

Abstract
Reactive oxygen species (ROS) have been implicated as one of the agents responsible for many neurodegenerative diseases. A critical target for ROS is DNA. Most oxidative stress-induced DNA damage in the nucleus and mitochondria is removed by the base excision repair pathway. Apn1 is a yeast enzyme in this pathway which possesses a wider substrate specificity and greater enzyme activity than its mammalian counterpart for removing DNA damage, making it a good therapeutic candidate. For this study we targeted Apn1 to mitochondria in a neuronal cell line derived from the substantia nigra by using a mitochondrial targeting signal (MTS) in an effort to hasten the removal of DNA damage and thereby protect these cells. We found that following oxidative stress, mitochondrial DNA (mtDNA) was repaired more efficiently in cells containing Apn1 with the MTS than controls. There was no difference in nuclear repair. However, cells that expressed Apn1 without the MTS showed enhanced repair of both nuclear and mtDNA. Both Apn1-expressing cells were more resistant to cell death following oxidative stress compared with controls. Therefore, these results reveal that the expression of Apn1 in neurons may be of potential therapeutic benefit for treating patients with specific neurodegenerative diseases.
Keywords: DNA repair, gene therapy, mitochondrial DNA, neuronal cell line, yeast apurinic/apyrimidinic endonuclease
The level of reactive oxygen species (ROS), a byproduct of oxidative phosphorylation, has been found to be elevated in cells from postmortem specimens taken from patients with chronic neurodegenerative diseases (Jenner 2003; Wu et al. 2003). ROS can pose a threat to cellular homeostasis, function and lifespan by oxidizing lipids (Gutteridge and Halliwell 1990), proteins (Starke-Reed and Oliver 1989), and nucleic acids (Dizdaroglu 1992). With respect to nucleic acids the amount of DNA adducts has been found to be increased (Jenner 2003), which suggests that ROS-induced DNA damage may be at least in part responsible for neurodegeneration. Of the DNA found in the cell, it has been reported that mitochondrial DNA (mtDNA) is more sensitive to oxidative stress than nuclear DNA (nDNA) (Yakes and Van Houten 1997), likely because of its close proximity to the production of ROS. Although some investigators have disputed this increased sensitivity (Lim et al. 2005), it is clear that there are various consequences which can result from the failure to protect mtDNA against ROS exposure or repair the resulting damage. These include the induction of mutations, altered gene expression and, eventually, cytostasis, cytotoxicity, or neoplastic growth (Wallace 1994, 2005; Evans and Cooke 2004). Thus, it is readily apparent that damage to mtDNA could have significant functional consequences. Therefore, it is essential that damage in both nuclear and mitochondrial DNA be repaired efficiently.
The short patch base excision repair (BER) pathway is required for repair of single-base modifications in DNA caused by ROS or spontaneous depurinations in both the nucleus and the mitochondrion. This BER pathway involves four basic enzymatic steps, with the flow of each step to the next having been described as the handing over of a baton (Wilson and Kunkel 2000), such that the intermediate products, which may often be mutagenic, are passed from one enzyme to the next. The first step is the recognition and removal of the damaged nucleotide by a glycosylase, resulting in the generation of an abasic site (AP site). The predominant group of glycosylases which specifically recognize oxidatively damaged bases is the bifunctional glycosylases (Hazra et al. 2003). With these glycosylases, the intrinsic class I AP endonuclease, known as AP lyase, performs the second step of cleaving the DNA sugar phosphate backbone 3′ to the AP site, generating either a 3′-blocking end, such as a 3′-α, β-unsaturated aldehyde via β-elimination or a 3′- phosphate via β, δ-elimination. These 3′-blocked terminals are subsequently removed by the 3′-phosphodiesterase activity associated with class II AP endonucleases, generating a 3′-hydroxyl group. Alternatively, spontaneously generated or monofunctional glycosylase produced AP sites can be cleaved by class II AP endonucleases 5′ to the AP site, generating a 5′-deoxyribose phosphate which requires a 5′-deoxyribose phosphatase (5′-dRPase) activity for its removal to form a 5′-phosphate. The third step in BER is the nucleotide synthesis step and is performed by either DNA polymerase β in the nucleus or polymerase γ in mitochondria. The final step is the ligation of the DNA strands which is accomplished by a DNA ligase. The particular sub-pathway for repair is determined by the type of DNA glycosylase utilized for the removal of the modified base (Fortini et al. 1999).
Apn1, a homologue of E. coli Endonuclease IV, is the major class II AP endonuclease in Saccharomyces cerevisiae. This 1104 bp gene has a bi-partite nuclear localization signal (NLS) which targets Apn1 to the nucleus (Ramotar et al. 1993). Although the Apn1 gene does not have a mitochondrial targeting signal (MTS), the Apn1 protein is sent to mitochondria in yeast through its association with the Pir1 protein (Vongsamphanh et al. 2001). In addition to its class II AP endonuclease and 3′-phosphodiesterase activities, Apn1 also participates in nucleotide incision repair (Ischenko and Saparbaev 2002), which is a glycosylase independent repair mechanism for repairing DNA damage generated by ionizing radiation, such as α-2′-deoxynucleosides, 5,6-dihydro-2′-deoxyuridine, 5,6-dihydrothymidine, and 5-hydroxy-2′-deoxyuridine (Gros et al. 2004), without producing mutagenic AP sites.
As mentioned above, oxidative stress-induced DNA damage is often recognized and cleaved by bi-functional glycosylases, generating an intermediate product that contains 3′-blocking groups, which require enzymes with 3′-phosphodiesterase activity for their removal. Kinetic studies have revealed that the 3′-phosphodiesterase activity of Apn1 is more robust than that of its human counterpart, AP endonuclease APE1 (Johnson and Demple 1988; Suh et al. 1997), which is the main enzyme responsible for this activity in humans (Parsons et al. 2004). In addition, Apn1 is active whereas APE1 is insufficient for removing 3′-phosphate groups (Wiederhold et al. 2004), which must be removed by polynucleotide kinase (PNK) in the nucleus (Wiederhold et al. 2004). Also, others have reported that Apn1 is more effective than APE1 at enhancing cell survival (Wilson et al. 1995; Tomicic et al. 1997). Therefore, Apn1 has been the subject of interest for developing therapeutic tools against neurodegenerative diseases that may be caused by heightened oxidative stress (Mol et al. 2000).
Despite the higher enzyme efficiency (Frosina 2001) and wider substrate specificity (Ischenko and Saparbaev 2002; Ishchenko et al. 2004) of Apn1 compared with APE1, and the higher mutation rate observed in mtDNA than nDNA following oxidative stress, there has been only a few reports of attempted expression of Apn1 in mammalian cells (Tomicic et al. 1997; He et al. 2001; Roth et al. 2003), and none has addressed the question as to whether targeting Apn1 to mitochondria in mammalian cells could further enhance DNA repair and cell survival. Herein we report the first study, to our knowledge, to target Apn1 to mitochondria in mammalian cells. RCSN-3 cells transfected with HA-tagged Apn1, either with, or without an MTS and enhanced mtDNA repair and improved cell survival were observed following oxidative stress. The results of this study suggest that some neurodegenerative processes can be attenuated by targeting Apn1 to either the nucleus or mitochondria.
