub to the ε-amino group of a Lys residue of a substrate protein. Repeated conjugation of Ub results in a polyubiquitylated substrate that is recognized by the proteolytic machinery of the UPS, the 26S proteasome (Figure 2). In mammals, more than 500 Ub ligases mediate polyubiquitylation of substrates through the recognition of degrons. Degradation of certain substrates require an additional component, E4, which binds short Ub chains and allows the formation of longer chains.
Figure 2. The ubiquitin-proteasome system.

The substrates are ubiquitylated through multiple rounds of a linear reaction catalyzed by E1, E2 and E3. Shown as an example is the N-end rule pathway.
The structure and components of the N-end rule pathway
N-recognins recognize a set of basic (type 1; Arg, Lys, and His) and bulky hydrophobic (type 2; Phe, Tyr, Trp, Leu, and Ile) N-terminal residues as a degradation determinant (Bachmair et al., 1986; Tasaki et al., 2005) (Figure 3A). In addition to N-terminal residues, a functional N-degron requires an internal Lys residue (the site of poly-Ub chain formation) and a characteristic conformational feature appropriate for ubiquitylation. A destabilizing N-terminal residue can be created by modifying a pre-N-degron (Asn, Gln, Cys, Asp, or Glu) through an enzymatic cascade (Kwon et al., 2000; 2001; 2002). In mammals, N-terminal asparagine (Asn) and glutamine (Gln) are conditionally destabilizing through deamidation into aspartate (Asp) and glutamate (Glu), which are respectively mediated by two distinct amidohydrolases (Grigoryev et al., 1996; Kwon et al., 2000). N-terminal Asp and Glu are arginylated by ATE1-encoded R-transferase, a universal eukaryotic posttranslational modification that creates the type-1 substrate Arg (Kwon et al., 1999a).
Figure 3. The structure of the N-end rule pathway and the creation of destabilizing N-terminal residues.

(A) The mammalian N-end rule pathway.
(B, C) The creation of destabilizing N-terminal residues through the removal of N-terminal Met (B) or the endoproteolytic cleavage of a protein (C).
Posttranslational arginylation and a sensor for oxygen and nitric oxide
N-terminal arginylation requires Arg from Arg-tRNAArg of the protein synthesis machinery, defining a tRNA-dependent Ub system (Varshavsky, 1996). In contrast to S. cerevisiae, in mammal, N-terminal cysteine (Cys) as well as Asp and Glu is conditionally destabilizing through arginylation (Kwon et al., 2002). However, in contrast to Asp and Glu, arginylation of N-terminal Cys requires oxidation prior to arginylation (Lee et al., 2005; Hu et al., 2005). In the presence of O2 (or its derivatives) and NO, Cys is oxidized into CysO2(H) or CysO3(H), which is recognized by ATE1, perhaps based on the structural similarity to Asp. ATE1-deficient embryos die associated with defects in cardiac development and angiogenesis (Kwon et al., 2002), which was later attributed to failure to degrade multiple Regulator of G-protein Signaling (RGS) proteins (RGS4, RGS5, and RGS16) (Lee et al., 2005; Hu et al., 2005). Following the cleavage of N-terminal Met by Met aminopeptidases (MetAPs), Cys-2 of these RGS proteins is N-terminally exposed and subsequently undergoes oxidation and arginylation to produce the destabilizing residue Arg (Figure 3B). As RGS4 and RGS5 play a critical role in Gq-dependent proliferation and signaling in cardiomyocytes and vascular smooth muscle cells, respectively, it has been proposed that the Ub system targeting these RGS proteins controls homeostasis in cardiovascular signaling by sensing O2 and NO (Lee et al., 2005). In addition to these RGS proteins, it has been reported that numerous proteins can be arginylated at N-terminal or internal residues (Karakozova et al., 2006; Wong et al., 2007). Thus, the failure in nondegradable arginylation may also contribute to cardiovascular null phenotypes in ATE1-deficient embryos.
Creation of the N-degron
As newly synthesized proteins bear N-terminal Met in eukaryotes (fMet in prokaryotes), a functional N-degron must be created by a posttranslational modification (Tasaki and Kwon, 2007). One way to create an N-end rule substrate is to expose the second residue at the N-terminus by MetAPs, which removes the N-terminal Met when the second residue is either Val, Gly, Pro, Ala, Ser, Thr, or Cys (Lee et al., 2005; Kendall and Bradshaw, 1992) (Figure 3B). Amongst these, Cys can be converted into a primary destabilizing residue through oxidation and arginylation, whereas the rest of the residues are stabilizing. Thus, in mammals N-degrons can be created via the removal of N-terminal Met when the second residue is Cys. The mammalian genome encodes at least 502 proteins bearing an N-terminal Met-Cys sequence (Y. Jiang and Y.T.K., unpublished data); it remains to be tested how many of these produce N-degrons after exposing Cys-2 at the N-terminus. Another way to create an N-degron is via an endoproteolytic cleavage of a long-lived polypeptide, which produces a short-lived C-terminal fragment bearing a destabilizing N-terminal residue (Figure 3C). Intracellular endopeptidases (e.g., caspases, separases and calpains) can create a C-terminal fragment bearing a tertiary or secondary destabilizing N-terminal residue (Asn, Gln, Cys, Asp, or Glu in mammals) or a primary destabilizing residue (Arg, Lys, His, Leu, Phe, Trp, Tyr, or Ile in mammals).
Physiological substrates and functions of the N-end rule pathway
In addition to RGS proteins, several proteins are known to be targeted by the N-end rule pathway. In Drosophila melanogaster, caspase-dependent cleavage of DIAP1 produces a C-terminal fragment with the N-terminal Asn (Ditzel et al., 2003), which is subsequently deamidated into the second destabilizing residue Asp. In S. cerevisiae, the cohesin component SCC1 is cleaved by Separase at the metaphase-to-anaphase transition to produce a C-terminal fragment bearing the destabilizing residue Arg, which is indispensable for chromosome stability (Rao et al., 2001). The N-end rule pathway is known to control half-lives of several viral and bacterial proteins that are exposed in the cytoplasm of the host cell during the life cycle. The human immunodeficiency virus type-1 (HIV-1) integrase, produced from the Gag-Pol precursor, catalyzes the insertion of viral genome into host chromosome (Pommier et al., 2005). The integrase, bearing the type-2 destabilizing residue Phe, is degraded in mammalian cells by the N-end rule pathway (Mulder and Muesing, 2000; Tasaki et al., 2005). The bacterium Listeria monocytogenes is a life-threatening pathogen that infects the cytosol of host cells through the activity of a pore-forming toxin, listeriolysin O. Because of its potential cytotoxicity, the activity of this virulence factor is controlled in part through ubiquitylation by the N-end rule pathway (Schnupf et al., 2007). N-recognins recognize not only N-degrons but also internal degrons embedded in the substrate's body. The latter class of substrates include S. cerevisiae CUP9 (a transcriptional repressor of the peptide transporter PTR2), S. cerevisiae GPA1 (the Gα subunit that controls signal transduction during mating) and mammalian c-Fos (reviewed in Tasaki and Kwon, 2007).
