

Abstract
Double transgenic mice [rat insulin promoter (RIP)-tumor necrosis factor (TNF) and RIP-CD80] whose pancreatic β cells release TNF and bear CD80 all develop an acute early (6 wk) and lethal diabetes mediated by CD8 T cells. The first ultrastructural changes observed in β cells, so far unreported, are focal lesions of endoplasmic reticulum swelling at the points of contact with islet-infiltrating lymphoblasts, followed by cytoplasmic, but not nuclear, apoptosis. Such double transgenic mice were made defective in either the perforin, Fas, or TNF pathways. Remarkably, diabetes was found to be totally independent of perforin and Fas. Mice lacking TNF receptor (TNFR) II had no or late diabetes, but only a minority had severe insulitis. Mice lacking the TNF-lymphotoxin (LTα) locus (whose sole source of TNF are the β cells) all had insulitis comparable to that of nondefective mice, but no diabetes or a retarded and milder form, with lesions suggesting different mechanisms of injury. Because both TNFR II and TNF-LTα mutations have complex effects on the immune system, these data do not formally incriminate membrane TNF as the major T cell mediator of this acute autoimmune diabetes; nevertheless, in the absence of involvement of the perforin or Fas cytotoxic pathways, membrane TNF appears to be the likeliest candidate.
Type I diabetes mellitus (T1DM) is a disorder caused by T cell-mediated pancreatic β-cell destruction. Direct T cell cytotoxicity is known to involve two major pathways: one involves perforin-granzyme B release and the other depends on FasL molecules borne by cytotoxic lymphocytes (CTL) interacting with Fas borne by the target cells. Other mechanisms have been described that depend on different effectors such as tumor necrosis factor (TNF), lymphotoxin (LT, a membrane molecule made of LT-α and -β heterotrimers), other TNF family molecules, or cytokines like IFN-γ and IL1 that are toxic for the target cells. For instance, IL1 is reported to kill β cells in vitro (1, 2). Indeed, the most frequently incriminated mechanisms of β-cell damage in T1DM and in animal models of that disease have involved perforin, Fas, and IL1 (3–8).
We (9) and others (10) have developed a transgenic mouse model for T1DM by creating mice expressing two rat insulin promoter (RIP)-driven transgenes, one leading to β-cell expression of mouse TNF and the other to β-cell surface expression of mouse CD80 (CD80, or B7–1, a costimulator molecule of T cell activation). This T1DM model is characterized by an early age of onset (all are diabetic by 6 wk of age) and by complete penetrance. Transgenic mice bearing either of the transgenes but without the other appear completely healthy and typically conserve normal β-cell numbers [although a very small minority, less than 5%, of the RIP-CD80 mice do spontaneously develop insulitis and diabetes (11)]. Further, although RIP-CD80 mice have normal islets, all RIP-TNF mice display a severe lymphocytic insulitis affecting all islets, indicating that insulitis is not diabetogenic by itself (12). In mice expressing both transgenes, the insulitis is accompanied by massive β-cell destruction. We have previously shown that this diabetes is entirely CD4 T cell-independent, CD8 T cell depletion by monoclonal antibodies fully protecting all TNF/CD80 transgenic mice from diabetes (9).
Given the rapid progression toward diabetes displayed by the TNF/CD80 transgenic mice, they represent a unique opportunity to study the mechanisms by which autoreactive CD8 T cells may induce β-cell destruction. We now report the breeding of the TNF/CD80 double transgenic mice to create mice expressing both transgenes but genetically unable to express either perforin, Fas, or TNF-R II receptors, or unable to express TNF or lymphotoxin-α (LTα) in their lymphocytes. The phenotype displayed by these mice suggests mechanisms that may be involved in the insulitis development and in CD8 T cell-mediated β-cell killing.
Materials and Methods
(i) Mice. Genotyping of the Different Mutant Alleles.
The mice used in this study with genetically absent expression of perforin, Fas, or TNF genes had been maintained in the C57BL/6 genetic background for at least 10 generations (backcrosses). Mice were housed under aseptic conditions in the animal facility of the University of Geneva School of Medicine. All other animals were kept in standard conditions at the same facility. All were fed and watered ad libitum. Euthanasia was performed by cervical dislocation. The different genotypes were determined by PCR on genomic DNA prepared from tail biopsies following standard protocols, i.e., with proteinase-K digestion, phenol-chloroform-isoamyl alcohol extraction, and isopropanol precipitation. The different oligonucleotides to detect each genetic trait are listed below:
RIP-TNF transgene.
5′-TAA GGC TAA GTA GAG GTG T-3′ (on RIP) and 5′-GAG AAG AGG CTG AGA CAT AG-3′ (on TNF).
RIP-CD80 transgene.
5′-CAA ACA ACA GCC TTA CCT TCG-3′, and 5′-GCC TCC AAA ACC TAC ACA TCC-3′.
Perforin knockout allele.
5′-AGC CCT CCC CCG CAA CTT TAA CAG CTC C-3′ (exon 3, upstream of the neo cassette), 5′- TGG GCA GCA GTC CTG GTT GGT GAC CTT-3′ (exon 3, downstream of the neo cassette), and 5′-ATT CGC AGC GCA TCG CCT TCT ATC GCC-3′ (on neo gene).
Lymphoproliferation (lpr) mutation.
5′-GAT TCC ATT TGC TGC TGT GTC-3′ (on intron 2, upstream of ETn), 5′-TCC AGG CAG AAC TAT TGA GC-3′ (on ETn), and 5′-AGA GAT GCT AAG CAG CAG CCG-3′ (on intron 2, downstream of ETn).
TNF-RII inactivated allele.
As suggested by H. Bluethmann: 5′-CCT CTC ATG CTG TCC CGG ATT-3′, 5′-AGC TCC AGG CAC AAG GGC GGG-3′, and 5′-GCG CAT CGC CTT CTA TCG CC-3′.