Materials and methods
Materials
Dulbecco's modified Eagle medium (DMEM), Hanks’ Balanced Salt Solution (HBSS), RadPrime DNA Labeling System Geneticin (G418) were from Invitrogen (Grand Island, NY, USA). Fetal bovine serum was purchased from Hyclone (Logan, UT, USA). Gentamycin sulfate, menadione bisulfate, a derivative of menadione (Morrison et al. 1984; Frei et al. 1986), hematoxylin, and NP-40 were from Sigma (St Louis, MO, USA). FuGENE 6 Transfection Reagent, restriction endonucleases, and Rnase A were from Roche Molecular Biochemicals (Indianapolis, IN, USA). PolyFect Transfection Reagent was from Qiagen (Valencia, CA, USA). Oligonucleotides, purified E. coli Fpg, human APE, and Comet Assay kit were from Trevigen (Gaithersburg, MD, USA). T4 polynucleotide kinase (PNK) was from Promega (Madison, WI, USA). Chemiluminescent reagents were from SuperSignal, Pierce (Rockford, IL, USA). Zeta-Probe GT nylon membranes were from Bio-Rad (Hercules, CA, USA). Antibodies were obtained from the following sources: the antibody against Apn1 was a generous gift from Dr. D. Ramotar (University of Montreal, Montreal, Quebec, Canada); the antibody against flavoprotein (complex II in mitochondria) was from Molecular Probe (Eugene, OR, USA), anti-cytochrome c from BD PharMingen (San Diego, CA, USA); anti- proliferating cell nuclear antigen (PCNA), anti-tyrosine hydroxylase and horseradish peroxidase-conjugated anti-mouse or anti-rabbit IgG antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA); anti-HA antibody was from Sigma.
Plasmid constructs
To obtain a plasmid with the Apn1 gene connected to the MTS from the human MnSOD gene (Dobson et al. 2000) and hemagglutinin (HA) 5′ to Apn1 (MTS/HA/APN1), a BamHI/XbaI fragment containing the Apn1 gene was cloned into the BamHI/XbaI site of pcDNA3.neo plasmid containing an elongation factor 1 alpha promoter (EF1a) and a MTS/HA sequence. To generate HA/APN1 plasmid, the same BamHI/XbaI fragment containing the Apn1 gene was cloned into the BamHI/XbaI site of pcDNA3.neo plasmid containing an EF1a/HA sequence. The constructs obtained were sequenced to verify the integrity of the reading frame and the fidelity of the sequence.
Cell culture and transfection
Raul Caviedes Substantia Nigra (RCSN-3) cells, a neuronal cell line derived from 4-month-old normal Fischer 344 rat substantia nigra pars compacta (Paris et al. 2001), were cultured in DMEM/HAM-F12 (1 : 1), supplemented with 10% fetal bovine serum and 40 mg/L gentamycin sulfate, and incubated in 5% CO2 at 37°C in a humidifier chamber. The MTS/HA/Apn1 and HA/Apn1 plasmids were transfected into RCSN-3 cells using FuGENE 6 Transfection Reagent or PolyFect Transfection Reagent, respectively, following the manufacturer's recommendations. As a negative control, cells were transfected with the pcDNA3.neo vector alone using FuGENE. Colonies of transfected RCSN-3 cells were selected with 0.6 mg/mL G418 and clones were picked and maintained with 0.5 mg/mL G418 in normal growth media thereafter. The insertion of the plasmids was verified by Southern blot analysis, and the expression of the recombinant protein was verified by Western blot analysis.
Subcellular protein fractionation
Nuclear and mitochondrial extracts were isolated as previously described with some modifications (Storrie and Madden 1990). Transfected RCSN-3 cells grown to confluence in four 150 mm culture dishes were harvested using a rubber policeman and collected by centrifugation. All procedures were conducted on ice or at 4°C. Cells were washed twice in 2 mL of divalent cation-free phosphate-buffered saline (PBS) (0.14 mol/L NaCl, 0.01 mol/L KCl and 0.01 mol/L NaH2PO4) and homogenized in a PBS containing 0.25 mol/L sucrose by 15 strokes in a Dounce homogenizer (Kontes Glass Company, Vineland, NJ, USA). The lysate was examined with a light microscope to ensure 90−95% cell lysis. The nuclear pellet was collected by centrifugation at 1300 g for 10 min twice, and washed in nuclear wash solution (0.25 mol/L sucrose, 10 mmol/L NaCl, 10 mmol/L Tris–HCl and 1.5 mmol/L MgCl2), vortexed in detergent (one part 10% sodium deoxycholate and two parts NP-40) and collected by centrifugation at 1300 g for 5 min.
The supernatants containing mitochondria were pooled and centrifuged first at 1300 g for 10 min to remove any nuclear contaminant, and then at 13 000 g for 20 min to pellet mitochondria. The mitochondrial pellet was then air dried and resuspended in sucrose–Tris/EDTA buffer (50 mmol/L Tris pH 7.4, 5 mmol/L EDTA and 20% sucrose) and carefully loaded onto a 1.0 mol/L/ 1.5 mol/L discontinuous sucrose gradient, of which the latter was frozen at −70°C the previous night. The mitochondrial suspension was centrifuged at 50 000 g for 15 min in a Beckman SW 50.1 rotor (Beckman Coulter, Fullerton, CA, USA). Mitochondrial pellets were collected at the interphase, washed in mannitol–sucrose buffer (210 mmol/L mannitol, 70 mmol/L sucrose, 5 mmol/L EDTA, 5 mmol/L Tris pH 7.5), and centrifuged for 20 min at 13 000 g. The mitochondrial and nuclear pellets were dried and lysed with lysis buffer containing protease inhibitors. Protein concentrations were determined using the Bio-Rad protein dye micro-assay following the manufacturer's recommendations (Bradford method).
Drug treatment
The redox cycler menadione bisulfate (MEN), a derivative of menadione (Morrison et al. 1984; Frei et al. 1986), was dissolved in HBSS to a 10 mmol/L stock solution and then diluted to appropriate concentration (100−900 μmmol/L). Exposure to menadione or HBSS alone was limited to 1 h in 5% CO2 at 37°C for all studies. Cells were approximately 80−90% confluent, unless otherwise stated. Cells were processed immediately or regular culture media was replaced and cells were allowed various periods of time to repair.
Southern blot analysis
Cells were lysed in 10 mmol/L Tris–HCl (pH 8.0), 1 mmol/L EDTA, 0.5% SDS and 0.3 mg/mL proteinase K, and incubated overnight at 37°C, followed by the addition of 0.25 volume of 5 mol/L NaCl. High molecular weight DNA was extracted with equal volumes of chloroform/isoamyl alcohol (24 : 1) three times, followed by precipitation with 11 mol/L ammonium acetate and 100% ethanol. DNA was then resuspended in distilled H2O, treated with Rnase A (final concentration, 1 mg/mL) for 2 h, and digested with restriction enzyme overnight at 37°C. For insert confirmation, BamHI and XbaI were used for Apn1(+)MTS cells and XhoI and XbaI for Apn1(−)MTS cells. BamHI was used to digest total DNA in mtDNA repair studies. Digested samples were precipitated, resuspended in TE buffer (10 mmol/L Tris, pH 8.0, 1 mmol/L EDTA), and quantified using calf thymus DNA as standards in a Hoefer DyNA Quant 200 fluorometer (GE Healthcare, Piscataway, NJ, USA). Samples for mtDNA repair studies were heated for 15 min at 70°C and cooled to 22°C. NaOH was added to a final concentration of 0.1 N, and incubated at 37°C for 20 min followed by the addition of alkaline loading dye. Samples were loaded onto a horizontal 0.6% alkaline agarose gel for mtDNA repair studies, 1% neutral gel for insert confirmation, and electrophoresed at 30 V (1.5 V/cm gel length) for 16 h. After ethidium bromide staining to confirm even loading and standard gel washes, the DNA was transferred to Zeta-Probe GT nylon membranes (Bio-Rad) and cross-linked in a GS Gene Linker UV Chamber (Bio-Rad). To confirm Apn1 insertion, membranes were hybridized with a 32P-labeled HA/Apn1 fragment generated by a RadPrime DNA Labeling System, and the plasmids were used as a positive control. For mtDNA repair studies, the membranes were hybridized with a 32P-labeled rat mtDNA-specific PCR-generated probe. Hybridization and subsequent washes were performed according to the manufacturer's recommendations. Mitochondrial DNA damage and repair were determined as previously described (Driggers et al. 1993, 1997).