Heterovalent Inhibitors of the N-End Rule Pathway
To explore the model of heterovalent interaction targeting an intracellular pathway, Lee et al. (2008) recently designed the heterodivalent molecule RF-C11 whose type-1 and type-2 ligands bind to multiple N-recognins. Heterovalent interaction to N-recognins was demonstrated to be an efficient way to control the function of this posttranslational modification pathway in vitro and in mammalian cells, such as cardiomyocytes. RF-C11 is a prototype compound, in which each of four replaceable components can be further optimized in affinity, stability and cell permeability. The techniques described here are likely to be useful for finding and developing multivalent compounds that modulate the function of other intracellular pathways in vitro and in vivo.
The N-recognin family as a target of heterodivalent molecules
Known mammalian N-recognins, termed UBR1, UBR2, UBR4 and UBR5, are characterized by the UBR box, a ∼70-residue zinc finger-like domain that functions as a general substrate binding domain (Kwon et al., 1998; Tasaki et al., 2005) (Figure 4). The UBR box provides a structural element for binding to N-termini, in which specific residues in the UBR box (for type 1) or the N-domain (for type 2) provide substrate selectivity through interaction with the side group of an N-terminal residue (Tasaki and Kwon, 2007; Tasaki et al., 2008). UBR box-containing fragments of UBR1 exhibit moderate affinity and high selectivity to destabilizing N-terminal residues with Kd of 1.6-3.4 μM (Xia et al., 2008; Tasaki et al., 2008). This moderate affinity allows an appropriate balance between substrate selectivity and enzymatic processivity, ensuring both selective binding to a substrate and rapid dissociation from the N-terminus for an optimal rate of polyubiquitylation.
Figure 4. The UBR box protein family.
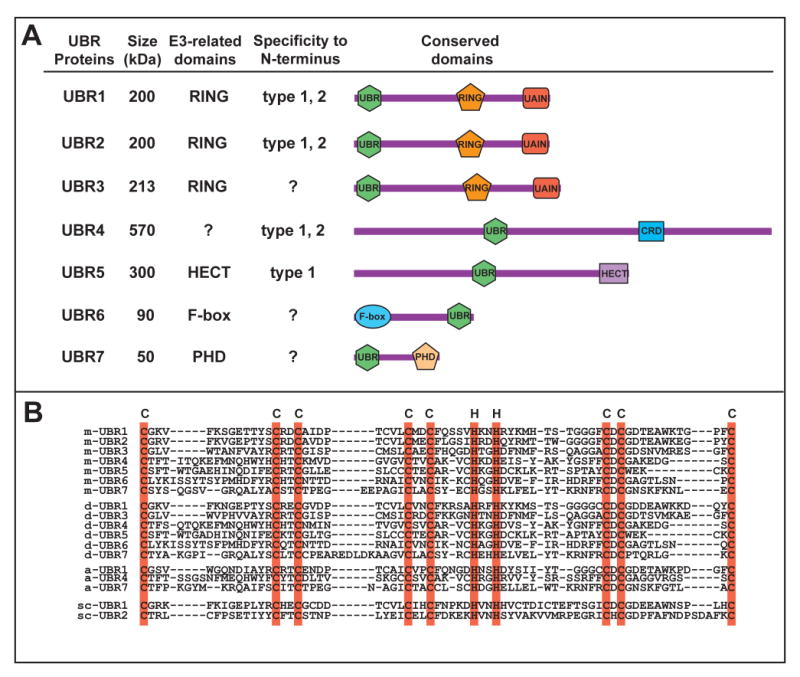
(A) A schematic diagram of UBR box proteins. UBR, UBR box; RING, RING finger; CRD, cysteine rich domain; HECT, HECT domain; PHD, plant homeodomain finger; UAIN, UBR-specific autoinhibitory domain.
(B) A sequence alignment of the UBR boxes from four species. Shown are the ∼70 amino acid regions where conserved Cys and His residues are highlighted (cyan). m, Mus musculus; d, D. melanogaster; a, A. thaliana; sc, S. cerevisiae.
Mammalian genome encodes at least seven UBR box proteins, termed UBR1 through UBR7 (Tasaki et al., 2005). It has been proposed that the UBR box acts as a receptor for small molecules whose structures are homologous to type-1 and type-2 ligands as part of a small molecule-modulated feedback mechanism (Tasaki et al., 2007). UBR box proteins are generally heterogeneous in size and sequence but contain, with the exception of UBR4, specific signatures unique to E3s or a substrate recognition subunit of the E3 complex (Figure 4A). UBR1 and UBR2 are 200-kDa RING-finger E3s with 46% similarity that form E3-E2 complexes with the Ub conjugating enzyme HR6A or HR6B and exhibit similar enzymatic specificities to N-degrons (Kwon et al., 2001; Tasaki et al., 2005; An et al., 2006). Mutations in human UBR1 cause Johanson-Blizzard syndrome (JBS; OMIM 243800), an autosomal recessive disorder characterized by exocrine pancreatic insufficiency and multiple malformations (Zenker et al., 2005). UBR1-deficient mice also develop JBS-like phenotypes, including pancreatic exocrine insufficiency (Zenker et al., 2005). Regardless of biochemical similarity between UBR1 and UBR2, UBR2-deficient mice exhibit distinct phenotypes: male-specific infertility and female-specific lethality (Kwon et al., 2003). Weakly homologous to UBR1 and UBR2, 213 kDa-UBR3 does not exhibit affinity to N-degrons (Tasaki et al., 2007). UBR3 is prominently expressed in sensory nervous cells critical for five major senses (smell, touch, vision, hearing, and taste), and UBR3-deficient neonatal pups die associated with anosmia (Tasaki et al., 2007). 570-kDa UBR4 can bind to type-1 and type-2 N-termini, interacts with E7 oncoprotein and retinoblastoma protein, and has been implicated in anchorage-independent growth and cellular transformation (DeMashi et al., 2005). The functions of other UBR proteins are discussed in Tasaki and Kwon (2007).
