

Abstract
Mature myeloid cells (macrophages and CD11b+ dendritic cells) form a prominent component of neuroinflammatory infiltrates in multiple sclerosis and experimental autoimmune encephalomyelitis (EAE). The mechanism by which these cells are replenished during relapsing and chronic neuroinflammation is poorly understood. Here we demonstrate that CD11b+CD62L+Ly6Chi monocytes with colony-forming potential are mobilized into the bloodstream by a granulocyte-macrophage colony-stimulating factor-dependent pathway immediately before EAE relapses. Circulating Ly6Chi monocytes traffic across the blood-brain barrier, up-regulate proinflammatory molecules, and differentiate into central nervous system dendritic cells and macrophages. Enrichment of Ly6Chi monocytes in the circulating pool is associated with an earlier onset and increased severity of clinical EAE. Our studies indicate that granulocyte-macrophage colony-stimulating factor–driven release of Ly6Chi precursors from the bone marrow prevents exhaustion of central nervous system myeloid populations during relapsing or chronic autoimmune demyelination, suggesting a novel pathway for therapeutic targeting.
Introduction
Myeloid cells, such as macrophages and dendritic cells (DCs), are a prominent constituent of inflammatory infiltrates in the central nervous system (CNS) during multiple sclerosis (MS) and experimental autoimmune encephalomyelitis (EAE).1,2 These cells not only serve as antigen-presenting cells for the reactivation of infiltrating myelin-reactive CD4+ T cells but are thought to directly inflict tissue damage through secretion of toxic factors, such as reactive oxygen species, proteases, and tumor necrosis factor-α (TNF-α).3,4 They might also recruit naive myelin-reactive T cells into the effector pool in the context of epitope spreading.5 We and others have demonstrated that bone marrow–derived CD11c+ major histocompatibility complex (MHC) class II+ DCs accumulate in the CNS during EAE and have the capacity to polarize naive T cells along encephalitogenic Th1 and Th17 lineages.2,6 However, the circulating cell that gives rise to CNS-infiltrating DCs and macrophages has yet to be defined. The specific chemokine pathways and adhesion molecule interactions required for infiltration of the CNS by myeloid cells will depend on whether they cross the blood-brain barrier as immature monocytes or as macrophages and DCs. Therefore, identification of the differentiation status of the migrating cell holds implications regarding candidate therapeutic targets in neuroinflammatory diseases, such as multiple sclerosis (MS).
Under steady-state conditions, mature myeloid lineages are maintained within lymphoid and peripheral tissues through controlled release of bone marrow progenitors/precursors into the peripheral circulation.7 In the setting of infection or injury, myeloid cell mobilization is accelerated to meet the demands imposed by the increased turnover of macrophages and DCs at the site of inflammation.8,9 The pathways underlying expansion of peripheral myeloid cell pools under stress are thought to serve an adaptive role by reinforcing host protection against infectious agents and by promoting wound healing.8 Conversely, leukocyte-mobilizing pathways might be subverted to sustain target organ inflammation during relapsing or chronic autoimmune disease. For example, the number of macrophages and DCs in the CNS contracts during remissions and rebounds during exacerbations of EAE, suggesting that myeloid precursors might be released at a heightened rate before, or in concert with, clinical disease activity.10,11
Recently, it was shown that CCL2 expression by nonhematopoetic (likely glial) cells is important for the accumulation of proinflammatory DCs in the CNS during acute EAE.12 Furthermore, transgenic animals that simultaneously express CCL2 in the CNS and Fms-like tyrosine kinase 3 ligand in the periphery spontaneously develop meningeal and perivascular inflammation in association with an ascending paralysis. The neuroinflammation in this model appears to be primarily driven by myeloid cells and occurs independent of T and B lymphocytes.13 The receptor for CCL2, CCR2, is expressed on a subset of so-called inflammatory monocytes that express the cell surface phenotype CD62L+Ly6ChiCX3CR1low.14 These cells have been shown to traffic to sites of inflammation and to play a physiologic role in animal models of infection and atherosclerosis.14–16 We considered this subset of monocytes to be a logical candidate for the precursors of CNS DCs and/or macrophages during autoimmune demyelination.
In the current study, we found that the frequency of granulocyte/monocyte colony-forming units (GM-CFU) rises in the circulation of myelin-immunized mice immediately before clinical episodes of EAE in a granulocyte-macrophage colony-stimulating factor (GM-CSF)–dependent fashion. GM-CFU activity is contained within the CD11b+CD62L+Ly-6Chi subset of peripheral monocytes. Furthermore, we demonstrate that circulating CD11b+Ly-6Chi white blood cells migrate to the CNS during EAE and up-regulate CD11c and MHC class II in situ. EAE is more severe and occurs after a shorter latency under conditions favoring the enrichment of circulating CD11b+Ly-6Chi cells. Furthermore, administration of recombinant GM-CSF to GM-CSF–deficient animals triggers CD11b+Ly-6Chi mobilization and confers susceptibility to EAE. Collectively, our results indicate that a GM-CSF–dependent pathway is used during autoimmune demyelinating disease to augment the release of CD11b+Ly-6Chi cells from the bone marrow that, in turn, act as precursors of CNS DCs and macrophages.
Methods
Mice
SJL, C57BL/6, and B6-LY5.2/Cr mice were obtained from the National Cancer Institute (Frederick, MD). GM-CSF−/− mice, provided by Bruce Trapnell (Cincinnati Children's Hospital Medical Center, Cincinnati, OH), were bred in our laboratory. Animals were housed under specific pathogen-free conditions. All experiments were performed under University of Rochester Committee on Animal Resources and University of Michigan Committee on Animal Use and Care of Animals–approved protocols.