Western blot analysis
A total of 30 μg of protein from whole cell, nuclear, or mitochondrial fractions was denatured and resolved by 12.5% SDS-PAGE and transferred to polyvinylidene difluoride (PVDF) membrane using standard procedures. The membrane was blocked in 5% non-fat dry milk/Tris–buffered saline (TBS) overnight at 4°C, and immunoblotted with primary antibody in 0.05% Tween 20/2% non-fat dry milk/TBS (TBST) for 1 h. Following two 5 min washes in TBST, the membrane was probed with a horseradish peroxidase-conjugated anti-mouse or anti-rabbit IgG antibody for 30 min. After two more 5 min TBST washes, the immuno-complex was detected using chemiluminescent reagents. The antibody against flavoprotein (complex II in mitochondria) and cytochrome c were used to detect mitochondrial contaminant in nuclear extracts, and anti-PCNA antibody was used to detect nuclear contaminant in mitochondrial extracts.
Oligonucleotide cleavage assay
The AP endonuclease and 3′-phosphodiesterase activities of Apn1 were evaluated by an oligonucleotide cleavage assay. In brief, a 21-mer oligonucleotide with an abasic site (AP) at the 10th position was 5′-end labeled. The labeling reaction consisted of 20 pmol of the single stranded oligonucleotide, 5 pmol of 33P-ATP, T4 polynucleotide kinase (PNK), and appropriate kinase buffer in a total volume of 20 μL, incubated for 45 min at 37°C and 10 min at 68°C. Complementary oligonucleotide was then added at 22°C to form duplex DNA. Activity assays contained 0.5 pmol of labeled duplex oligonucleotide, 1X REC Buffer (100 mmol/L HEPES-KOH pH 7.4, 100 mmol/L KCl), 1 μg of protein extract in a 10 μL reaction volume and were incubated with increasing time points at 37°C. The oligonucleotide used to detect only the AP endonuclease activity, and not AP lyases in the extract contained a synthetic AP site, a tetrahydrofuran (THF). The substrate for 3′-phosphodiesterase activity was generated by digesting an AP oligonucleotide with E. coli Fpg, and 0.5 pmol of the resulting 5′-end labeled 3′-phosphate oligonucleotide was incubated with protein extracts as described above. Purified human APE was used to produce positive controls for identifying the location of cleavage products. Two microliters of the reaction products from assaying AP endonuclease activity were resolved in 20% polyacrylamide gel with 7 mol/L urea, and from assaying 3′-phosphodiesterase activity were resolved in 8% polyacrylamide sequencing gel with 7 mol/L urea. Wet gels were autoradiographed at −70°C.
Single cell gel electrophoresis/Comet assay
Total DNA damage was assessed by single cell gel electrophoresis, or Comet assay using a Comet Assay kit according to the manufacturer's protocol. In brief, transfected cells were exposed to 0, 300, 600 or 900 μmol/L menadione. Cells that were collected by mechanical dislodging and centrifugation at 300 g for 5 min at 22°C were washed and resuspended in ice cold PBS at a concentration of 1 × 105 cells/mL. Next, cells were added to low melting agarose and spread into a thin layer on two different microscope slides. After solidification, cells were lysed on ice for 45 min. Following incubation at 37°C for 30 min, slides were incubated in an alkali solution (pH 12.1) for 30 min to unwind the double-stranded DNA followed by neutralization in TBE buffer. Electrophoresis was conducted in TBE at 1 V/cm between the two electrodes for 20 min. The slides were dipped in 100% cold ethanol for 5 min and air dried. Finally, the DNA was stained with SYBR green and observed using fluorescent microscopy with an Olympus BX-FLA System (Olympus, Center Valley, PA, USA) and photographed using Magna Fire 2.1C software (Optronics, Goleta, CA, USA). The tail length, or the distance of DNA migration, was defined as the distance between the leading edge of the nucleus and the end of the tail. This is proportional to the level of single stranded breaks and abasic sites in DNA. The data were analyzed from a minimum of 65 cells per cell type per dose using the Fuji Image Gauge software (Fuji Film Life Science, Stamford, CT, USA). Data represent three separate experiments.
Colony formation assay
Cell viability was determined by a colony formation assay. Due to a difference in cell attaching efficiency, 500, 700, or 600 cells (vector only controls, Apn1(+)MTS, or Apn1(−)MTS cells, respectively) were plated in 60-mm dishes and exposed to MEN at various concentrations for 1 h. Cells were allowed to proliferate in culture media for 10 days following drug treatment, with fresh media replacement every 3 days. On the 10th day, the culture media was removed, and cells were fixed in methanol : acetate (3 : 1) for 10 min and stained with hematoxylin for 10 min. Visible colonies were counted following several gentle washes with distilled H2O.
Results
Transfection of RCSN-3 cells with Apn1
To determine whether HA/Apn1 plasmids with or without the MTS were inserted correctly into the RCSN-3 genome, qualitative Southern blot analyses were performed using total genomic DNA from transfected RCSN-3 cells. Blots were hybridized using a 32P-labeled Apn1 fragment as the probe. For cells transfected with the MTS/HA/Apn1 containing plasmid [Apn1(+)MTS], clone two of the three clones that showed full length insertion of the plasmid was used for subsequent analyses (Fig. 1a). For cells transfected with the HA/Apn1 containing plasmid [Apn1(−)MTS], clone four of the four clones identified to contain the full length insert was used for subsequent analyses (Fig. 1b).
Fig. 1.
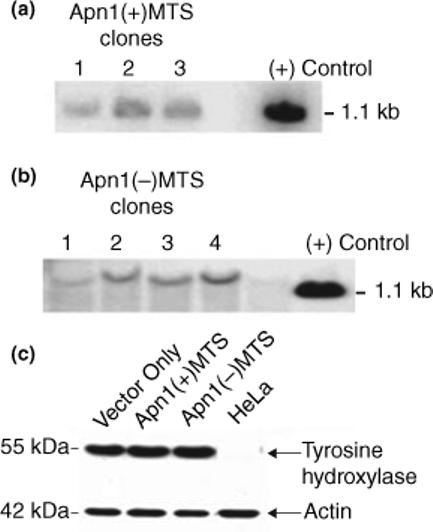
Apn1 transfection in RCSN-3 cells and their dopaminergic property. (a and b) Southern blot analyses showing that the HA-tagged Apn1 plasmids were incorporated into the RCSN-3 genome. (a) DNA from three clones containing the Apn1 plasmid with a mitochondrial targeting sequence from the human MnSOD gene. (b) DNA from four clones containing the Apn1 plasmid without the mitochondrial targeting sequence. Total DNA was isolated and hybridized with the Apn1(+)MTS plasmid serving as the probe. Due to the different sites of restriction enzyme cleavage, the probe hybridized to a fragment of a larger size in Apn1(−)MTS clones. (c) A Western blot showing the expression of tyrosine hydroxylase in whole cell lysates of cells transfected with vector only controls, Apn1(+)MTS or Apn1(−) MTS cells. Lysates from HeLa cells were used as a negative control. Actin was used as a loading control.
RCSN-3 cells retain the ability to express tyrosine hydroxylase
To ensure that the transfected RCSN-3 cells were still functional, whole cell lysates from the transfected cells were immunoblotted against tyrosine hydroxylase, the rate-limiting enzyme in the synthesis of dopamine using Western blot analysis. Lysates from HeLa cells were used as a negative control. Fig. 1(c) shows that transfection did not alter the cells’ ability to express tyrosine hydroxylase.