Design of the heterodivalent inhibitor RF-C11 of the N-recognin family
Taking advantage of the two-site architecture of N-recognin, Kwon et al. (1999b) tested whether coexpression of two metabolically stabilized N-end rule substrates, Arg-βgal (type 1) and Leu-βgal (type 2), would competitively inhibit degradation of short-lived substrates in S. cerevisiae. In a βgal tetramer, two N-termini of each dimer are spatially close, exposed, and oriented to the same direction so that one heterodimer bearing N-terminal Arg and Leu is expected to be present in a βgal tetramer. Although moderate in efficacy, this proof-of-concept inhibitor was demonstrated to inhibit the N-end rule pathway.
Based on the protein-based heterodivalent inhibitor, Lee et al. (2008) designed and characterized the synthetic heterovalent inhibitor RF-C11, whose two different ligands bind to two binding sites of the N-recognin family (Figure 5). RF-C11 was synthesized as one of the model compounds, L1L2-Cn, which are composed of four replaceable components: ligand (L1L2), linker (Cn), core (lysine) and tag (e.g., biotin) (Figure 5A). The amino acid lysine was chosen as the core component as it has trifunctional groups, among which ε-amine and α-amine are conjugated to two identical hydrocarbon chain linkers. In RF-C11, two C11 hydrocarbon chains were conjugated to the type-1 substrate Arg and the type-2 substrate Phe. Two homodivalent compounds, RR-C11 (bearing Arg at its termini) and FF-C11 (bearing Phe at its termini), were synthesized to compare heterodivalent vs. homodivalent interactions. The structural control GV-C11, with the stabilizing residues Gly and Val at its termini, was synthesized to evaluate the potential interaction of the linkers (Figure 5B). The linker length is an important parameter in heterovalent interaction. As the structures of N-recognins were unknown, the guanidium group of Arg and the phenyl group of Phe were designed to be ∼45Å apart to simultaneously reach the entire binding pocket of the UBR box-like domain, which was deduced from the crystal structure of mouse zinc finger protein 665 (Lee et al., 2008). When the direct interaction of L1L2-C11 to N-recognins was evaluated, the reactive carboxylic acid end of the core component was conjugated by a tag, biotin.
Figure 5. The heterodivalent inhibitor RF-C11 and its control compounds.

(A) A space-filling model of RF-C11.
(B) Structures of RF-C11 and its control compounds. Terminal moieties are indicated by colored background.
Inhibition of the N-end rule pathway using heterovalent interaction to N-recognins
Polyubiquitylation involves an enzymatic cascade comprising E1, E2, E3, and the proteasome, in which crosstalk between E3-substrate interaction spatiotemporally modulates the metabolic stability of a short-lived protein. Accordingly, various assays are needed to verify biochemical and functional interaction of a small molecule to the N-end rule pathway (Kwon et al., 2001; Lee et al., 2008). One efficient assay is to monitor the inhibitory efficacy of a small molecule on the degradation of an N-end rule substrate that is expressed in transcription-translation coupled reticulocyte lysates; this provides parameters concerning an empirical binding event (e.g., IC50) rather than the actual affinity. Model N-end rule substrates can be created by cotranslational cleavage of a Ub-protein fusion by deubiquitylating enzymes (DUBs), which yields a set of proteins bearing either type-1, type-2, or stabilizing residues (Bachmair et al., 1986). Using Arg-nsP4 (type 1) and Tyr-nsP4 (type 2) as model substrates, Lee et al. (2008) observed that the type-1 dipeptide Arg-Ala inhibited degradation of the type-1 substrate Arg-nsP4 with IC50 of 283 μM but showed no efficacy for the type-2 substrate. Reciprocally, the type-2 dipeptide Trp-Ala inhibited degradation of the type-2 substrate Tyr-nsP4 (IC50, 21 μM) but not type-1 substrates. In contrast to monovalent compounds, RF-C11 inhibited both type-1 and type-2 substrates and, moreover, with significantly higher efficacy (IC50, 16 μM for Arg-nsP4; 2.7 μM for Tyr-nsP4). RF-C11 also showed significantly higher efficacy compared to type-1 homodivalent RR-C11 (67 μM for Arg-nsP4) and type-2 homodivalent FF-C11 (151 μM for Tyr-nsP4). The activity of these L1L2-C11 compounds should be specific to ligands as the structural control GV-C11 did not affect the degradation. The possibility that the enhanced efficacy of RF-C11 is due to allosteric conformational change of binding sites was ruled out because mixtures of monovalent or homodivalent compounds did not give significantly additive effects. To further verify the effect of L1L2-C11 on the E3 activity of N-recognins, Lee et al. (2008) showed that RF-C11 inhibits in vitro ubiquitylation of N-end rule substrates with higher efficacy than homodivalent compounds, that RF-C11 directly binds to a 50-kDa UBR box-containing fragment of UBR1, and that RF-C11 can pulldown multiple endogenous N-recognins from rat testes extracts. These results provide experimental evidence that heterodivalent interaction to multiple N-recognins, in the midst of the mammalian proteome, leads to inhibition of both type-1 and type-2 N-end rule activities with higher efficacy compared to homodivalent or monovalent interaction.
Maly et al. (2000) showed that a heterodivalent inhibitor, composed of carbazole and catechol units linked by a flexible alkane chain, bound to the c-Src kinase with the heterodivalent IC50 of 0.064 μM, compared to the monovalent IC50 of ∼40 μM. Rao and Whitesides (1997) reported an enhancement factor of 103 for homodivalent vancomycin and D-Ala-D-Ala interaction. A relatively moderate enhancement factor of RF-C11 heterovalent interaction can be mainly attributed to the linker length and the ligand affinity to targets, if the off-target interaction of the linker and ligands with themselves or with other cellular macromolecules is ignored. As far as two ligands can reach their target binding sites, a shorter linker is generally favorable thermodynamically; a shorter linker is expected to result in a lower conformational entropic penalty on binding and a higher value of effective concentration (Ceff). Ceff can be better explained, in particular for biologists, by the enhanced local concentration of the ligands near the binding sites. Specifically, during RF-C11 interaction, the bound Phe ligand to the type-2 site will partially constrain the unbound Arg ligand of the same molecule within the hemisphere of radius equivalent to the linker length, and thereby increases the local Arg concentration in the proximity of the type-1 site. This will increase the probability of Arg binding to the type-1 site. Reciprocally, the bound Arg, whose binding has been facilitated by the bound Phe, in turn increases the local Phe concentration in the proximity of the type-2 site, further facilitating the Phe interaction to N-recognin. This mutual enhancement of local ligand concentrations is inversely correlated to the linker length, until the linker matches the distance between two targets (Figure 1D-1F). This was indeed experimentally observed with serial RF-Cn compounds with shorter linkers (S.M.S., R.B., and Y.T.K., unpublished data). The enhancement in potency obtained by using divalent ligands is determined by not only the linker length but also by the affinity of the ligands for their targets. Lee et al. (2008) found that the ligand Phe, linked to a nonproteinaceous C11 hydrocarbon chain in homodivalent FF-C11, exhibited much lower inhibitory efficacy compared to the dipeptide Phe-Ala that is thought to have Kd of low micromolar (Xia et al., 2008; Tasaki et al., 2008). Thus, the other way to increase heterovalent avidity is by enhancing the affinity of ligands, in particular the type-2 ligand, to the target. Recently, various Phe derivatives were synthesized and, a few of them were demonstrated to have higher inhibitory efficacy against N-recognins than the Phe ligand of RF-C11 (S.M.S., R. Kuruba, and Y.T.K., unpublished data). Future work will involve the design of amino acid derivatives with high affinity to achieve high monovalent enthalpy of binding as well as optimization of the linker length to achieve the maximal effective concentration without contributing significantly to the conformational entropic penalty.