Antibodies and reagents
The following monoclonal antibodies were purchased for flow cytometry: Ly6C (AL-21), MHC class II (7-16.17; BD Biosciences, San Jose, CA); and CD11c (N418), CD11b (M1/70), F4/80 (BM8), CD62L (MEL-14); and CD115 (M-CSF R; AFS98; eBioscience, San Diego, CA). Anti-GM-CSF monoclonal antibody (clone 22E9.11; ATCC, Manassas, VA) was purified from hybridoma supernatants on protein G columns (GE Healthcare, Little Chalfont, United Kingdom).
Induction and assessment of EAE
For active immunization, mice were immunized subcutaneously over the flanks with 100 μg PLP139-151 (SJL) or MOG35-55 (C57BL/6) in complete Freund adjuvant (CFA) containing 250 μg Mycobacterium tuberculosis H37RA (Difco, Detroit, MI). C57BL/6 mice were injected intraperitoneally with 300 ng Bordetella pertussis toxin (List Biological Laboratories, Campbell, CA) on days 0 and 2 after immunization. For adoptive transfer, C57BL/6 mice were immunized with MOG35-55 in CFA (1:1) by the subcutaneous route, but without injection of pertussis toxin. Twelve days later, draining lymph nodes (inguinal and axillary) were removed and processed as previously described. Cells were cultured in standard medium with myelin peptide (50 μg/mL myelin oligodendrocyte glycoprotein [MOG]) and murine rIL-12 (5 ng/mL; R&D Systems, Minneapolis, MN). At 96 hours, cells were harvested, washed, counted by trypan blue exclusion, and injected into naive syngeneic recipients (16 × 106 cells) intraperitoneally. Clinical assessment of EAE was performed according to the following criteria: 0, no disease; 1, limp tail; 2, hind-limb weakness; 3, partial hind-limb paralysis; 4, complete paralysis of one or more limbs; and 5, moribund state.
Colony-forming unit assays
Peripheral blood was collected via the tail vein into BD microtainer tubes (BD Biosciences) coated with ethylenediaminetetraacetic acid. Red blood cells were lysed, and recovered leukocytes were plated in 40% methylcellulose cultures supplemented with recombinant GM-CSF (10 ng/mL) and stem cell factor (60 ng/mL; PeproTech, Rocky Hill, NJ). GM-CFU (defined by a cluster of 8 or more cells) were quantified after 7 days of culture.
CNS cell isolation
Mice were anesthetized with avertin and perfused with phosphate-buffered saline (PBS) by the intracardiac route. Spinal cords were minced and digested with 2 mg/mL of collagenase (CLS-4; Worthington Biochemical, Freehold, NJ) and 1 mg/mL DNase I (DN25; Sigma-Aldrich, St Louis, MO) for 1 hour at 37°C. Pooled spinal cord cells were layered over a discontinuous Percoll gradient, centrifuged for 20 minutes at 1455g, and collected from the 30%/70% interface.
Flow cytometric analysis
Peripheral blood and CNS mononuclear cells were isolated as described under “CNS cell isolation” and incubated with Fc Block (hybridoma 2.4G2; ATCC) followed by biotinylated or fluorochrome-conjugated antibodies for 25 minutes on ice. For secondary staining, cells were washed and incubated with peridinin chlorophyll protein– or allophycocyanin-conjugated streptavidin for an additional 25 minutes. Data acquisition was performed on a flow cytometer (FACSCalibur or FACS CantoII; BD Biosciences) and analyzed with FlowJo software (TreeStar, Ashland, OR). For cell sorting experiments, blood leukocytes were prepared from 20 donors, stained with the antibodies described, and sorted using a FACSAria (BD Biosciences). Some CD11b+Ly-6Chi blood cells were analyzed for nuclear morphology using the HEMA3 stain set (Fisher Scientific, Pittsburgh, PA).
Real-time reverse-transcribed–polymerase chain reaction
RNA was isolated with TRIZOL (Invitrogen, Carlsbad, CA) according to the manufacturer's protocol. cDNA was synthesized with a reverse transcription kit (QuantiTect; QIAGEN, Valencia, CA). Polymerase chain reaction (PCR) was performed using a single-color real-time PCR detection system (MyiQ; Bio-Rad, Hercules, CA). IL-12p40, IL-12p35, interleukin-10 (IL-10), TNF-α, and IL-6 primers and probes were designed with Beacon Designer software (Premier Biosoft International, Palo Alto, CA). IL-23p19, EBI3, inducible nitric oxide synthase (iNOS), and MMP9 primers and probes were purchased from Applied Biosystems (Foster City, CA). Samples were amplified over 40 cycles according to the following protocol: 15 seconds at 95°C, 1 minute at 60°C. Target gene expression was normalized to glyceraldehyde-3-phosphate dehydrogenase.
Treatment with clodronate liposomes
To enrich Ly6Chi cells during EAE, mice were injected intravenously with 0.25 mL clodronate liposomes (Roche Diagnostics, Mannheim, Germany) or PBS liposomes 24 hours after transfer of myelin-primed CD4+ lymph node cells.
To deplete monocytes during the effector phase of EAE, mice were injected with 0.25 mL of clodronate liposomes on days 8, 10, and 12 after transfer of lymph node cells.
Cl2MDP (or clodronate) was a gift of Roche Diagnostics.