TNF silenced locus.
5′-TGA CAA GCC TGT AGC CCA CGT CGT AGG-3′ (exon 3), 5′-CTG GAA GAC TCC TCC CAG GTA TAT GGG-3′ (exon 4), 5′-ACC TGT CCG GTG CCC TGA ATG-3′ (on neo gene), and 5′-CAA CGC TAT GTC CTG ATA GCG-3′ (on neo gene).
(ii) Measurement of Blood Glucose.
Blood glucose concentrations were measured weekly by using Hemo-Glucotest strips (20–800R, Boehringer Mannheim) on retroorbital samples harvested from mice fasted for at least 4 hr. Mice were considered diabetic if they had blood glucose values ≥14 mM (300 mg/dl) for two consecutive measurements.
(iii) Histological Analysis: Optical and Electron Microscopy and Immunohistochemistry.
Once harvested, all pancreata were divided into three pieces. One piece was fixed in formalin and embedded in paraffin, one was fixed in 2.5% glutaraldehyde and embedded in epon 812, and one was frozen in methylbutane/liquid nitrogen and stored at −80°C before being cut in a cryostat. The aldehyde-fuchsin staining method was used to reveal β cells as well as collagen and elastin fibers (13); immunostaining with antiinsulin antibodies, anti-CD4 (GK1.5), anti-CD8 (H-35), and the FITC-conjugated antibodies was described elsewhere (12). Ultrastructural analysis was done by using standard copper grids in a JEOL JEM-100CX electron microscope. Terminal deoxynucleotidyl-transferase-mediated dUTP nick-end labeling (TUNEL) assays were performed by using a kit (ApopTag, Appligene Oncor, Strasbourg, France; no. S7100) on formalin-fixed paraffin sections. Islet-infiltrating CD8+ T cells were quantified by using nine different islet sections from six diabetic TNF/CD80 mice, 5 to 8 wk of age, and 12 islet sections from three TNF/CD80/TNF-LTα-knockout mice (one of which was diabetic), 6, 12, and 13 wk of age.
Results
(i) Diabetes Is Not Prevented in RIP-TNF/CD80 Transgenic Mice Lacking Perforin.
Single transgenic RIP-TNF and RIP-CD80 mice were crossed with perforin-knockout mice (14) to obtain mice heterozygous for the perforin null mutation and bearing either transgene. Such mice were then intercrossed, and eight TNF/CD80 mice lacking a functional perforin gene were obtained at the expected 1/16 ratio. As predicted, one-fourth of the TNF/CD80 double transgenic mice obtained were perforin −/− (i.e., 8 mice of 33). All these mice became diabetic by 6 wk of age, as did the 25 TNF/CD80 littermates that were either wild type or perforin +/−. Islets from all the mice, whether perforin deficient or not, displayed comparable pathology by histology, immunofluorescence, and electron microscopy (detailed below).
(ii) Diabetes Is Not Prevented in RIP-TNF/CD80 Mice Bearing the lpr Mutation.
TNF/CD80 mice lacking a functional Fas pathway were obtained by introducing the two transgenes in C56BL/6-lpr/lpr mice by using the same breeding scheme as outlined above. lpr is an insertion of an early transposable element in intron 2 of the Fas gene (15). All of 13 lpr mice bearing both transgenes became diabetic as did all double transgenic mice with one or two normal Fas alleles. Histological lesions were identical in the two groups.
(iii) Diabetes Is Prevented or Severely Delayed in TNF/CD80 Transgenic Mice Lacking the TNF Receptor Type II (TNFR II) (TNFR75, CD120b) (Table 1).
Table 1.
TNF/CD80/TNFR II knockout mice
| Mouse no. | Diabetes | Age at death, mo | Histology |
|---|---|---|---|
| 38 | No | 6.5 | Severe insulitis |
| 39 | No | 2.5 | Periinsulitis |
| 48 | Yes | 6 | Insulitis, atrophy |
| 54 | No | 2.5 | Mild insulitis |
| 127 | No | 5 | Normal islets |
| 134 | No | 4 | Mild insulitis |
| 137 | No | 5 | Normal islets |
The diabetic mouse was sacrificed immediately after the detection of hyperglycemia (weekly tests).
It has been proposed that TNFR II is the main receptor of membrane TNF (mTNF) and the main mediator of cell-mediated cytotoxicity in TNF sensitive cells (16), whereas the TNFR I receptor is primarily responsible for soluble TNF (sTNF) signaling, although recent evidence suggests that the respective roles of these two receptors may be more complex (17). No gross phenotype is apparent in TNFR II null mice (18). None of seven double transgenic mice on the TNFR II null background, obtained by intercrossing as described above, developed diabetes by 6 wk of age. These mice were killed over the ensuing months, whether diabetic or not. Only one became hyperglycemic at 6 mo of age; the six others remained normoglycemic until they were killed at either 2.5, 4, 5, or 6.5 mo of age; insulitis in these mice varied from severe to mild or absent (Table 1), an unexpected finding because the effects of TNF on endothelial cells appear to be mediated by TNFR I (19). In contrast, all double transgenic mice heterozygous for the TNFR II mutation developed diabetes by 6 wk of age. Although the variable degree of insulitis make this model insufficiently suitable for more extensive study, these results nevertheless show that, even after CD80-mediated costimulation, islet-infiltrating T cells cannot efficiently kill β cells when TNFR II is missing.
(iv) TNF/CD80 Mice Lacking Functional TNF and LTα Genes Display Typical Insulitis but Diabetes Onset Is Delayed (Table 2).
Table 2.