In vivo application of heterovalent inhibitors
As the N-end rule pathway is mediated by a set of functionally overlapping N-recognins, pharmaceutical inhibitors are a useful tool to dissect the function of the entire pathway. Dipeptides bearing destabilizing N-terminal residues have been widely used as competitive inhibitors in biochemical and physiological analyses of the N-end rule pathway (Tasaki and Kwon, 2007). However, these monovalent compounds are at best weak inhibitors, often used at mM concentrations, and, moreover, are highly unstable due to the cleavage of the peptide bond by endopeptidases (Kwon et al., 2001), making it ineffective for mammalian cells. Lee et al. (2008) demonstrated that RF-C11 is capable of inhibiting the degradation of a physiological N-end rule substrate, RGS4, in mammalian cells. The in vivo efficacy of RF-C11 and its derivatives opens up an avenue to new physiological functions of the N-end rule pathway. Using RF-C11 and its structural control, Lee et al. (2008) revealed a cell-autonomous function of the N-end rule pathway in cardiac proliferation and hypertrophy. It has been shown that mouse embryos lacking ATE1 R-transferase die associated with defects in cardiac development and angiogenesis, which was also observed in animals lacking two downstream E3 components, UBR1 and UBR2 (Kwon et al., 2002; An et al., 2006). In an attempt to determine a cell-autonomous function of these components, Lee et al. (2008) found that RF-C11 significantly reduces cardiac proliferation and hypertrophy in primary cardiomyocytes isolated from mouse embryonic hearts. In contrast, the structural control GV-C11 exhibited no detectible efficacy. In humans, myocardial hypertrophy, associated with hypertension, cardiac valvular disease, or ischemia, is typically followed by serious myocardial diseases which account for the leading causes of death in western society. As such, N-recognins may be a therapeutic target for heterovalent inhibitors to control pathophysiological conditions in cardiovascular signaling.
The Ubiquitin-Proteasome Pathway as a Potential Target for Multivalent Ligands
In addition to the N-end rule pathway, recent advances in structural understanding of the UPS components reveal several potential targets for multivalent interaction. The 26S proteasome, composed of the 19S regulatory particle and the 20S core particle, is an abundant supracomplex with the concentration of 1-20 μg/mg of soluble protein (Kuehn et al., 1986). The 19S particle consists of the 9-protein ‘lid’ that recognizes polyubiquitin and the 10-protein ‘base’ with the ATPase activity that binds to the α ring of the 20S core particle (Glickman et al., 1998). The 19S particle deubiquitylates, unfolds, and transfers polyubiquitylated substrates into the 20S particle (Verma et al., 2004). The 20S particle is a stack of four rings of heptameric complexes composed of two different types of subunits; α subunits are gatekeepers for the proteolytic core composed of β subunits (Groll et al., 1997). Inside the 20S cylinder, subunits β1, β2, and β5 of two stacked β-rings expose their proteolytically active sites to execute postglutamyl peptide hydrolysing, trypsin-like and chymotrypsin-like activities, respectively (Groll and Clausen, 2003). The multimeric nature of the 20S particle, the availability of various monovalent inhibitors with distinct inhibitory mechanisms, and the short distances of catalytic sites of the β-subunits, ranging from 28 to 64 Å, together make its internal surface as an ideal supramolecular array for heterodivalent interaction. One feasible approach would be to link two nonoverlapping monovalent inhibitors in a way that does not interfere with the activity of the monovalent molecules and ensures the simultaneous binding to two binding sites of the β-subunits. Various small molecule inhibitors have been developed to target the 26S proteasome, mostly the inner surface of the 20S particle, with IC50 values ranging from low nM to 100 μM (Kisselev, 2008). Velcade (bortezomib), a dipeptide boronate with affinity to the N-terminal threonine hydroxyl group of β5, has been approved by the FDA in 2003 for the treatment of multiple myeloma and mantle cell lymphoma and in 2006 for the treatment of mantle cell lymphoma (Kisselev, 2008). Salinosporamide A (NPI-0052), a β-lactone derivative, inhibits all three peptidase activities of the 20S particle and is in Phase I clinical trials for the patients with solid tumors and lymphomas resistant to Velcade treatment (Chauhan et al., 2006). Also developed were other proteasome inhibitors categorized into aldehydes (Tyropeptin A, Fellutamide B, and MG132), epoxyketones (Epoxomicin, Eponemycin, and Carfilzomib), vinyl sulfones (NLVS and ZLVS) and macrocyclic vinyl ketones (Syringolin A and Glidobactin A) (Kisselev, 2008).
Although less well characterized compared to the 20S particle, the p53-MDM2 interface is also worthy of attention (reviewed in Dömling, 2008). The tumor suppressor p53 is a transcription factor that plays a critical role in maintaining genomic integrity. The level of p53 is tightly controlled by the Ub ligase MDM2 that binds p53 with Kd of 60-700 nM to mediate ubiquitylation and to inhibit the transcriptional activity of p53. Mutations of p53 is involved in approximately half of all known cancers, and overexpression of MDM2 is found in many cancers as well, including soft tissue sarcomas, osteosarcomas, and breast tumors (Momand et al., 1998). In contrast to most other protein-protein interactions where the large, undefined interface area hampers the design of small molecules inhibitors, X-ray structures indicate that the p53-MDM2 interface is confined to a pretty small area of 809 and 660 (Å)2 for p53 and MDM2, respectively (Chene, 2003). Accordingly, various small molecule inhibitors of p53 have been designed as anti-cancer agents, including Nutlins, Ke-43, 5-deazaflavin derivatives, rhodamine derivatives and tricyclic derivatives (Berg, 2008), some of which show high activity to induce apoptosis and inhibit cancer cell proliferation. Thus, the well characterized interface and the availability of various monovalent inhibitors associated with clinical importance together make the p53-MDM2 interface a potential target for heterodivalent interaction.