Labeling of circulating Ly6Chi cells in vivo
Mice were injected with 0.25 mL of 0.5 μm Fluoresbrite fluorescein isothiocyanate (FITC)–dyed (YG) plain microspheres (2.5% solids [wt/vol]; Polysciences, Warrington, PA), diluted 1:25 in PBS, 20 hours after clodronate or PBS liposome treatment to label Ly-6C+ or Ly-6C− monocyte subsets, respectively.11
Statistical analysis
Real-time reverse-transcribed (RT) PCR data were analyzed by the pairwise fixed reallocation randomization test with REST-XL software, version 2. In all other cases, statistical significance was assessed with the unpaired Student t test.
Results
Mobilization of myeloid precursors is accelerated before clinical episodes of EAE
We bled myelin-immunized SJL mice at serial time points according to clinical stage and measured the frequency of GM-CFU in methylcellulose culture with GM-CSF and stem cell factor. As shown in Figure 1A, the frequency of circulating GM-CFU consistently rose shortly before the clinical onset and relapse of EAE but fell during remission. To identify the cellular source of GM-CFU in the circulation of myelin-immunized mice, methylcellulose cultures were set up with leukocytes sorted for cell surface profiles indicative of different maturation stages (Figure 1B).17 Relatively immature Ly6ChiCD11b+ cells, as opposed to Ly-6Cint or Ly-6Cneg cells, gave rise to GM-CFU at a high frequency (Figure 1C). The majority of colonies contained 8 to 50 cells, indicative of 3 to 5 cell divisions (I.L.K. and B.M.S., unpublished data, January 25, 2006). Ly-6Chi blood leukocytes had bean-shaped nuclei typical of the monocyte/macrophage lineage (Figure 1D) and uniformly expressed the macrophage colony-stimulating factor receptor, CD115 (Figure 1E). Ly-6Chi precursors also expressed CD62L, 7/4, and F4/80, but were Ly-6G−, indicative of an inflammatory monocyte phenotype (Figure 1E; and I.L.K. and B.M.S., unpublished data, March 23, 2008).17 Furthermore, the circulating Ly-6Chi cells have forward- and side-scatter characteristics that fall within a monocyte, as opposed to a granulocyte, gate (I.L.K. and B.M.S., unpublished data, March 23, 2008).
Figure 1.

Myeloid precursor cells expand in the blood before clinical episodes of EAE and are contained within the CD11b+Ly-6Chi population. (A) SJL mice were immunized with PLP139-151 in CFA. Mice were bled at various time points during relapsing-remitting EAE, and peripheral blood leukocytes were plated in methylcellulose cultures supplemented with GM-CSF and stem cell factor. GM-CFUs were counted on day 7. The data shown represent the mean (± SD) of 3 similar experiments. The frequency of GM-CFU at each time point represents the mean of at least 3 mice/group (*P < .05 comparing the frequency of GM-CFU before onset of EAE with their frequency during disease). (B) C57/B6 mice were immunized with MOG/CFA. Six days later, peripheral blood cells were stained for CD11b and Ly-6C and sorted into Ly-6Chi, Ly-6Cint, and Ly-6− subsets. (C) Sorted cells were plated in methylcellulose cultures as described in panel A, and GM-CFUs were counted on day 7. The data represent mean (± SE) of five experiments. (D) Hematoxylin and eosin staining of sorted CD11b+Ly-6Chi cells. (E) CD115, CD62L, and Ly6G levels were measured on gated CD11b+Ly-6Chi cells by flow cytometric analysis. Broken lines represent isotype controls. (B-E) Data are representative of at least 2 experiments.
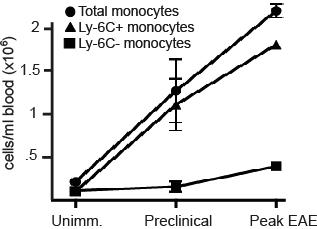
Consistent with the findings illustrated in Figure 1, the frequency of circulating Ly-6Chi cells increased in parallel to GM-CFU before and during EAE exacerbations. This was true both in PLP139-151-immunized SJL mice undergoing a relapsing-remitting course and MOG35-55-immunized C57BL/6 mice undergoing a chronic course (Figure 2A,B; Figure S1, available on the Blood website; see the Supplemental Materials link at the top of the online article). The characterization of Ly-6Chi monocytes as bona fide DC precursors was validated by their expression of MHC class II and CD11c in response to short-term stimulation with GM-CSF (Figure 2C).
Figure 2.

Ly-6C+ cells accumulate in the blood and CNS before the onset of EAE. (A) PBMCs from naive and PLP139-151-immunized SJL mice were analyzed by flow cytometry. Immunized mice were bled on the day before expected clinical onset and during peak EAE. Dot plots are gated on CD11b+CD115+ cells; percentages of each subset among total blood leukocytes are indicated. (B) Absolute numbers of circulating monocytes per milliliter of blood in naive and PLP139-151-immunized SJL mice (*P < .05, **P < .02 by comparison to naive). (C) Ly-6C+ monocytes were sorted from immunized SJL mice and analyzed for MHC class II and CD11c expression either immediately ex vivo or after a 48-hour culture with GM-CSF. Data shown are representative of 5 separate experiments. (D) Spinal cord mononuclear cells were harvested from naive and PLP139-151-immunized SJL mice and subjected to flow cytometric analysis. The lowest panels show CD11c and MHC class II staining of CD11b+LyChi-gated PBMCs from mice in the preclinical (left) or symptomatic (right) stages of EAE. The gates used are depicted in the middle panels. Data shown are representative of 4 separate experiments.