TNF/CD80/TNF-LTα knockout mice
| Mouse no. | Diabetes | Age at death, wk | Histology |
|---|---|---|---|
| 120 | Yes | 12 | Insulitis |
| 122 | Yes | 12 | Insulitis |
| 224 | No | 6 | Insulitis |
| 317 | No | 13 | Insulitis, pancreatitis |
| 347 | Yes | 12 | Insulitis |
| 445 | No | 11 | Insulitis, pancreatitis |
| 446 | yes | 9 | Insulitis |
| 464 | Yes | 9 | Insulitis |
| 476 | Yes | 11.5 | Insulitis, pancreatitis |
| 481 | Yes | 8 | Insulitis, pancreatitis |
| 483 | No | 13.5 | Insulitis, pancreatitis |
| 487 | Yes | 19 | Insulitis, pancreatitis |
| 502 | No | 9 | Insulitis, pancreatitis |
| 503 | No | 9 | Insulitis |
| 506 | Yes | 13 | Insulitis |
| 510 | No | 9 | Insulitis |
Diabetic mice were killed immediately after the detection of hyperglycemia (weekly tests).
The RIP-TNF and RIP-CD80 transgenes were introduced as described above into mice whose TNF–LTα locus had been inactivated (20). The main phenotype of the TNF-LTα −/− mice is the lack of lymph nodes, whose development requires the expression of LT molecules in developing lymphoid structures. In both double transgenic and null mutant mice, the only significant source of TNF is the pancreatic β cell, where the RIP-TNF transgene is expressed. Among the 16 double transgenic and −/− mice obtained, none was diabetic at 6 wk of age. These mice were subsequently killed at different times or immediately after the occurrence of hyperglycemia, when it occurred. In these last cases (eight mice), the hyperglycemia was mild (200–300 mg/dl) (Table 2), in contrast to TNF/CD80 mice being TNF-LTα +/− or +/+, all of which rapidly developed severe hyperglycemia (values >500 mg/ml). In all the 16 TNF/CD80/TNF-LTα −/− mice, whether diabetic or not, and killed as early as 6 wk, mesenteric lymphoid nodes had developed (but peripheral nodes remained absent); this presumably resulted from the high local concentration of sTNF released from β cells, because permanent TNF release favors formation of lymphoid structures (21). In all these 16 mice, a severe insulitis was observed, entirely comparable in intensity to that of the RIP-TNF single transgenic or TNF/CD80 double transgenic mice with a normal TNF-LTα locus, and with a similar cellular composition, i.e., a predominance of CD4+ cells over CD8+ cells, B cells, and macrophages (Fig. 1) (9, 12). Quantification of CD8+ T cell frequency in several double transgenic mice, either TNF-LTα +/+ or −/− (and in this last case, either diabetic or not) showed respectively 48 ± 17 and 43 ± 30 cells per islet section. In addition to insulitis, some of the TNF/CD80/TNF-LTα −/− mice displayed a marked fibrosis that dissociated the islets (Table 2), resulting in endocrine cell dissemination into small clusters mixed with infiltrating lymphocytes and accumulated collagen (Fig. 2 C and D). This fibrosis was severe in 7 of the 16 mice but did not correlate with diabetes (Table 2). In the most affected specimens, the fibrosis extended deeply in the adjacent pancreatic tissue and was associated with the appearance of intrapancreatic adipocytes (Fig. 2D).
Figure 1.
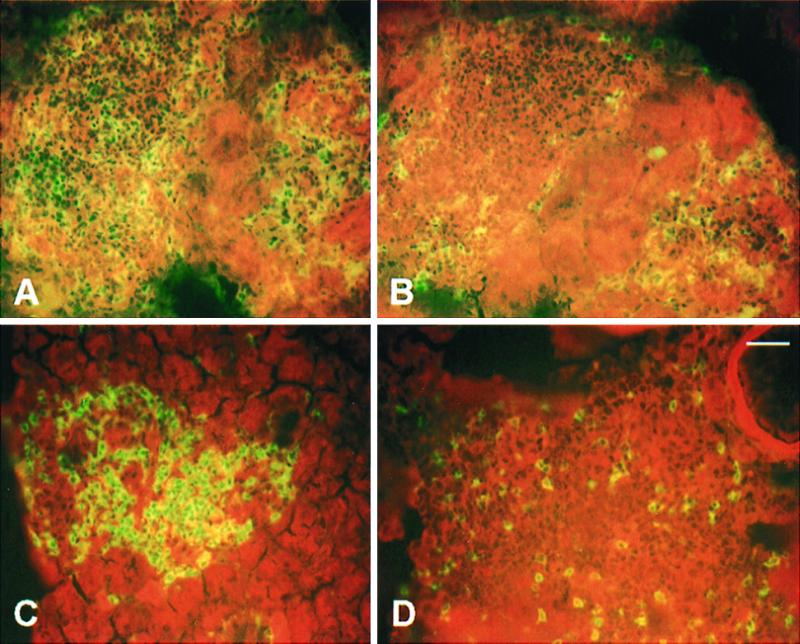
Eight micrometer-thick cryostat sections, stained by immunofluorescence, of pancreata from a TNF/CD80 mouse (6.5 wk old, hyperglycemic: A and B) and from a TNF/CD80 mouse with a mutated TNF-LT-α locus (13.5 wk, normoglycemic: C and D). A and C show islets stained for CD4+ cells; CD8+ cells are labeled in B and D. The proportion of CD4+ and CD8+ is similar in mutant and nonmutant transgenic mice. Bar = 16 μm (same magnification in A–D).
Figure 2.