Concluding Remarks
The purpose of this review was to introduce the N-end rule pathway and related intracellular signaling pathways as a model for multivalent molecules. Intracellular proteins communicate with other proteins or ligands in part through structurally and functionally distinct domains such as the UBR box of the N-end rule pathway or the F-box of the SCF E3 pathway, the latter being an adaptor between a substrate and the SKP1/CUL1 E3 complex (Bai et al., 1996). As demonstrated with the UBR box, multivalent ligands targeting multiple sites located in one or multiple domains may provide a tool to probe protein-protein interactions and to identify new physiological functions of a specific signaling pathway. This approach will be particularly useful when a mechanistically distinct pathway is mediated by a set of functionally overlapping proteins with the same domain, such as the N-end rule pathway and the SCF-type E3 systems. Some proteins form a transient or long-lasting complex with a multivalent array, such as the 26S proteasome or the APC E3 complex (Frescas and Pagano, 2008). A multivalent molecule targeting two subunits may be utilized to probe their assembly, disassembly and spatial localization within the complex, to probe their interactions with peripheral interactors, or to selectively inhibit the complex's activities. The concept of multivalency has been recently adopted in FBDD, and we are now witnessing a number of compounds entering into phase II clinical trials (Congreve et al., 2008). The concept of heterodivalency also may be exploited in drug repositioning (Ashburn and Thor, 2004), an approach to develop new use for an existing drug, in which two appropriate drugs are linked to yield higher efficacy or lower adverse effects, provided that tethering of the drugs does not adversely affect the pharmacokinetic properties. Future strategy includes identifying appropriate target molecules, which will require advances in structural and functional understanding on biological interactions. The linkers and ligands will need to be optimized in cell penetration, solubility and in vivo stability. New thermodynamic models may be needed to better explain the interactions of the linkers and ligands with themselves and other molecules within the cell.
Figure 6. The control of cardiac signaling and hypertrophy by RF-C11.

Shown is a model where RGS4, RGS5, and RGS16 are cotranslationally degraded through serial Cys-2 modifications (see the main text). In this model, the heterovalent interaction of RF-C11 to the N-recognin family inhibits the degradation of these RGS proteins in cardiomyocytes, leading to their metabolic stabilization and inactivation of G protein signaling.
Acknowledgments
We are grateful to Takafumi Tasaki, Min Jae Lee, Ramalinga Kuruba, and Xiang Gao for helpful discussions. This work was supported by the NIH grants to Y.T.K. (GM69482, GM074000 and HL083365) and R.S.K. (AI056546 and EB007295) and the American Heart Association grant to Y.T.K.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alarcón B, Swamy M, van Santen HM, Schamel WW. T-cell antigen-receptor stoichiometry: pre-clustering for sensitivity. EMBO Rep. 2006;7:490–495. doi: 10.1038/sj.embor.7400682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An JY, Seo JW, Tasaki T, Lee MJ, Varshavsky A, Kwon YT. Impaired neurogenesis and cardiovascular development in mice lacking the E3 ubiquitin ligases UBR1 and UBR2 of the N-end rule pathway. Proc Natl Acad Sci USA. 2006;103:6212–6117. doi: 10.1073/pnas.0601700103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburn TT, Thor KB. Drug repositioning: identifying and developing new uses for existing drugs. Nat Rev Drug Disc. 2004;3:673–682. doi: 10.1038/nrd1468. [DOI] [PubMed] [Google Scholar]
- Bachmair A, Finley D, Varshavsky A. In vivo half-life of a protein is a function of its amino-terminal residue. Science. 1986;234:179–186. doi: 10.1126/science.3018930. [DOI] [PubMed] [Google Scholar]
- Bai C, Sen P, Hofmann K, Ma L, Goebl M, Harper JW, Elledge SJ. SKP1 connects cell cycle regulators to the ubiquitin proteolysis machinery through a novel motif, the F-box. Cell. 1996;86:263–274. doi: 10.1016/s0092-8674(00)80098-7. [DOI] [PubMed] [Google Scholar]
- Banerjee AL, Eiler D, Roy BC, Jia X, Haldar MK, Mallik S, Srivastava DK. Spacer-based selectivity in the binding of “two-prong” ligands to recombinant human carbonic anhydrase I. Biochemistry. 2005;44:3211–3224. doi: 10.1021/bi047737b. [DOI] [PubMed] [Google Scholar]
- Basha S, Rai P, Poon V, Saraph A, Gujraty K, Go MY, Sadacharan S, Frost M, Mogridge J, Kane RS. Polyvalent inhibitors of anthrax toxin that target host receptors. Proc Natl Acad Sci USA. 2006;103:13509–13513. doi: 10.1073/pnas.0509870103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg T. Small-molecule inhibitors of protein-protein interactions. Curr Opin Drug Discov Devel. 2008;11:666–74. [PubMed] [Google Scholar]
- Chauhan D, Hideshima T, Anderson KC. A novel proteasome inhibitor NPI-0052 as an anticancer therapy. Brit J Cancer. 2006;95:961–965. doi: 10.1038/sj.bjc.6603406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chene P. Inhibiting the p53-MDM2 interaction: an important target for cancer therapy. Nat Rev Cancer. 2003;3:102–109. doi: 10.1038/nrc991. [DOI] [PubMed] [Google Scholar]
- Choi SK, Mammen M, Whitesides GM. Monomeric inhibitors of influenza neuraminidase enhance the hemagglutination inhibition activities of polyacrylamides presenting multiple C-sialoside groups. Chem Biol. 1996;3:97–104. doi: 10.1016/s1074-5521(96)90285-9. [DOI] [PubMed] [Google Scholar]