Ly-6Chi CD11b+ cells first began accumulating in the blood and CNS during the preclinical stage of disease (Figure 2A,D). Immediately before the onset of clinical disease, the majority of Ly-6Chi CD11b+ cells in the spinal cord were CD11c− MHC class II−/lo. However, the CNS Ly-6Chi CD11b+ cell population was predominantly CD11c+ and MHC class IIlo/hi during the symptomatic phase (Figure 2D bottom panels). Hence, immature monocytes that accumulate in the CNS during the preclinical stage either differentiate in situ or are replaced by a more mature subset as EAE evolves.
Ly-6C+ monocytes traffic to the CNS and express a proinflammatory phenotype
The aforementioned studies show that Ly-6Chi CD11b+ cells are mobilized into the circulation at an increased rate before EAE exacerbations, accumulate in the CNS during the preclinical phase of EAE, and differentiate into DCs on stimulation in vitro. We speculated that circulating Ly-6Chi cells home to the CNS and give rise to mature myeloid populations during EAE. To directly test that hypothesis, we capitalized on the previous observation that peripheral blood monocytes, transiently depleted with clodronate liposomes, are replaced within 18 to 24 hours by bone marrow mononuclear cells that are heavily enriched in the Ly-6Chi CD11b+ subset.18
Under homeostatic conditions, intravenous injection of FITC-conjugated microspheres results in preferential labeling of Ly6C− CD11b+ cells because of their relatively high phagocytic capacity. By contrast, the microspheres are selectively taken up by Ly6Chi cells when injected into clodronate-treated mice at the time of peripheral leukocyte reconstitution.18 We injected host mice with clodronate liposomes and FITC-conjugated microspheres 24 and 42 hours after the adoptive transfer of myelin-specific T cells, respectively. Subsequent fluorescence-activated cell sorter analysis confirmed that the majority of FITC+ peripheral blood mononuclear cells (PBMCs) from the clodronate-treated group were CD11b+Ly-6Chi, whereas FITC+ PBMCs from control mice treated with PBS liposomes were predominantly CD11b+Ly-6C− (Figure 3A).
Figure 3.

Ly-6C+ monocytes migrate to the CNS during EAE and up-regulate CD11c and MHC class II. Mice were treated with clodronate or PBS liposomes 24 hours after transfer of encephalitogenic T cells. Eighteen hours after liposome treatment, animals were injected with FITC-labeled microspheres. (A) Left: Expression of FITC and CD11b by CD115+-gated cells 5 days after liposome treatment and 4 days after FITC microsphere injection. Right: Ly-6C expression on FITC+CD115+ blood monocytes from clodronate versus PBS liposome-treated animals. Histograms are based on cells that fall within the R1 gate (left panel). (B) Spinal cord mononuclear cells were harvested from mice treated with clodronate (left) or PBS liposome (right) during peak EAE and analyzed for FITC expression. (C) The cell surface phenotype of CNS-infiltrating FITC+ cells (shown in the gate of panel B left) was determined by FACS. (D) CD11b+MHC class II+Ly-6C+ cells were sorted from the blood and CNS of mice with EAE and analyzed by real-time RT-PCR for the genes shown. The data are shown as fold expression in CNS-infiltrating cells over circulating cells. (A-D) All experiments shown were repeated 3 times with similar results. *P < .05, comparing mRNA levels in CNS versus blood Ly-6C+ monocytes.
By the time of peak EAE, a significant proportion of FITC+ cells had infiltrated the CNS of the adoptive transfer hosts that received clodronate liposomes, but not the CNS of hosts that had received PBS liposomes (Figure 3B). Virtually all of the CNS-infiltrating FITC+ cells were F4/80+Ly6Chi MHC class IIhi and CD11cint or CD11c− (Figure 3C), implying that circulating Ly-6Chi CD11b+ cells give rise to the mature macrophages and myeloid DCs that populate EAE lesions. These results were corroborated by parallel studies in which CD45.2+ Ly-6Chi blood monocytes, transferred into CD45.1 congenic hosts with active EAE, developed into Ly6Chi MHC class IIhi CD11cint/CD11c− CNS mononuclear cells (Figure S2).
By comparison to Ly-6Chi cells in the blood, Ly-6C+MHC class II+ cells in the CNS up-regulated mRNA encoding p40 (the common subunit of IL-12 and IL-23), p19 (the IL-23–specific subunit), and IL-6 (Figure 3D).19 Each of these genes has previously been shown to be indispensable for the induction of EAE. CNS Ly-6C+MHC class II+ cells also expressed elevated levels of TNF-α and iNOS (Figure 3D). Collectively, these data demonstrate that Ly-6Chi blood monocytes are a source of mature CNS-infiltrating myeloid cells during EAE and that they acquire a proinflammatory genetic profile having crossed the blood-brain barrier.
Increasing the frequency of Ly-6Chi precursors during the effector phase of EAE accelerates the onset and severity of disease
As previously described, intravenous treatment of mice with with clodronate liposomes results in enrichment of Ly-6C+ cells in the circulating monocyte pool but does not affect other leukocyte subsets.18 Ly6Chi cells comprise the majority of CD115+ cells in the blood for 7 to 10 days after the intervention (Figure 4A). They are CD11b+F4/80+CD62L+Ly6G−, consistent with the cell surface profile of inflammatory monocytes. Based on our data that Ly6Chi blood cells develop into CNS Ly-6C+MHC class II+ cells with a proinflammatory profile, we speculated that administration of clodronate liposomes shortly after adoptive transfer would exacerbate the clinical course of EAE.
Figure 4.