Recombinant Apn1 proteins are expressed
To verify that the recombinant Apn1 protein was targeted to either the nucleus or the mitochondrion, pure subcellular fractions were immunoblotted for the presence of HA or Apn1 by Western blot analysis. To ensure that the fractions were free of contamination, they were probed for the presence of flavoprotein, a subunit forming complex II of the electron transport chain in the inner membrane of mitochondria; cytochrome c, a mitochondrial protein found in the intermembrane space; and PCNA, a nuclear protein (Fig. 2a). Mitochondrial fractions from all three cell types (vector only, Apn1(+)MTS and Apn1(−)MTS) were free of nuclear contaminants (Fig. 2a, lanes 1−3). The nuclear fractions of all three cell types were free of mitochondrial contaminants (Fig. 2a, lanes 4−6).
Fig. 2.

Expression of Apn1 in subcellular and whole cell extracts. (a) Pure subcellular fractions. Purified mitochondrial (lanes 1−3) and nuclear (lanes 4−6) fractions from vector only control (lanes 1 and 4), Apn1(+)MTS (lanes 2 and 5), and Apn1(−)MTS (lanes 3 and 6) cells were immunoblotted against flavoprotein from complex II of the inner membrane of mitochondria, PCNA, a nuclear protein, or cytochrome c, which resides in the inter-membrane space of mitochondria. (b) Western blots showing Apn1 expression in purified nuclear, mitochondrial extracts, and whole cell lysates from vector only controls, Apn1(+)MTS or Apn1(−)MTS cells. The extracts were probed with anti-Apn1 antibody in nuclear extract and anti-HA antibody in mitochondrial and whole cell lysates. The expected size of Apn1 is 40.5 kDa.
Whole cell, mitochondrial, and nuclear fractions were obtained and the presence of the recombinant Apn1 was verified by Western blot analysis using either HA or Apn1 antibodies (Fig. 2b). As expected, Apn1 was not detectable in the whole cell, mitochondrial, or nuclear fractions from vector only controls. In Apn1(−)MTS cells, Apn1 was present in whole cell lysate and the nuclear fraction, and was absent in the mitochondrial extract. In Apn1(+)MTS cells Apn1 was found in both mitochondrial and nuclear extracts, as well as the whole cell lysates. The reason for this dual localization likely is that a portion of Apn1 expressed in Apn1(+)MTS cells was imported to the nucleus by its inherent NLS.
Recombinant Apn1 proteins are active
Apn1 is a multi-functional protein (Popoff et al. 1990; Ischenko and Saparbaev 2002; Ishchenko et al. 2003, 2004, 2005). To determine whether it was functionally active in cells, the AP endonuclease and 3′-phosphodiesterase activities of Apn1 were assayed using an oligonucleotide cleavage assay. To evaluate AP endonuclease activity, a synthetic 21-mer oligonucleotide containing a THF modified AP-site at position 10 was 5′ end-labeled with P33, annealed with its complementary oligonucleotide, and incubated with equal amounts of either mitochondrial (Fig. 3a) or nuclear (Fig. 3b) extracts for increasing periods of time. Enzymes with AP endonuclease activity cleave the 21-mer oligo-nucleotide 5′ to the THF-AP site; the end product is a 9-mer fragment. In order to distinguish between the endogenous AP endonuclease activity from that of the recombinant Apn1, 10 mmol/L EDTA was added to both the lysis buffer and reaction buffer, which also contained 0.2 mmol/L ZnCl2, to inactivate endogenous AP endonuclease activity. As shown in Fig. 3, pure APE using the manufacturer's buffer (Fig. 3a lane 2) gave a product band at the 9-mer position, while pure APE with the reaction buffer containing 10 mmol/L EDTA produced a band with much less intensity (Fig. 3b lane 1).
Fig. 3.
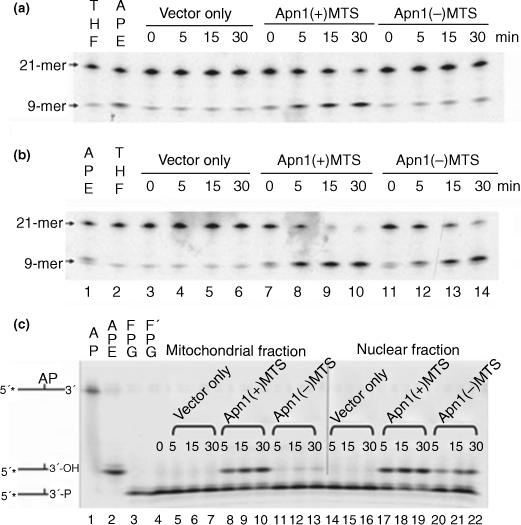
AP endonuclease and 3′-phosphodiesterase activity assays. Mitochondrial (a) and nuclear (b) extracts from vector only controls, Apn1(+)MTS, or Apn1(−)MTS cells were assayed for AP endonuclease activity. Extracts were incubated with 5′-end labeled oligonucleotide containing a modified AP site (THF lanes : lane 1 in panel a and lane 2 in panel b at 37°C for up to 30 min. Enzymes with AP endonuclease activity cleave the substrate 5′ to the AP site and the product appears as a 9-mer. The substrates for lane 2 of panel a and lane 1 of panel b were incubated with pure APE enzyme. The reaction buffer supplied by the manufacturer was used in lanes 1 and 2 of both panel a and lane 2 of panel b. Modified reaction buffer containing 10 mmol/L EDTA and 0.2 mmol/L ZnCl2 was used in the remaining lanes. (c) 3′-Phosphodiesterase activity assays. A 5′-end labeled oligonucleotide containing an AP site (lane 1) was pre-cleaved by FPG (lanes 3 and 4) to generate the 3′-phosphate substrate for 3′-phosphodiesterases. Mitochondrial (lanes 5−13) and nuclear (lanes 14−22) extracts from vector only controls (lanes 5−7 and 14−16), Apn1(+)MTS (lanes 8−10 and 17−19), or Apn1(−)MTS (lanes 11−13 and 20−22) cells were incubated with this substrate (lane 4) for 5, 15, or 30 min. The position of the hydrolyzed 3′-phosphate, 3′-hydroxyl, is identical to the product formed when pure APE cleaves the AP oligonucleotide (lane 2).
In the mitochondrial extract (Fig. 3a), there was not an increase in the intensity of product bands at the 9-mer position in vector only controls (lanes 3−6) nor in the Apn1(−)MTS extracts (lanes 11−14). However, AP endonuclease activity was apparent in extracts from Apn1(+)MTS cells (lanes 7−10). Similarly, in the nuclear extracts (Fig. 3b), bands at the 9-mer position were absent in vector only controls (Fig. 3b, lanes 3−6). However, in the nuclear extracts from both Apn1 expressing cells, either with or without the MTS, the 9-mer product appeared in a time-dependent manner (Fig. 3b, lanes 7−10 and 11−14, respectively).