- Choi SK. Synthetic multivalent molecules: concepts and biomedical applications. John Wiley & Sons; Hoboken: 2004. [Google Scholar]
- Congreve M, Chessari G, Tisi D, Woodhead AJ. Recent developments in fragment-based drug discovery. J Med Chem. 2008;51:3661–3680. doi: 10.1021/jm8000373. [DOI] [PubMed] [Google Scholar]
- Dam TK, Brewer CF. Effects of clustered epitopes in multivalent ligand-receptor interactions. Biochemistry. 2008;47:8470–8476. doi: 10.1021/bi801208b. [DOI] [PubMed] [Google Scholar]
- de Graffenried CL, Laughlin ST, Kohler JJ, Bertozzi CR. A small-molecule switch for Golgi sulfotransferases. Proc Natl Acad Sci USA. 2004;101:16715–16720. doi: 10.1073/pnas.0403681101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeMasi J, Huh KW, Nakatani Y, Münger K, Howley PM. Bovine papillomavirus E7 transformation function correlates with cellular p600 protein binding. Proc Natl Acad Sci USA. 2005;102:11486–11491. doi: 10.1073/pnas.0505322102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diestler DJ, Knapp EW. Statistical thermodynamics of the stability of multivalent ligand-receptor complexes. Phys Rev Lett. 2008;100:178101. doi: 10.1103/PhysRevLett.100.178101. [DOI] [PubMed] [Google Scholar]
- Ditzel M, Wilson R, Tenev T, Zachariou A, Paul A, Deas E, Meier P. Degradation of DIAP1 by the N-end rule pathway is essential for regulating apoptosis. Nat Cell Biol. 2003;5:467–473. doi: 10.1038/ncb984. [DOI] [PubMed] [Google Scholar]
- Dömling A. Small molecular weight protein-protein interaction antagonists: an insurmountable challenge? Curr Opin Chem Biol. 2008;12:281–291. doi: 10.1016/j.cbpa.2008.04.603. [DOI] [PubMed] [Google Scholar]
- Erlanson DA, Wells JA, Braisted AC. Tethering: fragment-based drug discovery. Annu Rev Biophys Biomol Struct. 2004;33:199–223. doi: 10.1146/annurev.biophys.33.110502.140409. [DOI] [PubMed] [Google Scholar]
- Frescas D, Pagano M. Deregulated proteolysis by the F-box proteins SKP2 and beta-TrCP: tipping the scales of cancer. Nat Rev Cancer. 2008;8:438–449. doi: 10.1038/nrc2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gargano JM, Ngo T, Kim JY, Acheson DWK, Lees WJ. Multivalent inhibition of AB5 toxins. J Am Chem Soc. 2001;123:12909–12910. doi: 10.1021/ja016305a. [DOI] [PubMed] [Google Scholar]
- Gestwicki JE, Kiessling LL. Inter-receptor communication through arrays of bacterial chemoreceptors. Nature. 2002;415:81–84. doi: 10.1038/415081a. [DOI] [PubMed] [Google Scholar]
- Gestwicki JE, Marinec PS. Chemical control over protein-protein interactions: beyond inhibitors. Comb Chem High Throughput Screen. 2007;10:667–675. doi: 10.2174/138620707782507296. [DOI] [PubMed] [Google Scholar]
- Glickman MH, Rubin DM, Coux O, Wefes I, Pfeifer G, Cjeka Z, Baumeister W, Fried VA, Finley D. A subcomplex of the regulatory particle required for ubiquitin-conjugate degradation and related to the COP9-signalosome and eIF3. Cell. 1998;94:615–623. doi: 10.1016/s0092-8674(00)81603-7. [DOI] [PubMed] [Google Scholar]
- Grigoryev S, Stewart AE, Kwon YT, Arfin SM, Bradshaw RA, Jenkins NA, Copeland NG, Varshavsky A. A mouse amidase specific for N-terminal asparagine. The gene, the enzyme, and their function in the N-end rule pathway. J Biol Chem. 1996;271:28521–28532. doi: 10.1074/jbc.271.45.28521. [DOI] [PubMed] [Google Scholar]
- Groll M, Ditzel L, Löwe J, Stock D, Bochtler M, Bartunik HD, Huber R. Structure of 20S proteasome from yeast at 2.4 Å resolution. Nature. 1997;386:463–471. doi: 10.1038/386463a0. [DOI] [PubMed] [Google Scholar]
- Groll M, Clausen T. Molecular shredders: how proteasomes fulfill their role. Curr Opin Stuct Biol. 2003;13:665–673. doi: 10.1016/j.sbi.2003.10.005. [DOI] [PubMed] [Google Scholar]
- Hajduk PJ. SAR by NMR: putting the pieces together. Mol Interv. 2006;6:266–272. doi: 10.1124/mi.6.5.8. [DOI] [PubMed] [Google Scholar]
- Haruki H, Nishikawa J, Laemmli UK. The anchor-away technique: rapid, conditional establishment of yeast mutant phenotypes. Mol Cell. 2008;31:925–932. doi: 10.1016/j.molcel.2008.07.020. [DOI] [PubMed] [Google Scholar]
- Houk KN, Leach AG, Kim SP, Zhang X. Binding affinities of host–guest, protein–ligand, and protein-transition-state complexes. Angew Chem Int Ed Engl. 2003;42:4872–4897. doi: 10.1002/anie.200200565. [DOI] [PubMed] [Google Scholar]
- Hu RG, Sheng J, Qi X, Xu Z, Takahashi TT, Varshavsky A. The N-end rule pathway as a nitric oxide sensor controlling the levels of multiple regulators. Nature. 2005;437:981–986. doi: 10.1038/nature04027. [DOI] [PubMed] [Google Scholar]
- Huskens J. Multivalent interactions at interfaces. Curr Opin Chem Biol. 2006;10:537–543. doi: 10.1016/j.cbpa.2006.09.007. [DOI] [PubMed] [Google Scholar]
- Janse DM, Crosas B, Finley D, Church GM. Localization to the proteasome is sufficient for degradation. J Biol Chem. 2004;279:21415–21420. doi: 10.1074/jbc.M402954200. [DOI] [PubMed] [Google Scholar]
- Karakozova M, Kozak M, Wong CC, Bailey AO, Yates JR, 3rd, Mogilner A, Zebroski H, Kashina A. Arginylation of β-actin regulates actin cytoskeleton and cell motility. Science. 2006;313:192–196. doi: 10.1126/science.1129344. [DOI] [PubMed] [Google Scholar]
- Kendall RL, Bradshaw A. Isolation and characterization of the methionine aminopeptidase from porcine liver responsible for the co-translational processing of proteins. J Biol Chem. 1992;267:20667–20673. [PubMed] [Google Scholar]
- Kiessling LL, Gestwicki JE, Strong LE. Synthetic multivalent ligands in the exploration of cell-surface interactions. Curr Opin Chem Biol. 2000;4:696–703. doi: 10.1016/s1367-5931(00)00153-8. [DOI] [PubMed] [Google Scholar]