Enrichment of Ly-6C+ cells in the circulating monocyte pool enhances EAE. (A) The absolute number of total monocytes and Ly6C+ monocytes per milliliter of blood in adoptive transfer recipients at serial time points after treatment with clodronate liposomes. (B) The absolute number of circulating Ly-6C+ monocytes per milliliter of blood 5 days after treatment with either clodronate or PBS liposomes. Mean (± SD) of 5 independent experiments; **P < .01 comparing frequency of Ly-6C+ monocytes in PBS versus clodronate treated mice. (C) Clinical course of mice treated with a single dose of clodronate or PBS liposomes 18 hours after the adoptive transfer of encephalitogenic cells. Mice in a third group received clodronate liposomes on days 8, 10, and 12 after T-cell transfer. Data shown represent the results from 3 separate experiments. Mean (± SD) of 3 independent experiments; P < .05 by comparison to PBS treated group.
Mice were injected with clodronate or PBS liposomes 18 hours after receiving encephalitogenic CD4+ T cells. As shown in Figure 4A, clodronate liposome treatment initially depleted all circulating CD115+ cells. Newly mobilized monocytes, which were skewed toward the immature Ly-6C+ subset, reconstituted the blood within 2 days. Consequently, from day 3 onward, clodronate liposome-treated mice had a significantly higher number of Ly-6C+ cells per milliliter of blood than PBS liposome-treated controls (Figure 4B and I.L.K. and B.M.S., unpublished data, June 1, 2008). Consistent with our hypothesis, administration of clodronate liposomes shortly after adoptive transfer resulted in an accelerated and more severe clinical course (Figure 4C). By contrast, repetitive treatment with clodronate liposomes beginning 8 days after T-cell transfer (a regimen that wholly depletes all monocytes from the circulation through out the effector stage) suppressed EAE and delayed its onset (Figure 4C).
GM-CSF triggers accelerated myelopoiesis during EAE
GM-CSF–deficient (−/−) mice and wild-type (WT) mice treated with neutralizing antibodies to GM-CSF are resistant to EAE.20 MOG-immunized GM-CSF−/− mice mount reduced IL-2 and interferon-γ recall responses, indicating that the cytokine plays a role in autoreactive CD4+ T-cell priming and/or Th differentiation, probably via indirect effects on antigen-presenting cells. However, GM-CSF has pleiotrophic functions, suggesting that it may act at multiple steps in autoimmune pathogenesis. It is well established that GM-CSF stimulates the mobilization of myeloid cells from the bone marrow during inflammation.21 Based on our earlier data, we speculated that one mechanism by which GM-CSF promotes EAE is to accelerate the release of bone marrow Ly-6Chi precursors that ultimately differentiate into CNS-infiltrating DCs and macrophages. Indeed, although circulating Ly-6C+ monocytes expanded more than 60-fold immediately before expected EAE onset in WT mice, their levels remained stable in GM-CSF−/− mice that were actively immunized in an identical fashion (Figure 5A). Similar results were obtained when myelin-immunized WT mice were treated with neutralizing antibodies to GM-CSF (Figure 5B). Conversely, administration of recombinant GM-CSF to immunized GM-CSF−/− mice triggered Ly-6Chi monocyte mobilization and restored susceptibility to EAE (Figure 5C).
Figure 5.

GM-CSF triggers accelerated myelopoiesis during EAE. (A) CD11b+CD115+Ly-6C+ or Ly-6C− blood cells (left) were enumerated 7 days after immunization of C57BL/6 WT and GM-CSF−/− mice with MOG peptide in CFA. The data are presented as the fold increase in each subset over their frequency in unimmunized counterparts. Frequencies of circulating Ly-6C+ and Ly-6C− monocytes did not differ significantly between unimmunized WT and unimmunized GM-CSF−/− mice. Clinical scores (right) of MOG-immunized WT and GM-CSF−/− mice. (B) Left: The number of circulating Ly-6C+ monocytes/mL of blood in anti–GM-CSF versus control antibody-treated WT mice on day 7 after immunization with MOG peptide. Mean (± SD) of 4 independent experiments; *P < .05 comparing frequency of Ly-6C+ monocytes in rat IgG versus αGM-CSF treated mice. Right: Clinical scores of MOG-immunized WT mice treated with either control antibody or anti–GM-CSF across a range of doses. (C) Left: Frequency of Ly6C+ blood monocytes on day 8 after active immunization of WT or GM-CSF−/− mice. Some GM-CSF−/− mice received 5 μg of recombinant mGM-CSF 8 hours before phlebotomy. Mean (± SD) of 3 independent experiments; *P < .05, **P < .01 by comparison to GM−/− mice. Right: Clinical scores of MOG-immunized WT and GM-CSF−/− mice. Some GM-CSF−/− mice received 5 μg of rmGM-CSF every day from days 0 to 16 after immunization.
Discussion
Numerous studies have demonstrated the importance of phagocytic cells, and CD11b+ DCs in particular, for the formation of CNS infiltrates and the manifestation of neurologic deficits during EAE. Accumulating evidence implicates myeloid DC in the pathogenesis of MS as well.1–3 EAE occurs in mice with MHC class II expression restricted to CD11c+ cells.3 Hence, DCs alone are sufficient for the priming and expansion of encephalitogenic T cells in vivo. Furthermore, we and others have found that CD11c+, but not CD11c−, CD11b+ cells isolated from the CNS of mice with EAE are particularly well equipped to polarize myelin-specific T cells toward the Th1 and Th17 pathways.2,22 These cells have also been implicated in epitope spreading in the context of relapsing disease.5 In addition, macrophages can secrete toxic factors and serve as local antigen-presenting cell in the CNS.4,23 Despite the critical role of mature myeloid cells in neuroinflammation, their origin is not well understood.