The 3′-phosphodiesterase activity of Apn1 also was assayed using an oligonucleotide cleavage method. To prepare the 3′-phosphate substrate for testing for 3′-phosphodiesterase activity, a synthetic 21-mer oligonucleotide containing an AP site at the 10th position (Fig. 3c; lane 1) was first 5′end-labeled with 33P and incubated with E. coli Fpg glycosylase (lanes 3 and 4). The AP lyase activity associated with this glycosylase cleaves the sugar phosphate backbone 3′ to the AP site, generating a 3′-blocking group. The position of the substrate is shown in lanes 3 and 4, with the difference being that the substrate in lane 4 was prepared in bulk to provide sufficient substrate for incubation with the cell extracts, while the substrate in lane 3 was prepared with only 0.5 pmol of the AP oligonucleotide. The end product, a 3′-hydroxyl containing 9-mer, generated by incubating purified human APE with the AP oligonucleotide (lane 2), is a slower migrating product (Ishchenko et al. 2003; Jilani et al. 2003). Mitochondrial and nuclear extracts from vector only controls showed no 3′-phosphodiesterase activity (lanes 5−7 and 14−16, respectively). However, strong 3′-phosphodiesterase activity was present in both the mitochondrial and nuclear extracts from Apn1(+)MTS cells (lanes 8−10 and 17−19, respectively). The mitochondrial extract from Apn1(−)MTS cells contained a barely detectable amount of 3′-phosphodiesterase activity (lanes 11−13), likely due to a minor undetected contamination, while the nuclear extract, as expected, had a prominent 3′-phosphodiesterase activity (lanes 20−22). Therefore, the results of the Apn1 activity assay show that the recombinant Apn1 protein has active AP endonuclease and 3′-phosphodiesterase activities. These results also support the Western blot results from Fig. 2(b) which showed that Apn1 was present in both nuclei and mitochondria in Apn1(+)MTS cells.
Apn1 expressing RCSN-3 cells have more efficient mtDNA repair capacity
The 3′-phosphodiesterase activity associated with Apn1 is responsible for removing the 3′-blocking group generated as a result of AP lyase activity from glycosylase/AP lyases which recognize oxidized DNA. As reactive oxygen species generated by MEN are directed via complex I of the electron transport chain to the matrix where mtDNA resides, we tested the hypothesis that mtDNA repair would be more efficient in cells expressing Apn1 targeted to mitochondria when compared with vector only controls and Apn1(−)MTS cells. The transfected cells were exposed to MEN for 1 h to a dose which generated a break frequency between 0.5 and one break per 10 kb restriction fragment of mtDNA. Subsequently, cells were allowed to repair for either 2 or 6 h in regular culture media. A representative autoradiograph of a quantitative alkaline Southern blot analysis evaluating mtDNA repair (Fig. 4a) and a graph summarizing the effects of three independent experiments (Fig. 4b) are shown. At 2 h, there was no significant difference in mtDNA repair between the three cell types. However, by 6 h, Apn1(+)MTS cells had repaired 54.5 ± 10.2% of the lesions in mtDNA compared with vector only controls which only had repaired 22.4 ± 3.3% of the lesion (p < 0.05). Apn1(−)MTS cells also repaired mtDNA more efficiently than vector only controls after 6 h (55.1 ± 3.4%; p < 0.05). There was no statistical difference in the efficiency of mtDNA repair between the two Apn1 expressing cells.
Fig. 4.

Apn1 expression enhances mtDNA repair efficiency. Mitochondrial DNA integrity was investigated in transfected RCSN-3 cells exposed to menadione following 2 or 6 h of repair. An equal amount of total DNA was probed for mtDNA and the percent of repair calculated. (a) Representative autoradiograph and (b) results of % repair (mean ± SE) from three independent experiments. The percent repair of either Apn1(+)MTS or Apn1(−)MTS cells differed significantly from vector only controls (*p < 0.05).
Nuclear DNA damage was less extensive in RCSN-3 cells expressing Apn1 without the MTS
Although MEN generates ROS primarily in mitochondria, damage also has been observed in nDNA from both human and rat cell lines exposed to this agent (Morrison et al. 1984; Martins and Meneghini 1990; Ngo et al. 1991). Therefore, we examined the extent of nDNA damage to see whether Apn1(+)MTS cells were able to reduce the amount of DNA damage following MEN treatment. Single cell gel electrophoresis, also known as the Comet assay, was used to analyze the extent of nDNA damage. In this technique, abasic sites and single strand breaks in DNA can be detected when the lysed cells undergo alkaline treatment followed by neutral gel electrophoresis. Figs 5 (a–f) show the results of the Comet assay when cells were treated to 600 μmol/L of MEN. Figs 5(a, c, and e) are the images obtained from fluorescent microscopy when DNA was stained with SYBR green. Figs 5(b, d, and f) are the corresponding images obtained from the software used to analyze the DNA. The distance of DNA migration (sometimes referred to as tail length), which is proportional to the level of single strand breaks and abasic sites in DNA (Tice et al. 2000), was measured by Image Gauge software. There were significantly fewer single strand breaks and abasic sites in Apn1(−)MTS cells at 300, 600, and 900 μmol/L MEN than either vector only controls or Apn1(+)MTS cells (Fig. 5g). There was no difference in the extent of nDNA damage between vector only controls and Apn1(+)MTS cells (Fig. 5g).
Fig. 5.

Apn1 expression in the nuclei protects DNA from single strand breaks. Nuclear DNA damage was assessed by single cell gel electrophoresis, or Comet assay, under alkaline treatment and neutral gel electrophoretic conditions. Migration distance of the SYBR green stained DNA, or tail length, which is proportional to the amount of DNA damage from vector only controls (a), Apn1(+)MTS (c), or Apn1(−)MTS (e) cells exposed to 600 μmol/L menadione was measured using analytical software (b,d and f). (g) Tail length (mean ± SE) from at least 65 cells was measured per cell type, dose, and experiment. Results were obtained from three independent experiments. *Indicates significant difference from vector only controls (p < 0.05).
Apn1 expression enhances cell survival
To determine whether mitochondrial targeting of Apn1 can reduce the cytotoxic effect of MEN, cell viability was assessed by a colony formation assay (Fig. 6). Both Apn1 expressing cells formed significantly more clones relative to cells containing only the vector following exposure to all the doses of MEN examined (Fig. 6). There was no difference in survival between either Apn1-expressing cells. Therefore, cell survival was enhanced in Apn1 expressing RCSN-3 cells following oxidative stress, regardless of whether Apn1 was expressed with or without the MTS.
Fig. 6.

Apn1 expression enhances cell survival. Cell viability was determined by examining the number of colonies formed 10 days following menadione exposure at various doses. *Indicates significant difference from vector only controls (p < 0.05). Results were obtained from three independent experiments.
Discussion
Many neurodegenerative diseases are associated with mutations in mtDNA (Wallace et al. 1998) suggesting a role for mtDNA damage in the pathogenesis of these diseases. In support of this notion it has been recently demonstrated that there are high levels of mtDNA deletions present in substantia nigra of older adults and patients with Parkinson's disease (Bender et al. 2006). Thus, it appears likely that damage to this DNA caused by oxidative stress which produces subsequent mutations could play a significant role in the pathogenesis of these diseases. Therefore, we sought to determine whether targeting the yeast AP/endonuclease Apn1 to mitochondria in a cell line derived from rat substantia nigra, would render them more resistant to oxidative stress. The results showed that the two Apn1 constructs, one with, and one without an MTS from human MnSOD, could be successfully transfected into RCSN-3 cells. The transfection process did not alter the dopaminergic properties of these cells, as evidenced by their ability to express tyrosine hydroxylase. Apn1 was found to be present and active in the nuclear fraction of Apn1(−)MTS cells whereas Apn1(+)MTS cells expressed active Apn1 in both the nuclear and mitochondrial fractions. The localization to the nucleus was likely due to the NLS inherent in the Apn1 gene. For future studies, truncating the NLS may help target Apn1 exclusively to mitochondria. However, it has been reported that even when the NLS was truncated from the Apn1 gene, there was still sufficient Apn1 entering into the nucleus, possibly via passive diffusion, to increase resistance to DNA damaging agents in yeast (Ramotar et al. 1993). Thus it appears likely that some nuclear localization cannot be avoided.