- Kiessling LL, Gestwicki JE, Strong LE. Synthetic multivalent ligands as probes of signal transduction. Angew Chem Int Ed Engl. 2006;45:2348–2368. doi: 10.1002/anie.200502794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kisselev AF. Joining the army of proteasome inhibitors. Chem Biol. 2008;15:419–421. doi: 10.1016/j.chembiol.2008.04.010. [DOI] [PubMed] [Google Scholar]
- Kitov PI, Bundle DR. On the nature of the multivalency effect: a thermodynamic model. J Am Chem Soc. 2003;125:16271–16284. doi: 10.1021/ja038223n. [DOI] [PubMed] [Google Scholar]
- Kitov PI, Sadowska JM, Mulvey G, Armstrong GD, Ling H, Pannu NS, Read RJ, Bundle DR. Shiga-like toxins are neutralized by tailored multivalent carbohydrate ligands. Nature. 2000;403:669–672. doi: 10.1038/35001095. [DOI] [PubMed] [Google Scholar]
- Kramer RH, Karpen JW. Spanning binding sites on allosteric proteins with polymer-linked ligand dimers. Nature. 1998;395:710–713. doi: 10.1038/27227. [DOI] [PubMed] [Google Scholar]
- Krishnamurthy VM, Estroff LA, Whitesides GM. Multivalency in ligand design. In fragment-based approaches in drug discovery. Weinheim, Germany: Wiley-VCH Verlag GmbH & Co; 2006. pp. 11–54. [Google Scholar]
- Krishnamurthy VM, Semetey V, Bracher PJ, Shen N, Whitesides GM. Dependence of effective molarity on linker length for an intramolecular protein-ligand system. J Am Chem Soc. 2007;129:1312–1320. doi: 10.1021/ja066780e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon YT, Reiss Y, Fried VA, Hershko A, Yoon JK, Gonda DK, Sangan P, Copeland NG, Jenkins NA, Varshavsky A. The mouse and human genes encoding the recognition component of the N-end rule pathway. Proc Natl Acad Sci USA. 1998;95:7898–7903. doi: 10.1073/pnas.95.14.7898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon YT, Kashina AS, Varshavsky A. Alternative splicing results in differential expression, activity, and localization of the two forms of arginyl-tRNA-protein transferase, a component of the N-end rule pathway. Mol Cell Biol. 1999a;19:182–193. doi: 10.1128/mcb.19.1.182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon YT, Levy F, Varshavsky A. Bivalent inhibitor of the N-end rule pathway. J Biol Chem. 1999b;274:18135–18139. doi: 10.1074/jbc.274.25.18135. [DOI] [PubMed] [Google Scholar]
- Kwon YT, Balogh SA, Davydov IV, Kashina AS, Yoon JK, Xie Y, Gaur A, Hyde L, Denenberg VH, Varshavsky A. Altered activity, social behavior, and spatial memory in mice lacking the NTAN1p amidase and the asparagine branch of the N-end rule pathway. Mol Cell Biol. 2000;20:4135–4148. doi: 10.1128/mcb.20.11.4135-4148.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon YT, Xia ZX, Davydov IV, Lecker SH, Varshavsky A. Construction and analysis of mouse strains lacking the ubiquitin ligase UBR1 of the N-end rule pathway. Mol Cell Biol. 2001;21:8007–8021. doi: 10.1128/MCB.21.23.8007-8021.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon YT, Kashina AS, Davydov IV, Hu RG, An JY, Seo JW, Varshavsky A. N-Terminal arginylation is essential for cardiovascular development. Science. 2002;297:96–99. doi: 10.1126/science.1069531. [DOI] [PubMed] [Google Scholar]
- Kwon YT, Xia ZX, An JY, Seo JW, Varshavsky A. Apoptosis of meiotic spermatocytes and female-specific lethality in mice lacking UBR2 ubiquitin ligase. Mol Cell Biol. 2003;23:8255–8271. doi: 10.1128/MCB.23.22.8255-8271.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuehn L, Dahlmann B, Reinauer H. Tissue distribution of the multicatalytic proteinase in rat: An immunological and enzymic study. Cienc Biol (Portugal) 1986;11:101–107. [Google Scholar]
- Lee RT, Lee YC. Affinity enhancement by multivalent lectin-carbohydrate interaction. Glycoconj J. 2000;17:543–551. doi: 10.1023/a:1011070425430. [DOI] [PubMed] [Google Scholar]
- Lee MJ, Tasaki T, An JY, Moroi K, Davydov IV, Kwon YT. RGS4 and RGS5 are in vivo substrates of the N-end rule pathway. Proc Natl Acad Sci USA. 2005;102:15030–15035. doi: 10.1073/pnas.0507533102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MJ, Pal K, Tasaki T, Roy S, Jiang Y, An JY, Banerjee R, Kwon YT. Synthetic heterovalent inhibitors targeting recognition E3 components of the N-end rule pathway. Proc Natl Acad Sci USA. 2008;105:100–105. doi: 10.1073/pnas.0708465105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda DY, Mahajan SS, Atkins WM, Zebala JA. Bivalent inhibitors of glutathione S-transferase: the effect of spacer length on isozyme selectivity. Bioorg Med Chem Lett. 2006;16:3780–3783. doi: 10.1016/j.bmcl.2006.04.041. [DOI] [PubMed] [Google Scholar]
- Mallet VO, Mitchell C, Guidotti JE, Jaffray P, Fabre M, Spencer D, Arnoult D, Kahn A, Gilgenkrantz H. Conditional cell ablation by tight control of caspase-3 dimerization in transgenic mice. Nat Biotechnol. 2002;20:1234–1239. doi: 10.1038/nbt762. [DOI] [PubMed] [Google Scholar]
- Mammen M, Choi SK, Whitesides GM. Polyvalent interactions in biological systems: implications for design and use of multivalent ligands and inhibitors. Angew Chem Int Ed Engl. 1998;37:2755–2794. doi: 10.1002/(SICI)1521-3773(19981102)37:20<2754::AID-ANIE2754>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- Matrosovich MN. Towards the development of antimicrobial drugs acting by inhibition of pathogen attachment to host cells: a need for polyvalency. FEBS Lett. 1989;252:1–4. doi: 10.1016/0014-5793(89)80879-8. [DOI] [PubMed] [Google Scholar]
- Maly DJ, Choong IC, Ellman JA. Combinatorial target-guided ligand assembly: Identification of potent subtype-selective c-Src inhibitors. Proc Natl Acad Sci USA. 2000;97:2419–2424. doi: 10.1073/pnas.97.6.2419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Momand J, Jung D, Wilczynski S, Niland J. The MDM2 gene amplification database. Nucleic Acids Res. 1998;26:3453–3459. doi: 10.1093/nar/26.15.3453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulder LCF, Muesing MA. Degradation of HIV-1 integrase by the N-end rule pathway. J Biol Chem. 2000;275:29749–29753. doi: 10.1074/jbc.M004670200. [DOI] [PubMed] [Google Scholar]