In the past, researchers have proposed that CNS DCs arise from transformed microglia based on in vitro experiments in which microglia up-regulated CD11c and acquired DC-like morphology after stimulation with GM-CSF.24 However, studies with bone marrow chimeric mice have shown that the majority of DCs in EAE lesions are derived from radiosensitive hematopoietic cells.2,3 Before the current report, the phenotype and maturation stage of the circulating cell that crosses the blood-brain barrier to become a CNS DC was unknown. Here we establish that CD11b+CD115+Ly-6Chi blood monocytes are a precursor of CNS DCs and macrophages in EAE lesions. These cells are dynamically regulated during the course of EAE, accumulating in the blood and CNS immediately before clinical episodes. Several groups have found that circulating monocytes can give rise to CD11b+CD45lo microglia in other experimental systems.25,26 We did not find that to be the case in our EAE model.
Although Ly-6C− monocytes migrate to the CNS under steady-state conditions, they do not tend to home to sites of inflammation.14 Therefore, we focused our studies on CD11b+Ly-6Chi precursors. However, our findings do not exclude a parallel role of Ly-6C− blood monocytes in EAE. A primitive c-Kit+CX3CR1+ bone marrow progenitor has been described that exclusively gives rise to macrophages and DCs (MDPs) in the lung and intestinal lamina propria during homeostasis.27 We are currently investigating whether such alternative cell types also migrate to CNS demyelinating lesions.
There are conflicting data in the literature as to whether CD11b+ Ly6Chi (or Gr-1+) cells act as regulators or inducers of inflammation. This controversy is reflected in the paradoxical use of the terms “myeloid derived suppressor cells” and “inflammatory monocytes” to describe cells bearing those markers.14,28 The former term has been most widely used in studies of antitumor immunity and allograft tolerance in reference to GM-CSF–driven CD11b+ Gr-1+ cells that inhibit protective CD8+ T-cell responses and/or trigger T-cell apoptosis.28 An analogous population of myeloid cells, expressing the phenotype CD11b+Ly6ChiF4/80+CD11c−, was recently found to expand in spleens of mice with EAE.29 Consistent with the cancer immunity literature, those cells suppressed polyclonal CD4+ and CD8+ T-cell proliferation and cytokine production and induced CD4+ T-cell apoptosis in vitro via an NOS2/arginase 1–dependent pathway.29 Of note, the authors did not determine whether their myeloid derived suppressor cells trafficked to the CNS, nor did they assess their effect on the clinical course.
On the other hand, Geissmann et al performed adoptive transfer studies to show that CD11b+CCR2+CD62l+Gr-1+ cells are selectively recruited to sites of inflammation (in this case, thioglycollate-induced peritonitis) and differentiate into DCs in situ.14 Similarly, CD11b+Ly6Chi cells traffic to sites of Listeria infection in response to CCR2 ligands and develop into TNF-α and iNOS-producing DCs, which are critical for efficient bacterial clearance.16,30 Furthermore, CD11b+Ly6Chi cells migrate to the CNS during Listeria infection.31 In our studies, CD11b+ Ly-6Chi cells in the circulation of myelin-immunized mice behaved in a manner more reminiscent of inflammatory monocytes than myeloid derived suppressor cells. They uniformly expressed CD62L (Figure 1E) and preferentially homed to the inflamed CNS on transfer into hosts with active EAE. On transgressing the blood-brain barrier, a significant percentage of these monocytes up-regulated CD11c as well as TNF-α and iNOS, thereby acquiring characteristics of TNF-α and iNOS-producing DCs.30 More importantly, enrichment/augmented mobilization of Ly-6Chi cells in the circulation (either by treatment of WT mice with clodronate liposomes or reconstitution of GM-CSF−/− mice with recombinant GM-CSF) exacerbated clinical EAE. Similarly, the administration of G-CSF to accelerate myelopoiesis in MS patients after bone marrow transplantation was associated with severe disease exacerbation.32,33
A unifying explanation of the aforementioned data is that recently exported CD11b+Ly6Chi cells are at a stage in which their differentiation potential remains flexible, allowing them to acquire opposing biologic functions based on signals from the local microenvironment. For example, there is evidence that neoplastic cells secrete factors that favor the differentiation of tumor-infiltrating monocytes into myeloid derived suppressor cells in situ.28 Conversely, we propose that the microenvironment established in the CNS during active EAE is conducive to development of proinflammatory DCs that promote Th1 and Th17 polarization. It is possible that the CNS microenvironment evolves during the course of disease, such that infiltrating CD11b+Ly6Chi are induced to adopt an innocuous, or even immunosuppressive, phenotype at times close to clinical remission. Indeed, we have previously found that CNS DCs lose immunostimulatory properties immediately before the onset of the remission phase.2
Our results provide insights into the mechanism of resistance of CCR2-deficient and CD62L-deficient mice to EAE, in that expression of both of those molecules by circulating CD11b+Ly6Chi cells could facilitate their migration to the CNS during active EAE.34,35 In support of this theory, reconstitution of CD62L-deficient mice with WT monocytes restored susceptibility.35 Resistance of GM-CSF–deficient mice may be, in part, secondary to impaired mobilization of CD11b+Ly6Chi cells from the bone marrow.16 By extension, agents that specifically block CD11b+Ly6Chi cells from trafficking into the circulation and/or peripheral tissues might be therapeutically useful in autoimmune diseases, such as MS, when administered during lesion formation. These agents probably impede the access of inflammatory monocytes to the target organ and their subsequent development into Th1/Th17-polarizing DCs. However, caution must be exercised in that the very same intervention could theoretically exacerbate disease by suppressing myeloid-derived suppressor activity if administered close to the time of remissions.
Supplementary Material
Acknowledgments
The authors thank Dr James Palis for advice and Anne Koniski for technical assistance in setting up methylcellulose CFU-GM assays.