Analysis of mtDNA repair following transfection revealed that it was enhanced following MEN treatment in Apn1(+)MTS cells compared with vector only controls. However, surprisingly, mtDNA repair also was enhanced in Apn1(−)MTS cells compared with vector only controls. As Apn1 was found in the nuclear fraction of Apn1(+)MTS cells, the extent of nDNA damage also was examined following MEN treatment. The presence of this enzyme in the nucleus did not lead to enhanced protection of the DNA in this organelle. Cell viability studies revealed that Apn1 expression in RCSN-3 cells, with or without the MTS, enhanced cell survival and proliferation following oxidative stress.
To explain these results, we propose the following model (Fig. 7). We postulate that oxidative damage to mtDNA and the resulting mutations cause defective electron transport by impairment of the mitochondrially encoded components of these chains. This defective electron transport results in heightened production of ROS, which further damage mtDNA and cause more mutations and thus initiates a vicious cycle of escalating mtDNA damage and ROS production. In support of this concept it has been shown that accumulation of mtDNA mutations increased ROS production in many cell types (de Grey 2005; Petros et al. 2005; Simmons et al. 2005). When mtDNA damage surpasses a specific threshold, it will act as the initiator of a stress signal from mitochondria, likely through the generation of ROS, which some researchers have referred to as the ‘retrograde signal’ (Mandavilli et al. 2002). This signal activates specific transcription factors and/or signal transduction pathways, which in turn activate and/or enhance DNA repair mechanisms in both the nucleus and mitochondria. This altered repair ultimately leads to enhanced cell survival. In order for these events to occur, the integrity of the genes which encode for all the factors involved must be intact. In support of this model, there is evidence implicating mitochondria as the source of the signal which activates transcription factors such as NFκB (Garcia-Ruiz et al. 1995; Hughes et al. 2005), and c-jun, AP-1, cAMP-response element binding protein (CREB), and nuclear respiratory factor 1 (Storz et al. 2005), as well as other signal transduction cascades, such as protein kinase D (Storz et al. 2005) and p38 MAPK (Emerling et al. 2005) through the generation of ROS. Furthermore, it has been reported that DNA repair can be modulated by phosphorylation (Gu and Lu 2001; Parker et al. 2003; Hu et al. 2005). Therefore, this and other post-translational modifications, activated by the mitochondrial signal, play a key role in regulating DNA repair enzyme activity, localization and stability (Hu et al. 2005) and thereby influence cellular resistance to oxidative stress.
Fig. 7.

Model for observed phenomenon. Oxidative damage to mtDNA is required to activate a retrograde signal from mitochondria which will ultimately lead to enhanced nDNA integrity, mtDNA repair and cell survival, if the genes for the transcription factors, signal transduction pathways and DNA repair enzymes involved were intact. This is demonstrated in Apn1(−)MTS cells. If the genes were not intact, such as in the case of vector only controls, then no mtDNA repair, nDNA integrity or cell survival enhancement would result. However, when mtDNA repair was directly enhanced, such as in Apn1(+)MTS cells, cell survival also would be enhanced, but nDNA integrity would not be enhanced because the retrograde signal was not activated.
To apply this model to the current study, we found that Apn1, which was stably expressed in Apn1(−)MTS cells, enhanced nuclear DNA repair. We hypothesize that this enhanced nuclear repair caused better preservation of nuclear genes which encode transcription factors, signal transduction pathways and DNA repair enzymes. Following MEN treatment, which caused oxidative damage to mtDNA, the retrograde signal was activated. This induced post-translational modification of DNA repair enzymes which led to their translocation and/or activation in both the nucleus and mitochondria. Due to the enhanced repair of mtDNA, the escalating vicious cycle of ROS production and mtDNA damage was aborted and overall cell survival was enhanced. In the case of vector only controls, although the retrograde signal had been activated due to mtDNA damage, the endogenous DNA repair enzymes and factors responsible for post-translational modifying these enzymes were insufficient to overcome the damage to the DNA, and therefore cell survival was not enhanced. In the case of the Apn1(+)MTS cells, the Apn1 present in mitochondria was directly responsible for enhancing mtDNA repair, thus blocking the initiation of the vicious cycle and enhancing cell survival. Although, Apn1 translocated to the nucleus, no enhanced protection of nDNA integrity was observed. We believe that this is because this enzyme was unable to participate in BER. The current concept of BER is that one enzyme hands over to the next enzyme in this process (Wilson and Kunkel 2000). Because Apn1(+)MTS has an MTS, this peptide prevents the Apn1 in the nucleus from handing over to the next enzyme (polymerase) in BER and repair is unable to proceed. Mitochondria often clip off the MTS after proteins are translocated into them. However, Western blot analysis revealed that this is not the case for the Apn1(+)MTS protein as the bands from the nuclear and mitochondrial fractions were the same size (data not shown). This surprising finding indicates that repair in mitochondria, likely due to the genome being much simpler, is not as complex as in the nucleus and handing off from one step in repair to the next is not necessary. We feel that the protective mechanism initiated by Apn1(+)MTS is superior to that of Apn1(−)MTS because it blocks the initiation of the vicious cycle rather than modulates the response to it.
In summary, to our knowledge, this is the first report of the expression Apn1 in mammalian cells derived from the substantia nigra. We have found enhanced DNA repair in a mammalian neuronal cell line expressing Apn1 with and without MTS. More importantly, this enhanced repair led to increased cell survival in neurons, whether Apn1 was directly targeted to the mitochondria or not. This indicates that Apn1 could be used as a possible treatment strategy for attenuating or slowing down the pathogenesis of neurodegenerative diseases. Future work in this laboratory will explore how protein transduction technology can be used to deliver this protein therapeutically.
Acknowledgments
We thank Dr Mikhail Alexeyev (University of South Alabama, Mobile, Alabama) for providing pcDNA3.neo/EF1a/MTS/HA and pcDNA3.neo/EF1a/HA plasmids. This research was supported by National Institutes of Health Grants ES03456 (GLW), AG19602 (GLW), ES05865 (SPL), and NS 041208 (SPL); and the Glenn/AFAR Grant for Biological Research in Aging from the American Federation for Aging Research.