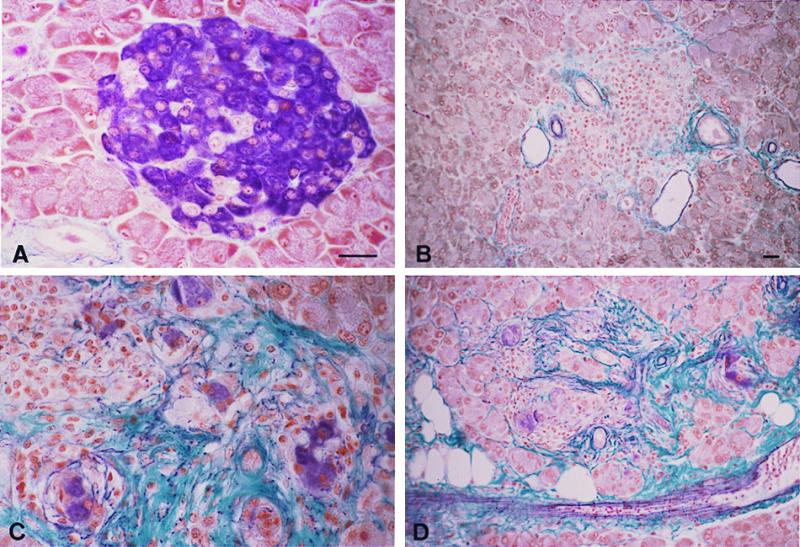
A severe pancreatic fibrosis develops in TNF/CD80 mice with a mutated TNF-LTα locus. Seven micrometer-thick pancreatic paraffin sections were stained with the aldehyde-fuchsin method to reveal insulin cells (in violet) as well as collagen and elastin fibers (green and violet, respectively). (A) Normal mouse islet; B–D, mouse islets from three different transgenic animals bearing the TNF-LTα mutation. B shows an atrophic islet in which no β cells remain; only infiltrating cells are seen, and the fibrotic reaction remains moderate, as usually observed in TNF/CD80 nonmutant transgenic mice. In C, several clusters of isolated β cells (violet) are surrounded by fibrosis (green), together with lymphocytes (Upper Left); unaffected adjacent exocrine cells are also seen (Upper Right). In D, fibrosis extends to the adjacent exocrine tissue, which is progressively replaced by adipocytes (white holes). Bars = 20 μm (same magnification in A–C and in B–D).
(v) Ultrastructural Studies: β-Cell Apoptosis Starts with Very Focal Cytoplasmic Lesions Localized Immediately Beneath the Cell Membrane.
A characteristic ultrastructural lesion of β cells was observed in TNF/CD80 mice around the time of diabetes occurrence. As is shown in Fig. 3 A and B, the endoplasmic reticulum (ER) located just beneath the β-cell membrane at a site of close contact with an infiltrating lymphoblast displayed a marked focal dilatation and rounding. The rest of the cytoplasm was otherwise normal (Fig. 3A), suggesting that this lesion is an early sign of β-cell distress. More damaged β cells (Fig. 3C) displayed a more generalized and marked ER swelling associated with signs of mitochondrial damage, i.e., features compatible with cytoplasmic apoptosis (22). Surprisingly, the characteristic nuclear features of apoptosis, i.e., chromatin condensation and margination, were absent, even in the most severely damaged cells (Fig. 3 A–C); nuclear apoptosis was on the contrary frequently observed in the infiltrating lymphoblasts. This point was confirmed on paraffin-embedded pancreatic sections by using the TUNEL technique and an antiinsulin antibody staining, to identify β-cell nuclei. Only lymphocytic nuclei were TUNEL + (128 positive cells scored, from three mice): among 663 β cells analyzed, none was TUNEL +. Similar observations were made in islets from RIP-TNF/CD80 mice lacking either perforin, Fas, or TNF-LTα, at different ages (not shown). Focal submembranous cytoplasmic lesions were present in all instances except, interestingly, in the TNF-LTα null mice (Fig. 3E), whereas general cytoplasmic damage and lack of nuclear apoptosis were a constant observation.
Figure 3.

β-cell damage in TNF/CD80 transgenic mice. Electron micrographs of damaged β cells in either TNF/CD80 double transgenic mice (A–C) or in transgenic mutant mice lacking either a functional perforin gene (D) or a normal TNF-LTα locus (E). Damaged β cells show lesions of submembranous focal apoptosis with dilation of ER cavities limited at the points of contact with activated lymphocytes (A and B, arrows) or extended throughout the whole cytoplasm (C, arrowheads, D and E) together with swelling of mitochondria (C, arrow). There is neither chromatin condensation nor nuclear fragmentation in any of these situations (see the nuclei of β cells, A–E). Bars = 500 nm (magnification is the same for B and D as well as for C and E).
Discussion
The RIP-TNF/CD80 Double Transgenic Mice as a Model of Autoimmune Diabetes.
In these mice, the occurrence of diabetes is most likely attributable to TNF-induced insulitis and increased β-cell MHC class I expression (23), coupled with CD80-triggered antigen-specific T cell activation via CD28. Consistent with this reasoning, RIP-TNF single transgenic mice develop an early and massive insulitis associated, in the islet capillaries, with endothelial changes involved in lymphocyte extravasation and recirculation (e.g., vascular cell adhesion molecule and glycosylated cell adhesion molecule expression) (ref. 12; P.H. and P.V., unpublished observations). Nevertheless, these mice never develop diabetes and are no more sensitive to some potentially diabetogenic treatments, indicating that exposure to TNF does not by itself make β cells more fragile in general (12). Similarly, RIP-CD80 single transgenic mice (11, 24) only very rarely develop diabetes and insulitis (less than a 5% lifetime incidence), indicating that potentially autoreactive T cells are harmless if they do not traffic to the tissue where they might encounter cognate antigen (i.e., “immune ignorance”), or if the cells that might express cognate antigen do so at low density (i.e., “immune indifference”). Thus, the RIP-TNF/CD80 double transgenic mouse model for T1DM has two interesting features: it results from autoantigenic stimulation, and the course of the disease is extremely rapid and predictable, with 100% prevalence. This makes it a model of autoimmune illness uniquely suited to study both the type of β-cell death and the mechanisms of CD8+ T cell-mediated β-cell destruction.
A Special Form of T Cell-Mediated β-Cell Apoptosis.