This study was supported by the National Multiple Sclerosis Society (grant RG 3866-A-3) and the National Institutes of Health (National Institute of Neurological Disorders and Stroke, NS047687-01A1). I.L.K. is a PhD candidate at the University of Rochester, and this work is submitted in partial fulfillment of the requirement for the PhD.
Footnotes
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: B.M.S. oversaw the design of all experiments and interpretation of the data that was generated and prepared the final manuscript; I.L.K. performed all experiments, contributed to their design and the interpretation of results, made the figures, and wrote the manuscript; and T.L.D. assisted with all experimental studies and helped in the development of protocols.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Benjamin M. Segal, Holtom-Garrett Program in Neuroimmunology, Department of Neurology, University of Michigan, Ann Arbor, MI 48109; e-mail: bmsegal@umich.edu.
References
- 1.Serafini B, Rosicarelli B, Magliozzi R, et al. Dendritic cells in multiple sclerosis lesions: maturation stage, myelin uptake, and interaction with proliferating T cells. J Neuropathol Exp Neurol. 2006;65:124–141. doi: 10.1097/01.jnen.0000199572.96472.1c. [DOI] [PubMed] [Google Scholar]
- 2.Deshpande P, King IL, Segal BM. Cutting edge: CNS CD11c+ cells from mice with encephalomyelitis polarize Th17 cells and support CD25+CD4+ T cell-mediated immunosuppression, suggesting dual roles in the disease process. J Immunol. 2007;178:6695–6699. doi: 10.4049/jimmunol.178.11.6695. [DOI] [PubMed] [Google Scholar]
- 3.Greter M, Heppner FL, Lemos MP, et al. Dendritic cells permit immune invasion of the CNS in an animal model of multiple sclerosis. Nat Med. 2005;11:328–334. doi: 10.1038/nm1197. [DOI] [PubMed] [Google Scholar]
- 4.Segal BM. CNS chemokines, cytokines, and dendritic cells in autoimmune demyelination. J Neurol Sci. 2005;228:210–214. doi: 10.1016/j.jns.2004.10.014. [DOI] [PubMed] [Google Scholar]
- 5.McMahon EJ, Bailey SL, Castenada CV, Waldner H, Miller SD. Epitope spreading initiates in the CNS in two mouse models of multiple sclerosis. Nat Med. 2005;11:335–339. doi: 10.1038/nm1202. [DOI] [PubMed] [Google Scholar]
- 6.Miller SD, McMahon EJ, Schreiner B, Bailey SL. Antigen presentation in the CNS by myeloid dendritic cells drives progression of relapsing experimental autoimmune encephalomyelitis. Ann N Y Acad Sci. 2007;1103:179–191. doi: 10.1196/annals.1394.023. [DOI] [PubMed] [Google Scholar]
- 7.Liu K, Waskow C, Liu X, Yao K, Hoh J, Nussenzweig M. Origin of dendritic cells in peripheral lymphoid organs of mice. Nat Immunol. 2007;8:578–583. doi: 10.1038/ni1462. [DOI] [PubMed] [Google Scholar]
- 8.Dunay IR, Damatta RA, Fux B, et al. Gr1(+) inflammatory monocytes are required for mucosal resistance to the pathogen Toxoplasma gondii. Immunity. 2008;29:306–317. doi: 10.1016/j.immuni.2008.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nahrendorf M, Swirski FK, Aikawa E, et al. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J Exp Med. 2007;204:3037–3047. doi: 10.1084/jem.20070885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Begolka WS, Vanderlugt CL, Rahbe SM, Miller SD. Differential expression of inflammatory cytokines parallels progression of central nervous system pathology in two clinically distinct models of multiple sclerosis. J Immunol. 1998;161:4437–4446. [PubMed] [Google Scholar]
- 11.Jee Y, Yoon WK, Okura Y, Tanuma N, Matsumoto Y. Upregulation of monocyte chemotactic protein-1 and CC chemokine receptor 2 in the central nervous system is closely associated with relapse of autoimmune encephalomyelitis in Lewis rats. J Neuroimmunol. 2002;128:49–57. doi: 10.1016/s0165-5728(02)00147-9. [DOI] [PubMed] [Google Scholar]
- 12.Dogan RN, Elhofy A, Karpus WJ. Production of CCL2 by central nervous system cells regulates development of murine experimental autoimmune encephalomyelitis through the recruitment of TNF- and iNOS-expressing macrophages and myeloid dendritic cells. J Immunol. 2008;180:7376–7384. doi: 10.4049/jimmunol.180.11.7376. [DOI] [PubMed] [Google Scholar]
- 13.Furtado GC, Pina B, Tacke F, et al. A novel model of demyelinating encephalomyelitis induced by monocytes and dendritic cells. J Immunol. 2006;177:6871–6879. doi: 10.4049/jimmunol.177.10.6871. [DOI] [PubMed] [Google Scholar]
- 14.Geissmann F, Jung S, Littman DR. Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity. 2003;19:71–82. doi: 10.1016/s1074-7613(03)00174-2. [DOI] [PubMed] [Google Scholar]
- 15.Tacke F, Alvarez D, Kaplan TJ, et al. Monocyte subsets differentially employ CCR2, CCR5, and CX3CR1 to accumulate within atherosclerotic plaques. J Clin Invest. 2007;117:185–194. doi: 10.1172/JCI28549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Serbina NV, Pamer EG. Monocyte emigration from bone marrow during bacterial infection requires signals mediated by chemokine receptor CCR2. Nat Immunol. 2006;7:311–317. doi: 10.1038/ni1309. [DOI] [PubMed] [Google Scholar]