Abbreviations used
- AP site
apurinic/apyrimidinic site
- APN1
yeast AP endonuclease
- APE1
mammalian AP endonuclease
- BER
base excision repair
- DMEM
Dulbecco's modified Eagle medium
- 5′-dRPase
5′-deoxyribose phosphatase
- EF1a
elongation factor 1 alpha
- FPG
formamidopyrimidine DNA glycosylase
- HA
hemagglutinin
- HBSS
Hanks’ Balanced Salt Solution
- MEN
menadione bisulfate
- MnSOD
manganese superoxide dismutase
- mtDNA
mitochondrial DNA
- MTS
mitochondrial targeting sequence
- nDNA
nuclear DNA
- NLS
nuclear localization signal
- PBS
phosphate-buffered saline
- PD
Parkinson's disease
- PCNA
proliferating cell nuclear antigen
- PNK
polynucleotide kinase
- RCSN-3
Raul Caviedes Substantia Nigra
- ROS
reactive oxygen species
- THF
tetrahydrofuran
References
- Bender A, Krishnan KJ, Morris CM, et al. High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat. Genet. 2006;38:515–517. doi: 10.1038/ng1769. [DOI] [PubMed] [Google Scholar]
- Dizdaroglu M. Oxidative damage to DNA in mammalian chromatin. Mutat. Res. 1992;275:331–342. doi: 10.1016/0921-8734(92)90036-o. [DOI] [PubMed] [Google Scholar]
- Dobson AW, Xu Y, Kelley MR, LeDoux SP, Wilson GL. Enhanced mitochondrial DNA repair and cellular survival after oxidative stress by targeting the human 8-oxoguanine glycosylase repair enzyme to mitochondria. J. Biol. Chem. 2000;275:37 518–37 523. doi: 10.1074/jbc.M000831200. [DOI] [PubMed] [Google Scholar]
- Driggers WJ, LeDoux SP, Wilson GL. Repair of oxidative damage within the mitochondrial DNA of RINr 38 cells. J. Biol. Chem. 1993;268:22 042–22 045. [PubMed] [Google Scholar]
- Driggers WJ, Holmquist GP, LeDoux SP, Wilson GL. Mapping frequencies of endogenous oxidative damage and the kinetic response to oxidative stress in a region of rat mtDNA. Nucleic Acids Res. 1997;25:4362–4369. doi: 10.1093/nar/25.21.4362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emerling BM, Platanias LC, Black E, Nebreda AR, Davis RJ, Chandel NS. Mitochondrial reactive oxygen species activation of p38 mitogen-activated protein kinase is required for hypoxia signaling. Mol. Cell Biol. 2005;25:4853–4862. doi: 10.1128/MCB.25.12.4853-4862.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans MD, Cooke MS. Factors contributing to the outcome of oxidative damage to nucleic acids. Bioessays. 2004;26:533–542. doi: 10.1002/bies.20027. [DOI] [PubMed] [Google Scholar]
- Fortini P, Parlanti E, Sidorkina OM, Laval J, Dogliotti E. The type of DNA glycosylase determines the base excision repair pathway in mammalian cells. J. Biol. Chem. 1999;274:15 230–15 236. doi: 10.1074/jbc.274.21.15230. [DOI] [PubMed] [Google Scholar]
- Frei B, Winterhalter KH, Richter C. Menadione- (2-methyl-1,4-naphthoquinone-) dependent enzymatic redox cycling and calcium release by mitochondria. Biochemistry. 1986;25:4438–4443. doi: 10.1021/bi00363a040. [DOI] [PubMed] [Google Scholar]
- Frosina G. Counteracting spontaneous transformation via over-expression of rate-limiting DNA base excision repair enzymes. Carcinogenesis. 2001;22:1335–1341. doi: 10.1093/carcin/22.9.1335. [DOI] [PubMed] [Google Scholar]
- Garcia-Ruiz C, Colell A, Morales A, Kaplowitz N, Fernandez-Checa JC. Role of oxidative stress generated from the mitochondrial electron transport chain and mitochondrial glutathione status in loss of mitochondrial function and activation of transcription factor nuclear factor-kappa B: studies with isolated mitochondria and rat hepatocytes. Mol. Pharmacol. 1995;48:825–834. [PubMed] [Google Scholar]
- de Grey AD. Reactive oxygen species production in the mitochondrial matrix: complications for the mechanism of mitochondrial mutation accumulation. Rejuvenation Res. 2005;8:13–17. doi: 10.1089/rej.2005.8.13. [DOI] [PubMed] [Google Scholar]
- Gros L, Ishchenko AA, Ide H, Elder RH, Saparbaev MK. The major human AP endonuclease (Ape1) is involved in the nucleotide incision repair pathway. Nucleic Acids Res. 2004;32:73–81. doi: 10.1093/nar/gkh165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Y, Lu AL. Differential DNA recognition and glycosylase activity of the native human MutY homolog (hMYH) and recombinant hMYH expressed in bacteria. Nucleic Acids Res. 2001;29:2666–2674. doi: 10.1093/nar/29.12.2666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutteridge JM, Halliwell B. The measurement and mechanism of lipid peroxidation in biological systems. Trends Biochem. Sci. 1990;15:129–135. doi: 10.1016/0968-0004(90)90206-q. [DOI] [PubMed] [Google Scholar]
- Hazra TK, Izumi T, Kow YW, Mitra S. The discovery of a new family of mammalian enzymes for repair of oxidatively damaged DNA, and its physiological implications. Carcinogenesis. 2003;24:155–157. doi: 10.1093/carcin/24.2.155. [DOI] [PubMed] [Google Scholar]
- He YH, II, Wu M, Kobune M, Xu Y, Kelley MR, Martin WJ. Expression of yeast apurinic/apyrimidinic endonuclease (APN1) protects lung epithelial cells from bleomycin toxicity. Am. J. Respir. Cell Mol. Biol. 2001;25:692–698. doi: 10.1165/ajrcmb.25.6.4550. [DOI] [PubMed] [Google Scholar]
- Hu J, Imam SZ, Hashiguchi K, de Souza-Pinto NC, Bohr VA. Phosphorylation of human oxoguanine DNA glycosylase (alpha-OGG1) modulates its function. Nucleic Acids Res. 2005;33:3271–3282. doi: 10.1093/nar/gki636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes G, Murphy MP, Ledgerwood EC. Mitochondrial reactive oxygen species regulate the temporal activation of nuclear factor kappaB to modulate tumour necrosis factor-induced apoptosis: evidence from mitochondria-targeted antioxidants. Biochem. J. 2005;389:83–89. doi: 10.1042/BJ20050078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ischenko AA, Saparbaev MK. Alternative nucleotide incision repair pathway for oxidative DNA damage. Nature. 2002;415:183–187. doi: 10.1038/415183a. [DOI] [PubMed] [Google Scholar]
- Ishchenko AA, Sanz G, Privezentzev CV, Maksimenko AV, Saparbaev M. Characterisation of new substrate specificities of Escherichia coli and Saccharomyces cerevisiae AP endonucleases. Nucleic Acids Res. 2003;31:6344–6353. doi: 10.1093/nar/gkg812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishchenko AA, Ide H, Ramotar D, Nevinsky G, Saparbaev M. Alpha-anomeric deoxynucleotides, anoxic products of ionizing radiation, are substrates for the endonuclease IV-type AP endonucleases. Biochemistry. 2004;43:15210–15216. doi: 10.1021/bi049214+. [DOI] [PubMed] [Google Scholar]
- Ishchenko AA, Yang X, Ramotar D, Saparbaev M. The 3′–>5′ exonuclease of Apn1 provides an alternative pathway to repair 7,8-dihydro-8-oxodeoxyguanosine in Saccharomyces cerevisiae. Mol. Cell Biol. 2005;25:6380–6390. doi: 10.1128/MCB.25.15.6380-6390.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenner P. Oxidative stress in Parkinson's disease. Ann. Neurol. 2003;53(Suppl 3):S26–S36. doi: 10.1002/ana.10483. discussion S36–S28. [DOI] [PubMed] [Google Scholar]
- Jilani A, Vongsamphanh R, Leduc A, Gros L, Saparbaev M, Ramotar D. Characterization of two independent amino acid substitutions that disrupt the DNA repair functions of the yeast Apn1. Biochemistry. 2003;42:6436–6445. doi: 10.1021/bi034163m. [DOI] [PubMed] [Google Scholar]
- Johnson AW, Demple B. Yeast DNA diesterase for 3′-fragments of deoxyribose: purification and physical properties of a repair enzyme for oxidative DNA damage. J. Biol. Chem. 1988;263:18009–18016. [PubMed] [Google Scholar]