β-Cell death in various T1DM models has been ascribed to apoptosis (25–29). In the RIP-TNF/CD80 double transgenic mice, electron microscopy revealed that the earliest detectable manifestation of β-cell damage was a very localized cytoplasmic process, occurring at the point of intimate contact between the β cell and the infiltrating lymphocyte and characterized by focal swelling and rounding of the most peripheral ER cavities (Fig. 3). This hitherto unreported observation of β-cell structural defects at multiple sites immediately beneath the cell membrane suggests an apoptotic process resulting from local signals received by the cell membrane. More damaged cells showed extensive ER dilatation comparable to that described for enucleated cells undergoing cytoplasmic apoptosis (22, 30). Surprisingly, the typical nuclear features associated with apoptosis (e.g., chromatin condensation and DNA fragmentation as revealed by the TUNEL technique) were not observed. It should be stressed that these nuclear changes are not necessary for apoptosis. Rather, nuclear apoptosis appears to result from caspase 3 activation, as suggested by its absence in the apoptotic cells of caspase 3 null mice (31, 32). It may well be that the caspase cascade leading to the accumulation of caspase 3, which is required to achieve marked nuclear damage, is not initiated at a proper pace or is slowed by some inhibitors in this form of β-cell cytotoxicity.
β-Cell Death Occurs in the Absence of Perforin or Fas: a mTNF-Induced Lesion?
When the two transgenes were introduced (by breeding) into mice genetically unable to express either perforin or Fas, diabetes onset occurred as rapidly and its course was as severe as in transgenic mice with intact perforin or Fas genes. It is thus obvious that neither the perforin nor the Fas pathways are required for the CD8+ T cell-mediated β-cell destruction in this model. Two alternative possibilities were considered. One is that the perforin and Fas pathways are operative but entirely redundant, each being able to replace the other with equal efficiency. This seems very unlikely because β-cells do not bear Fas unless stimulated in vitro with cytokines such as IL-1 (33–35). Although this is still disputed, Fas appears to play a minimal role in β-cell destruction in nonobese diabetic mice (4, 6, 8). An alternative is that CD8+ T cell-induced β-cell destruction may be mediated by mTNF molecules borne by the activated CD8+ T cells. mTNF-mediated cytotoxicity has indeed been detected in cytotoxic assays in vitro, but only when the perforin and Fas pathways are not operative, namely by using CTL obtained from perforin and gld double mutant mice and by performing more prolonged assays, which suggest that this cytotoxic mechanism acts more slowly (36). However, T cell TNF synthesis starts a few hours after TCR stimulation in vitro (37) and probably well before a significant amount of perforin is produced, judging from cytotoxic granule development kinetics. We therefore reasoned that incompletely differentiated CD8+ T cells, resulting from limited antigenic stimulation in vivo, might be able to destroy β cells through mTNF.
Lack of Diabetes or Delayed Disease in Double Transgenic Mice Defective in TNFR II or TNF.
It has been proposed that the main target of mTNF is TNFR II (16), TNFR I being the main receptor for sTNF (38). Although the action of these receptors may in fact be more complex (17), it is relevant in this respect to note that stimulation of TNFR II may induce apoptosis in cells insensitive to sTNF (39). We thus generated TNFR II null mice bearing both TNF and CD80 transgenes. We did not anticipate any interference with the sTNF-mediated insulitis because the effect of TNF on endothelial cells has been reported to occur exclusively via TNFR I (19). None of the seven TNF/CD80/TNFR II −/− mice developed hyperglycemia by 6 wk of age, and only one became diabetic at 6 mo, the others remaining normoglycemic until euthanasia for histological analysis at 2.5 and 6.5 mo of age. Surprisingly however, two of these six nondiabetic mice had no insulitis, and a severe insulitis was observed in only one. Thus, although suggesting a role for TNFR II in the β-cell destructive process in this T1DM model, these data further indicate that the lack of TNFR II may in some way interfere with endothelial activation. Therefore, this approach did not allow a reliable and reproducible way of exploring the putative cytotoxic role of mTNF in this diabetes.
TNF/LTα knockout mice bearing the two transgenes were then studied. In such mice, the only source of TNF is the β cell, through the action of the RIP-TNF transgene; lymphocytes can express neither mTNF nor mLT (which is made of LT-α and β molecules). Sixteen of these mice, either normoglycemic or mildly hyperglycemic, were killed between 6 and 19 wk of age. It is essential to stress that all displayed insulitis comparable in kinetics, severity, and T cell composition to that observed in mice with the wild-type TNF/LTα gene locus. This suggests that, in spite of the TNF/LTα null mutation (whose main phenotype is the lack of lymph nodes, the development of which requires the local expression of LT; ref. 20), these mice have a normal capacity to present local antigen, perhaps because they develop distinct mesenteric nodes (see Results). Because hyperglycemia onset was delayed several weeks or months, these data strongly suggest that mTNF plays a dominant role in the acute β-cell destruction observed in mice with an intact TNF/LTα gene locus. sTNF cannot be incriminated because its expression in the islets through the transgene shows that it is not, by itself, toxic for β cells (12). mLT, acting through the LT-β receptor (40), is not likely to be significantly involved, because its action would not have been prevented in the TNF-R II mutant mice. This interpretation requires some caveats, however. mTNF and LT may be necessary to obtain full CTL maturation or the stimulation of other cells (such as dendritic cells), which could be essential for the eventual complete β-cell destruction. Furthermore, prolonged exposure to sTNF has been reported to attenuate TCR signaling in vitro (41). Such a putative delay in CTL maturation in the defective mice might be accentuated by lower TCR signaling, which would result from permanent exposure to β-cell-released sTNF. This might explain why, although strongly retarded, diabetes can nevertheless occur; the mechanisms involved in this protracted diabetes may thus be those that are also operative in early onset diabetes. However, ultrastructural and histologic observations were not consistent with this hypothesis: lesions of focal submembranous apoptosis, so characteristic of the nondefective transgenic mice, were not observed in several of these mice studied at different ages, and histologic sections showed an unusual degree of islet fibrosis, indicative of a more chronic progressive process. It is thus possible that β-cell damage in these mice is mediated not by direct CTL membrane-induced cytotoxicity but through the action of cytokines, such as IL-1 β, which leads in vitro to the destruction of β cells, or IFN-γ, both acting by generating reactive oxygen intermediates, in particular nitric oxide (1, 2). Cell damage created by excessive generation of reactive oxygen intermediates would indeed be more consistent with fibrotic scars than destruction by conventional apoptosis, whose characteristic is to leave very few traces. In conclusion, because the TNFR II and TNF-LTα mutations obviously have complex effects on the immune system, it is not possible, on the sole basis of the observations made with mice, to formally incriminate mTNF borne by CTL as the major mechanism of β-cell damage in this acute diabetes. Nevertheless, mTNF is the likeliest candidate, or one would have to invoke the action of new membrane molecules of the TNF family.