- 17.Sunderkotter C, Nikolic T, Dillon MJ, et al. Subpopulations of mouse blood monocytes differ in maturation stage and inflammatory response. J Immunol. 2004;172:4410–4417. doi: 10.4049/jimmunol.172.7.4410. [DOI] [PubMed] [Google Scholar]
- 18.Tacke F, Ginhoux F, Jakubzick C, van Rooijen N, Merad M, Randolph GJ. Immature monocytes acquire antigens from other cells in the bone marrow and present them to T cells after maturing in the periphery. J Exp Med. 2006;203:583–597. doi: 10.1084/jem.20052119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Oppmann B, Lesley R, Blom B, et al. Novel p19 protein engages IL-12p40 to form a cytokine, IL-23, with biological activities similar as well as distinct from IL-12. Immunity. 2000;13:715–725. doi: 10.1016/s1074-7613(00)00070-4. [DOI] [PubMed] [Google Scholar]
- 20.McQualter JL, Darwiche R, Ewing C, et al. Granulocyte macrophage colony-stimulating factor: a new putative therapeutic target in multiple sclerosis. J Exp Med. 2001;194:873–882. doi: 10.1084/jem.194.7.873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hamilton JA. GM-CSF in inflammation and autoimmunity. Trends Immunol. 2002;23:403–408. doi: 10.1016/s1471-4906(02)02260-3. [DOI] [PubMed] [Google Scholar]
- 22.Bailey SL, Schreiner B, McMahon EJ, Miller SD. CNS myeloid DCs presenting endogenous myelin peptides “preferentially” polarize CD4+ T(H)-17 cells in relapsing EAE. Nat Immunol. 2007;8:172–180. doi: 10.1038/ni1430. [DOI] [PubMed] [Google Scholar]
- 23.Hickey WF, Kimura H. Perivascular microglial cells of the CNS are bone marrow-derived and present antigen in vivo. Science. 1988;239:290–292. doi: 10.1126/science.3276004. [DOI] [PubMed] [Google Scholar]
- 24.Re F, Belyanskaya SL, Riese RJ, et al. Granulocyte-macrophage colony-stimulating factor induces an expression program in neonatal microglia that primes them for antigen presentation. J Immunol. 2002;169:2264–2273. doi: 10.4049/jimmunol.169.5.2264. [DOI] [PubMed] [Google Scholar]
- 25.Ajami B, Bennett JL, Krieger C, Tetzlaff W, Rossi FM. Local self-renewal can sustain CNS microglia maintenance and function throughout adult life. Nat Neurosci. 2007;10:1538–1543. doi: 10.1038/nn2014. [DOI] [PubMed] [Google Scholar]
- 26.Mildner A, Schmidt H, Nitsche M, et al. Microglia in the adult brain arise from Ly-6ChiCCR2+ monocytes only under defined host conditions. Nat Neurosci. 2007;10:1544–1553. doi: 10.1038/nn2015. [DOI] [PubMed] [Google Scholar]
- 27.Fogg DK, Sibon C, Miled C, et al. A clonogenic bone marrow progenitor specific for macrophages and dendritic cells. Science. 2006;311:83–87. doi: 10.1126/science.1117729. [DOI] [PubMed] [Google Scholar]
- 28.Nagaraj S, Gabrilovich DI. Tumor escape mechanism governed by myeloid-derived suppressor cells. Cancer Res. 2008;68:2561–2563. doi: 10.1158/0008-5472.CAN-07-6229. [DOI] [PubMed] [Google Scholar]
- 29.Zhu B, Bando Y, Xiao S, et al. CD11b+Ly-6C(hi) suppressive monocytes in experimental autoimmune encephalomyelitis. J Immunol. 2007;179:5228–5237. doi: 10.4049/jimmunol.179.8.5228. [DOI] [PubMed] [Google Scholar]
- 30.Serbina NV, Salazar-Mather TP, Biron CA, Kuziel WA, Pamer EG. TNF/iNOS-producing dendritic cells mediate innate immune defense against bacterial infection. Immunity. 2003;19:59–70. doi: 10.1016/s1074-7613(03)00171-7. [DOI] [PubMed] [Google Scholar]
- 31.Drevets DA, Dillon MJ, Schawang JS, et al. The Ly-6Chigh monocyte subpopulation transports Listeria monocytogenes into the brain during systemic infection of mice. J Immunol. 2004;172:4418–4424. doi: 10.4049/jimmunol.172.7.4418. [DOI] [PubMed] [Google Scholar]
- 32.Nash RA, Bowen JD, McSweeney PA, et al. High-dose immunosuppressive therapy and autologous peripheral blood stem cell transplantation for severe multiple sclerosis. Blood. 2003;102:2364–2372. doi: 10.1182/blood-2002-12-3908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Openshaw H, Stuve O, Antel JP, et al. Multiple sclerosis flares associated with recombinant granulocyte colony-stimulating factor. Neurology. 2000;54:2147–2150. doi: 10.1212/wnl.54.11.2147. [DOI] [PubMed] [Google Scholar]
- 34.Fife BT, Huffnagle GB, Kuziel WA, Karpus WJ. CC chemokine receptor 2 is critical for induction of experimental autoimmune encephalomyelitis. J Exp Med. 2000;192:899–905. doi: 10.1084/jem.192.6.899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Grewal IS, Foellmer HG, Grewal KD, et al. CD62L is required on effector cells for local interactions in the CNS to cause myelin damage in experimental allergic encephalomyelitis. Immunity. 2001;14:291–302. doi: 10.1016/s1074-7613(01)00110-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}