- Lim KS, Jeyaseelan K, Whiteman M, Jenner A, Halliwell B. Oxidative damage to mitochondrial DNA is not extensive. Ann. NY Acad. Sci. 2005;1042:210–220. doi: 10.1196/annals.1338.023. [DOI] [PubMed] [Google Scholar]
- Mandavilli BS, Santos JH, Van Houten B. Mitochondrial DNA repair and aging. Mutat. Res. 2002;509:127–151. doi: 10.1016/s0027-5107(02)00220-8. [DOI] [PubMed] [Google Scholar]
- Martins EA, Meneghini R. DNA damage and lethal effects of hydrogen peroxide and menadione in Chinese hamster cells: distinct mechanisms are involved. Free Radic. Biol. Med. 1990;8:433–440. doi: 10.1016/0891-5849(90)90056-o. [DOI] [PubMed] [Google Scholar]
- Mol CD, Hosfield DJ, Tainer JA. Abasic site recognition by two apurinic/apyrimidinic endonuclease families in DNA base excision repair: the 3′ ends justify the means. Mutat. Res. 2000;460:211–229. doi: 10.1016/s0921-8777(00)00028-8. [DOI] [PubMed] [Google Scholar]
- Morrison H, Jernstrom B, Nordenskjold M, Thor H, Orrenius S. Induction of DNA damage by menadione (2-methyl-1,4-naphthoquinone) in primary cultures of rat hepatocytes. Biochem. Pharmacol. 1984;33:1763–1769. doi: 10.1016/0006-2952(84)90347-2. [DOI] [PubMed] [Google Scholar]
- Ngo EO, Sun TP, Chang JY, Wang CC, Chi KH, Cheng AL, Nutter LM. Menadione-induced DNA damage in a human tumor cell line. Biochem. Pharmacol. 1991;42:1961–1968. doi: 10.1016/0006-2952(91)90596-w. [DOI] [PubMed] [Google Scholar]
- Paris I, Dagnino-Subiabre A, Marcelain K, Bennett LB, Caviedes P, Caviedes R, Azar CO, Segura-Aguilar J. Copper neurotoxicity is dependent on dopamine-mediated copper uptake and one-electron reduction of aminochrome in a rat substantia nigra neuronal cell line. J. Neurochem. 2001;77:519–529. doi: 10.1046/j.1471-4159.2001.00243.x. [DOI] [PubMed] [Google Scholar]
- Parker AR, O'Meally RN, Sahin F, Su GH, Racke FK, Nelson WG, DeWeese TL, Eshleman JR. Defective human MutY phosphorylation exists in colorectal cancer cell lines with wild-type MutY alleles. J. Biol. Chem. 2003;278:47937–47945. doi: 10.1074/jbc.M306598200. [DOI] [PubMed] [Google Scholar]
- Parsons JL, Dianova II, Dianov GL. APE1 is the major 3′-phosphoglycolate activity in human cell extracts. Nucleic Acids Res. 2004;32:3531–3536. doi: 10.1093/nar/gkh676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petros JA, Baumann AK, Ruiz-Pesini E, et al. mtDNA mutations increase tumorigenicity in prostate cancer. Proc. Natl Acad. Sci. USA. 2005;102:719–724. doi: 10.1073/pnas.0408894102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popoff SC, Spira AI, Johnson AW, Demple B. Yeast structural gene (APN1) for the major apurinic endonuclease: homology to Escherichia coli endonuclease IV. Proc. Natl Acad. Sci. USA. 1990;87:4193–4197. doi: 10.1073/pnas.87.11.4193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramotar D, Kim C, Lillis R, Demple B. Intracellular localization of the Apn1 DNA repair enzyme of Saccharomyces cerevisiae. Nuclear transport signals and biological role. J. Biol. Chem. 1993;268:20 533–20 539. [PubMed] [Google Scholar]
- Roth TJ, Xu Y, Luo M, Kelley MR. Human-yeast chimeric repair protein protects mammalian cells against alkylating agents: enhancement of MGMT protection. Cancer Gene Ther. 2003;10:603–610. doi: 10.1038/sj.cgt.7700605. [DOI] [PubMed] [Google Scholar]
- Simmons RA, Suponitsky-Kroyter I, Selak MA. Progressive accumulation of mitochondrial DNA mutations and decline in mitochondrial function lead to beta-cell failure. J. Biol. Chem. 2005;280:28 785–28 791. doi: 10.1074/jbc.M505695200. [DOI] [PubMed] [Google Scholar]
- Starke-Reed PE, Oliver CN. Protein oxidation and proteolysis during aging and oxidative stress. Arch. Biochem. Biophys. 1989;275:559–567. doi: 10.1016/0003-9861(89)90402-5. [DOI] [PubMed] [Google Scholar]
- Storrie B, Madden EA. Isolation of subcellular organelles. Methods Enzymol. 1990;182:203–225. doi: 10.1016/0076-6879(90)82018-w. [DOI] [PubMed] [Google Scholar]
- Storz P, Doppler H, Toker A. Protein kinase D mediates mitochondrion-to-nucleus signaling and detoxification from mitochondrial reactive oxygen species. Mol. Cell Biol. 2005;25:8520–8530. doi: 10.1128/MCB.25.19.8520-8530.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh D, Wilson DM, III, Povirk LF. 3′-phosphodiesterase activity of human apurinic/apyrimidinic endonuclease at DNA double-strand break ends. Nucleic Acids Res. 1997;25:2495–2500. doi: 10.1093/nar/25.12.2495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tice RR, Agurell E, Anderson D, Burlinson B, Hartmann A, Kobayashi H, Miyamae Y, Rojas E, Ryu JC, Sasaki YF. Single cell gel/comet assay: guidelines for in vitro and in vivo genetic toxicology testing. Environ. Mol. Mutagen. 2000;35:206–221. doi: 10.1002/(sici)1098-2280(2000)35:3<206::aid-em8>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- Tomicic M, Eschbach E, Kaina B. Expression of yeast but not human apurinic/apyrimidinic endonuclease renders Chinese hamster cells more resistant to DNA damaging agents. Mutat. Res. 1997;383:155–165. doi: 10.1016/s0921-8777(96)00055-9. [DOI] [PubMed] [Google Scholar]
- Vongsamphanh R, Fortier PK, Ramotar D. Pir1p mediates translocation of the yeast Apn1p endonuclease into the mitochondria to maintain genomic stability. Mol. Cell Biol. 2001;21:1647–1655. doi: 10.1128/MCB.21.5.1647-1655.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace DC. Mitochondrial DNA sequence variation in human evolution and disease. Proc. Natl Acad. Sci. USA. 1994;91:8739–8746. doi: 10.1073/pnas.91.19.8739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace DC. The mitochondrial genome in human adaptive radiation and disease: on the road to therapeutics and performance enhancement. Gene. 2005;354:169–180. doi: 10.1016/j.gene.2005.05.001. [DOI] [PubMed] [Google Scholar]
- Wallace DC, Brown MD, Melov S, Graham B, Lott M. Mitochondrial biology, degenerative diseases and aging. Biofactors. 1998;7:187–190. doi: 10.1002/biof.5520070303. [DOI] [PubMed] [Google Scholar]
- Wiederhold L, Leppard JB, Kedar P, et al. AP endonuclease-independent DNA base excision repair in human cells. Mol. Cell. 2004;15:209–220. doi: 10.1016/j.molcel.2004.06.003. [DOI] [PubMed] [Google Scholar]
- Wilson SH, Kunkel TA. Passing the baton in base excision repair. Nat. Struct. Biol. 2000;7:176–178. doi: 10.1038/73260. [DOI] [PubMed] [Google Scholar]
- Wilson DM, III, Bennett RA, Marquis JC, Ansari P, Demple B. Trans-complementation by human apurinic endonuclease (Ape) of hypersensitivity to DNA damage and spontaneous mutator phenotype in apn1-yeast. Nucleic Acids Res. 1995;23:5027–5033. doi: 10.1093/nar/23.24.5027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu DC, Teismann P, Tieu K, Vila M, Jackson-Lewis V, Ischiropoulos H, Przedborski S. NADPH oxidase mediates oxidative stress in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine model of Parkinson's disease. Proc. Natl Acad. Sci. USA. 2003;100:6145–6150. doi: 10.1073/pnas.0937239100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yakes FM, Van Houten B. Mitochondrial DNA damage is more extensive and persists longer than nuclear DNA damage in human cells following oxidative stress. Proc. Natl Acad. Sci. USA. 1997;94:514–519. doi: 10.1073/pnas.94.2.514. [DOI] [PMC free article] [PubMed] [Google Scholar]