Why and How May Diabetogenic CTL Use mTNF Rather Than Perforin to Damage β Cells? Comparison with Other Models of Autoimmune Diabetes.
It is of special interest to compare the present model of diabetes with another experimental T1DM in which the antigen is well defined, namely the RIP-LCMV-GP transgenic studied by Zinkernagel, Hengartner, and their colleagues. These mice express in their pancreatic β cells the glycoprotein (GP) of the lymphocytic choriomeningitis virus (LCMV). They do not develop diabetes unless they are extrapancreatically stimulated by GP peptides, thus β cells in this situation act only as targets, not as antigenically stimulating cells (3, 5, 7, 24, 42–44). Inoculation of live LCMV represents a very strong antigenic challenge (42) in which CTLp stimulation is achieved by widely disseminated virally infected cells; most of these cells are not specialized antigen-presenting cells and bear no CD80 molecules, but high amounts of GP peptides. Activated CTL then induce diabetes by destroying the GP-bearing β cells by a mechanism that is entirely perforin dependent, because no diabetes occurs in transgenic mice that are also perforin defective (3, 5, 44). In the case of the RIP-TNF/CD80 transgenic mice, antigenic stimulation is very different because it occurs, or at least is initiated, in situ where CTLp present locally as the result of the TNF-mediated insulitis make repeated contacts with CD80-bearing β cells and putative autoantigenic peptides. These peptides are probably very diverse, and each is present in small amounts. The ensuing CTLp stimulation, initially made possible because of CD80 costimulation, may consequently be weak, leading to incompletely differentiated CTL that would express little or no perforin, but with a wide range of specificities, presumably much wider than in the case of the GP peptides, which elicit a very clonally restricted response.
In the contrasting conditions of antigenic stimulation between these two experimental T1DM, the rapid destruction of β cells probably results also from different mechanisms: in the case of GP-bearing β cells from a single lethal “kiss of death” given by highly differentiated CTLs acting through the release of perforin, and in the case of CD80-bearing β cells from infralethal but nevertheless harmful numerous hits of incompletely differentiated CTLs of diverse specificity acting through their increased expression of mTNF. This hypothesis of multiple sublethal hits, or “death by multiple little kisses,” is especially consistent with the striking observation of multiple submembranous sites of β-cell cytoplasmic apoptosis described above. On the other hand, when the RIP-GP transgenic mice are stimulated by repeated injections of dendritic cells bearing GP peptides (because of their expression of a GP transgene), diabetes can occur, depending on the timing and dose of the injections (7). In this situation, the antigenic stimulation is weaker than in LCMV-infected mice, as shown by a much lower CTL activity of, and CTLp frequency in, circulating lymphocytes. However, this protocol of immunization leads to a striking de novo formation of islet-associated lymphoid structures, necessary for diabetes to occur. In transgenic mice, also perforin defective, that are similarly stimulated, diabetes occurs in about half of the cases, showing that, in this model, diabetes is not entirely perforin dependent (7). It may be speculated that, in the cases where diabetes is perforin independent, its occurrence rests on the action of populations of CTLp expanded in the local lymphoid structures and acting through mTNF (not detectable in the conventional CTL and CTLp assays).
Might Mechanisms of mTNF-Mediated Cytotoxicity Exist in Other Conditions of Autoimmune Diabetes or of Autoimmunity in General?
Diabetes is largely perforin dependent in nonobese diabetic mice, because when perforin is lacking, the frequency of diabetes is markedly decreased and its appearance, when it occurs, markedly delayed (7). It may be argued that the transgenic expression of CD80, such as used in the present model, is but a very artificial condition to induce an immune response. However, constitutive expression of CD80 has been reported in a number of cell varieties, from keratinocytes (46) to thyroid follicular cells in patients with the autoimmune Hashimoto's disease, and CD80 can be induced in cells stimulated by both TNF and IFN-γ (45). It is thus conceivable that some conditions of prolonged local inflammation resulting in TNF release may prompt an autoimmune attack, for instance of epithelial cells in the thyroid gland or the skin, by some of the mechanisms discussed above. It has been reported in this respect that susceptibility to various autoimmune diseases is closely linked to the CD152 (formerly CTLA4) gene locus, which is a counter receptor for CD80 (46–50).
Acknowledgments
The authors are most grateful to Mr. Jordi Ritz for taking care of the different colonies of transgenic mice and to Ms. Danielle Ben-Nasr and Gissela Gallardo for performing histology and immunochemistry.