- Pommier Y, Johnson AA, Marchand C. Integrase inhibitors to treat HIV/AIDS. Nat Rev Drug Discov. 2005;4:236–248. doi: 10.1038/nrd1660. [DOI] [PubMed] [Google Scholar]
- Rai P, Padala C, Poon V, Saraph A, Basha S, Kate S, Tao K, Mogridge J, Kane RS. Statistical pattern matching facilitates the design of polyvalent inhibitors of anthrax and cholera toxins. Nat Biotechnol. 2006;24:582–586. doi: 10.1038/nbt1204. [DOI] [PubMed] [Google Scholar]
- Rao J, Whitesides GM. Tight binding of a dimeric derivative of vancomycin with dimeric L-Lys-D-Ala-D-Ala. J Am Chem Soc. 1997;119:10286–10290. [Google Scholar]
- Rao J, Lahiri J, Isaacs L, Weis RM, Whitesides GM. A trivalent system from vancomycin. D-Ala-D-Ala with higher affinity than avidin biotin. Science. 1998;280:708–711. doi: 10.1126/science.280.5364.708. [DOI] [PubMed] [Google Scholar]
- Rao J, Lahiri J, Weis RM, Whitesides GM. Design, synthesis, and characterization of a high-affinity trivalent system derived from vancomycin and L-Lys-D-Ala-D-Ala. J Am Chem Soc. 2000;122:2698–2710. [Google Scholar]
- Rao H, Uhlmann F, Nasmyth K, Varshavsky A. Degradation of a cohesin subunit by the N-end rule pathway is essential for chromosome stability. Nature. 2001;410:955–960. doi: 10.1038/35073627. [DOI] [PubMed] [Google Scholar]
- Sabatini DM, Erdjument-Bromage H, Lui M, Tempst P, Snyder SH. RAFT1: a mammalian protein that binds to FKBP12 in a rapamycin-dependent fashion and is homologous to yeast TORs. Cell. 1994;78:35–43. doi: 10.1016/0092-8674(94)90570-3. [DOI] [PubMed] [Google Scholar]
- Schnupf P, Zhou J, Varshavsky A, Portnoy DA. Listeriolysin O secreted by Listeria monocytogenes into the host cell cytosol is degraded by the N-end rule pathway. Infect Immun. 2007;75:5135–5147. doi: 10.1128/IAI.00164-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz EC, Saez L, Young MW, Muir TW. Post-translational enzyme activation in an animal via optimized conditional protein splicing. Nat Chem Biol. 2007;3:50–54. doi: 10.1038/nchembio832. [DOI] [PubMed] [Google Scholar]
- Shuker SB, Hajduk PJ, Meadows RP, Fesik SW. Discovering high-affinity ligands for proteins: SAR by NMR. Science. 1996;274:1531–1534. doi: 10.1126/science.274.5292.1531. [DOI] [PubMed] [Google Scholar]
- Spaltenstein A, Whitesides GM. Polyacrylamides bearing pendant α-sialoside groups strongly inhibit agglutination of erythrocytes by influenza virus. J Am Chem Soc. 1991;113:686–687. doi: 10.1021/jm00046a027. [DOI] [PubMed] [Google Scholar]
- Tasaki T, Mulder L, Iwamatsu A, Lee MJ, Varshavsky A, Muesing M, Kwon YT. A family of mammalian E3 ubiquitin ligases that contain the UBR box motif and recognize N-degrons. Mol Cell Biol. 2005;25:7120–7136. doi: 10.1128/MCB.25.16.7120-7136.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasaki T, Sohr R, Hellweg R, Hörtnagl H, Varshavsky A, Kwon YT. Biochemical and genetic studies of UBR3, a ubiquitin ligase with a function in olfactory and other sensory systems. J Biol Chem. 2007;282:18510–18520. doi: 10.1074/jbc.M701894200. [DOI] [PubMed] [Google Scholar]
- Tasaki T, Kwon YT. The mammalian N-end rule pathway: new insights into its components and physiological roles. Trends Biochem Sci. 2007;32:520–528. doi: 10.1016/j.tibs.2007.08.010. [DOI] [PubMed] [Google Scholar]
- Tasaki T, Zakrzewska A, Dudgeon DD, Jiang Y, Lazo JS, Kwon YT. The substrate recognition domains of the N-end rule pathway. J Biol Chem. 2008 doi: 10.1074/jbc.M803641200. Epub ahead of print, November 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolkatchev D, Vinogradova A, Ni F. Transforming bivalent ligands into retractable enzyme inhibitors through polypeptide-protein interactions. Bioorg Med Chem Lett. 2005;15:5120–5123. doi: 10.1016/j.bmcl.2005.08.085. [DOI] [PubMed] [Google Scholar]
- Varshavsky A. The N-end rule: functions, mysteries, uses. Proc Natl Acad Sci USA. 1996;93:12142–12149. doi: 10.1073/pnas.93.22.12142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varshavsky A. The ubiquitin system. Trends Biochem Sci. 1997;22:383–387. doi: 10.1016/s0968-0004(97)01122-5. [DOI] [PubMed] [Google Scholar]
- Verma R, Oania R, Graumann J, Deshaies RJ. Multiubiquitin chain receptors define a layer of substrate selectivity in the ubiquitin-proteasome-system. Cell. 2004;118:99–110. doi: 10.1016/j.cell.2004.06.014. [DOI] [PubMed] [Google Scholar]
- Weigelt J, Wikström M, Schultz J, van Dongen MJ. Site-selective labeling strategies for screening by NMR. Comb Chem High Throughput Screen. 2002;5:623–630. doi: 10.2174/1386207023329978. [DOI] [PubMed] [Google Scholar]
- Wong CC, Xu T, Rai R, Bailey AO, Yates JR, 3rd, Wolf YI, Zebroski H, Kashina A. Global analysis of posttranslational protein arginylation. PLoS Biol. 2007;5:e258. doi: 10.1371/journal.pbio.0050258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia Z, Webster A, Du F, Piatkov K, Ghislain M, Varshavsky A. Substrate-binding sites of UBR1, the ubiquitin ligase of the N-end rule pathway. J Biol Chem. 2008;283:24011–24028. doi: 10.1074/jbc.M802583200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zenker M, Mayerle J, Lerch MM, Tagariello A, Zerres K, Durie PR, Beier M, Hülskamp G, Guzman C, Rehder H, et al. Deficiency of UBR1, a ubiquitin ligase of the N-end rule pathway, causes pancreatic dysfunction, malformations and mental retardation (Johanson-Blizzard syndrome) Nat Genet. 2005;37:1345–1350. doi: 10.1038/ng1681. [DOI] [PubMed] [Google Scholar]