Abbreviations
- CTL
cytotoxic lymphocytes
- TNF
tumor necrosis factor
- LT
lymphotoxin
- T1DM
type 1 diabetes mellitus
- RIP
rat insulin promoter
- TUNEL
terminal deoxynucleotidyl-transferase-mediated dUTP nick-end labeling
- TNFR
TNF receptor
- mTNF
membrane TNF
- sTNF
soluble TNF
- ER
endoplasmic reticulum
- GP
glycoprotein
- LCMV
lymphocytic choriomeningitis virus
- lpr
lymphoproliferation
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Stassi G, De Maria R, Trucco G, Rudert W, Testi R, Galluzzo A, Giordano C, Trucco M. J Exp Med. 1997;186:1193–1200. doi: 10.1084/jem.186.8.1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Suarez-Pinzon W L, Szabo C, Rabinovitch A. Diabetes. 1997;46:907–911. doi: 10.2337/diab.46.5.907. [DOI] [PubMed] [Google Scholar]
- 3.Kagi D, Odermatt B, Ohashi P S, Zinkernagel R M, Hengartner H. J Exp Med. 1996;183:2143–2152. doi: 10.1084/jem.183.5.2143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Itoh N, Imagawa A, Hanafusa T, Waguri M, Yamamoto K, Iwahashi H, Moriwaki M, Nakajima H, Miyagawa J, Namba M, et al. J Exp Med. 1997;186:613–618. doi: 10.1084/jem.186.4.613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kagi D, Odermatt B, Seiler P, Zinkernagel R M, Mak T W, Hengartner H. J Exp Med. 1997;186:989–997. doi: 10.1084/jem.186.7.989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chervonsky A V, Wang Y, Wong F S, Visintin I, Flavell R A, Janeway C A, Jr, Matis L A. Cell. 1997;89:17–24. doi: 10.1016/s0092-8674(00)80178-6. [DOI] [PubMed] [Google Scholar]
- 7.Ludewig B, Odermatt B, Landmann S, Hengartner H, Zinkernagel R M. J Exp Med. 1998;188:1493–1501. doi: 10.1084/jem.188.8.1493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Allison J, Strasser A. Proc Natl Acad Sci USA. 1998;95:13818–13822. doi: 10.1073/pnas.95.23.13818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Herrera P L, Harlan D M, Fossati L, Izui S, Huarte J, Orci L, Vassalli J D, Vassalli P. Diabetologia. 1994;37:1277–1279. doi: 10.1007/BF00399802. [DOI] [PubMed] [Google Scholar]
- 10.Guerder S, Picarella D E, Linsley P S, Flavell R A. Proc Natl Acad Sci USA. 1994;91:5138–5142. doi: 10.1073/pnas.91.11.5138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Harlan D M, Barnett M A, Abe R, Pechhold K, Patterson N B, Gray G S, June C H. Diabetes. 1995;44:816–823. doi: 10.2337/diab.44.7.816. [DOI] [PubMed] [Google Scholar]
- 12.Higuchi Y, Herrera P, Muniesa P, Huarte J, Belin D, Ohashi P, Aichele P, Orci L, Vassalli J D, Vassalli P. J Exp Med. 1992;176:1719–1731. doi: 10.1084/jem.176.6.1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lillie R D, Fullmar H D. Histopathologic Technic and Practical Histochemistry. New York: McGraw–Hill; 1976. pp. 700–701. ; 714–715. [Google Scholar]
- 14.Lowin B, Beermann F, Schmidt A, Tschopp J. Proc Natl Acad Sci USA. 1994;91:11571–11575. doi: 10.1073/pnas.91.24.11571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Adachi M, Watanabe-Fukunaga R, Nagata S. Proc Natl Acad Sci USA. 1993;90:1756–1760. doi: 10.1073/pnas.90.5.1756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grell M, Douni E, Wajant H, Lohden M, Clauss M, Maxeiner B, Georgopoulos S, Lesslauer W, Kollias G, Pfizenmaier K, et al. Cell. 1995;83:793–802. doi: 10.1016/0092-8674(95)90192-2. [DOI] [PubMed] [Google Scholar]
- 17.Grell M, Zimmermann G, Gottfried E, Chen C-M, Grünwald U, Huang D C S, Wu Lee Y-H, Dürkop H, Engelmann H, Scheurich P, et al. EMBO J. 1999;18:3034–3043. doi: 10.1093/emboj/18.11.3034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Erickson S L, de Sauvage F J, Kikly K, Carver-Moore K, Pitts-Meek S, Gillett N, Sheehan K C, Schreiber R D, Goeddel D V, Moore M W. Nature (London) 1994;372:560–563. doi: 10.1038/372560a0. [DOI] [PubMed] [Google Scholar]
- 19.Mackay F, Loetscher H, Stueber D, Gehr G, Lesslauer W. J Exp Med. 1993;177:1277–1286. doi: 10.1084/jem.177.5.1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Eugster H P, Muller M, Karrer U, Car B D, Schnyder B, Eng V M, Woerly G, Le Hir M, di Padova F, Aguet M, et al. Int Immunol. 1996;8:23–36. doi: 10.1093/intimm/8.1.23. [DOI] [PubMed] [Google Scholar]
- 21.Koni P A, Sacca R, Lawton P, Browning J L, Ruddle N, Flavell R A. Immunity. 1997;6:491–500. doi: 10.1016/s1074-7613(00)80292-7. [DOI] [PubMed] [Google Scholar]
- 22.Jacobson M D, Burne J F, Raff M C. EMBO J. 1994;13:1899–1910. doi: 10.1002/j.1460-2075.1994.tb06459.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Campbell I L, Oxbrow L, West J, Harrison L C. Mol Endocrinol. 1988;2:101–107. doi: 10.1210/mend-2-2-101. [DOI] [PubMed] [Google Scholar]
- 24.Harlan D M, Hengartner H, Huang M L, Kang Y H, Abe R, Moreadith R W, Pircher H, Gray G S, Ohashi P S, Freeman G J, et al. Proc Natl Acad Sci USA. 1994;91:3137–3141. doi: 10.1073/pnas.91.8.3137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.O'Brien B A, Harmon B V, Cameron D P, Allan D J. Diabetes. 1997;46:750–757. doi: 10.2337/diab.46.5.750. [DOI] [PubMed] [Google Scholar]
- 26.Kurrer M O, Pakala S V, Hanson H L, Katz J D. Proc Natl Acad Sci USA. 1997;94:213–218. doi: 10.1073/pnas.94.1.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Augstein P, Elefanty A G, Allison J, Harrison L C. Diabetologia. 1998;41:1381–1388. doi: 10.1007/s001250051080. [DOI] [PubMed] [Google Scholar]
- 28.Augstein P, Stephens L A, Allison J, Elefanty A G, Ekberg M, Kay T W, Harrison L C. Mol Med. 1998;4:495–501. [PMC free article] [PubMed] [Google Scholar]
- 29.Giordano C, Richiusa P, Sbriglia M S, Pizzolanti G. Diabetes Metab Rev. 1998;14:194–195. doi: 10.1002/(sici)1099-0895(199806)14:2<194::aid-dmr8210>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 30.Schulze-Osthoff K, Walczak H, Droge W, Krammer P H. J Cell Biol. 1994;127:15–20. doi: 10.1083/jcb.127.1.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Woo M, Hakem R, Soengas M S, Duncan G S, Shahinian A, Kagi D, Hakem A, McCurrach M, Khoo W, Kaufman S A, et al. Genes Dev. 1998;12:806–819. doi: 10.1101/gad.12.6.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zheng T S, Schlosser S F, Dao T, Hingorani R, Crispe I N, Boyer J L, Flavell R A. Proc Natl Acad Sci USA. 1998;95:13618–13623. doi: 10.1073/pnas.95.23.13618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yamada K, Takane-Gyotoku N, Yuan X, Ichikawa F, Inada C, Nonaka K. Diabetologia. 1996;39:1306–1312. doi: 10.1007/s001250050574. [DOI] [PubMed] [Google Scholar]
- 34.Loweth A C, Williams G T, James R F, Scarpello J H, Morgan N G. Diabetes. 1998;47:727–732. doi: 10.2337/diabetes.47.5.727. [DOI] [PubMed] [Google Scholar]
- 35.Reimers J I. Dan Med Bull. 1998;45:157–180. [PubMed] [Google Scholar]
- 36.Lee R K, Spielman J, Zhao D Y, Olsen K J, Podack E R. J Immunol. 1996;157:1919–1925. [PubMed] [Google Scholar]
- 37.Millet I, Ruddle N H. J Immunol. 1994;152:4336–4346. [PubMed] [Google Scholar]
- 38.Grell M, Wajant H, Zimmermann G, Scheurich P. Proc Natl Acad Sci USA. 1998;95:570–575. doi: 10.1073/pnas.95.2.570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lazdins J K, Grell M, Walker M R, Woods-Cook K, Scheurich P, Pfizenmaier K. J Exp Med. 1997;185:81–90. doi: 10.1084/jem.185.1.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Force W R, Walter B N, Hession C, Tizard R, Kozak C A, Browning J L, Ware C F. J Immunol. 1995;155:5280–5288. [PubMed] [Google Scholar]
- 41.Cope A P, Liblau R S, Yang X D, Congia M, Laudanna C, Schreiber R D, Probert L, Kollias G, McDevitt H O. J Exp Med. 1997;185:1573–1584. doi: 10.1084/jem.185.9.1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ohashi P S, Oehen S, Buerki K, Pircher H, Ohashi C T, Odermatt B, Malissen B, Zinkernagel R M, Hengartner H. Cell. 1991;65:305–317. doi: 10.1016/0092-8674(91)90164-t. [DOI] [PubMed] [Google Scholar]
- 43.Ohashi P S, Oehen S, Aichele P, Pircher H, Odermatt B, Herrera P, Higuchi Y, Buerki K, Hengartner H, Zinkernagel R M. J Immunol. 1993;150:5185–5194. [PubMed] [Google Scholar]
- 44.Kagi D, Ledermann B, Burki K, Seiler P, Odermatt B, Olsen K J, Podack E R, Zinkernagel R M, Hengartner H. Nature (London) 1994;369:31–37. doi: 10.1038/369031a0. [DOI] [PubMed] [Google Scholar]
- 45.Pechhold K, Patterson N B, Craighead N, Lee K P, June C H, Harlan D M. J Immunol. 1997;158:4921–4929. [PubMed] [Google Scholar]
- 46.Yanagawa T, Hidaka Y, Guimaraes V, Soliman M, DeGroot L J. J Clin Endocrinol Metab. 1995;80:41–45. doi: 10.1210/jcem.80.1.7829637. [DOI] [PubMed] [Google Scholar]
- 47.Donner H, Braun J, Seidl C, Rau H, Finke R, Ventz M, Walfish P G, Usadel K H, Badenhoop K. J Clin Endocrinol Metab. 1997;82:4130–4132. doi: 10.1210/jcem.82.12.4406. [DOI] [PubMed] [Google Scholar]
- 48.Esposito L, Hill N J, Pritchard L E, Cucca F, Muxworthy C, Merriman M E, Wilson A, Julier C, Delepine M, Tuomilehto J, et al. Diabetes. 1998;47:1797–1799. doi: 10.2337/diabetes.47.11.1797. [DOI] [PubMed] [Google Scholar]
- 49.Donner H, Seidl C, Braun J, Siegmund T, Herwig J, Seifried E, Usadel K H, Badenhoop K. Diabetes. 1998;47:1158–1160. doi: 10.2337/diabetes.47.7.1158. [DOI] [PubMed] [Google Scholar]
- 50.Awata T, Kurihara S, Iitaka M, Takei S, Inoue I, Ishii C, Negishi K, Izumida T, Yoshida Y, Hagura R, et al. Diabetes. 1998;47:128–129. doi: 10.2337/diab.47.1.128. [DOI] [PubMed] [Google Scholar]