

Abstract
Expression of γ-glutamyl transpeptidase (GGT) in tumors contributes to resistance to radiation and chemotherapy. GGT is inhibited by glutamine analogues that compete with the substrate for the γ-glutamyl binding site. However, the glutamine analogues that have been evaluated in clinical trials are too toxic for use in humans. We have used high throughput screening to evaluate small molecules for their ability to inhibit GGT and have identified a novel class of inhibitors that are not glutamine analogues. These compounds are uncompetitive inhibitors, binding the γ-glutamyl enzyme complex. OU749, the lead compound, has an intrinsic Ki of 17.6 μm. It is a competitive inhibitor of the acceptor glycyl-glycine, which indicates that OU749 occupies the acceptor site while binding to the γ-glutamyl substrate complex. OU749 is more than 150-fold less toxic than the GGT inhibitor acivicin toward dividing cells. Inhibition of GGT by OU749 is species-specific, inhibiting GGT isolated from human kidney with 7–10-fold greater potency than GGT isolated from rat or mouse kidney. OU749 does not inhibit GGT from pig cells. Human GGT expressed in mouse fibroblasts is inhibited by OU749 similarly to GGT from human cells, which indicates that the species specificity is determined by differences in the primary structure of the protein rather than species-specific, post-translational modifications. These studies have identified a novel class of inhibitors of GGT, providing the basis for further development of a new group of therapeutics that inhibit GGT by a mechanism distinct from the toxic glutamine analogues.
The mechanism of inherent and acquired resistance of tumors to many forms of treatment involves glutathione. Elevated glutathione levels in tumors have been shown to contribute to resistance to chemotherapy and radiotherapy and prevent the initiation of the apoptotic cascade in tumor cells (1–5). The enzyme γ-glutamyl transpeptidase (GGT,2 EC 2.3.2.2), which is localized to the cell surface, cleaves the γ-glutamyl bond of extracellular glutathione, enabling the cell to use extracellular glutathione as a source of cysteine for increased synthesis of intracellular glutathione (6). GGT is induced in many human tumors, enhancing their resistance to chemotherapy (7, 8). Inhibiting GGT prior to chemotherapy or radiation would sensitize GGT-positive tumors to treatment. However, all known GGT inhibitors are too toxic for use in humans (9, 10).
GGT plays an essential role in releasing cysteine from extracellular glutathione. Most cells are unable to take up intact glutathione (6). In GGT knock-out mice, the absence of GGT in the renal proximal tubules results in the excretion of glutathione in the urine (11). In these mice, the glutathione in the glomerular filtrate cannot be cleaved into its constituent amino acids for reabsorption. GGT knock-out mice have a 2400-fold elevation of glutathione in their urine relative to their GGT-wild-type littermates. GGT knock-out mice grow slowly and die by 10 weeks of age due to a cysteine deficiency.
Inhibiting GGT for as little as 2 h lowers the intracellular cysteine concentration in GGT-positive tumors (3). Inhibitors of GGT activity could be used prior to the administration of chemotherapy to limit the supply of cysteine to the tumor, thereby blocking the ability of the tumor to maintain high levels of intracellular glutathione.
GGT catalyzes the cleavage of γ-glutamyl compounds and the transfer of the γ-glutamyl group to an acceptor substrate by a ping-pong kinetic mechanism (12). Glutathione and glutathione S conjugates are the most common physiologic substrates of GGT. They serve as the γ-glutamyl donor in the initial reaction. In the first reaction, the γ-glutamyl bond of the initial substrate is cleaved, the γ-glutamyl group becomes covalently bound to the enzyme, and the remainder of the substrate is released as the first product. With glutathione as the substrate, cysteinyl-glycine is released and is subsequently cleaved into cysteine and glycine by cell surface dipeptidases. In the second reaction of GGT transpeptidation, the γ-glutamyl group is transferred from the γ-glutamyl-GGT complex to the second substrate (the acceptor). Dipeptides and amino acids have the highest Km as acceptors. The second substrate with the covalently bound γ-glutamyl group is released as the second product from the enzyme.
Compounds that inhibit GGT include the glutamine analogues acivicin, 6-diazo-5-oxo-l-norleucine, and azaserine (Fig. 1) (13). Rational design of GGT inhibitors based on studies of the active site has led to the identification of additional γ-glutamyl analogues. Lherbet et al. (14, 15) have designed sulfur derivatives of l-glutamic acid that inhibit GGT. Han et al. (16, 17) have synthesized and tested a series of γ-(monophenyl)phosphono glutamate analogues that also functioned as inhibitors of GGT.
FIGURE 1.

Glutathione and glutamine analogues that inhibit GGT. Shown are the structures of glutathione (1) and the GGT inhibitors azaserine (2) and acivicin (3).
Evaluation of several of the glutamine analogues that inhibit GGT has shown them to be toxic (9, 10). Acivicin, the most potent inhibitor of GGT that has been tested clinically, is a neurotoxin (18). The neurotoxicity of the glutamine analogues may be due to interference with glutamine in recycling the neurotransmitter glutamate via the glutamate-glutamine cycle. Glutamine is also involved in the synthesis of several nucleotides and complex polysaccharides. Inhibition of these essential synthetic pathways can be toxic to dividing cells. Acivicin, 6-diazo-5-oxo-l-norleucine, and azaserine all cause bone marrow suppression (9). There is no known GGT inhibitor that can be used clinically.
Rather than design glutamine analogues, we have used physiologic conditions to screen libraries of small molecules to identify new inhibitors of GGT. This effort has lead to the identification of a novel class of uncompetitive inhibitors of GGT that are structurally distinct from and less toxic than the glutamine analogues. This new class of compounds occupies the acceptor site, not the γ-glutamyl site. The lead compound is species-specific, inhibiting human GGT, but with weak to no inhibitory activity toward GGT from mice, rats, and pigs.
EXPERIMENTAL PROCEDURES
High Throughput Screening—A high throughput method was developed to screen for inhibitors of GGT. The assays were conducted in 96-well plates. The final volume in each well was 100 μl. The assay buffer contained: 100 mm Na2HPO4, pH 7.4, with 3.2 mm KCl, 1.8 mm KH2PO4, and 27.5 mm NaCl. Each reaction contained 1 mm l-glutamic acid γ-4-nitroanilide HCl (GpNA) and 40 mm glycyl-glycine (glygly) (19). GGT is expressed on the cell surface, and the assay was initiated by the addition of 104 786-O cells (ATCC CRL-1932, a GGT-positive human renal cell adenocarcinoma line). Formation of the product, p-nitroaniline, at 37 °C was monitored continuously at A405 by a Bio-Rad model 680 microplate reader with Microplate Manager 5.2 software (Bio-Rad). One unit of GGT activity was defined as the amount of GGT that released 1 μmol of p-nitroaniline/min at 37 °C.
Screening Chemical Library—A 10,000 compound chemical library from the DIVERSet collection (ChemBridge Corp., San Diego, CA) was screened for inhibitors of GGT. The library was formatted in 96-well plates with 0.25 μmol of test compound per well. Stock solutions were prepared by the addition of 25 μl of dimethyl sulfoxide (DMSO) per well. For each test compound, 5 μl were added to the assay mixture, resulting in a final concentration of 500 μm. Glutathione is a competitive inhibitor of the GpNA substrate, and wells containing 400 μm oxidized glutathione were included on each 96-well plate as a positive control. Compounds that inhibited GGT activity to the same extent or greater than glutathione were scored as positive hits. The positive hits were retested at several concentrations for inhibition of GGT activity. For further testing, individual compounds and their structural analogues were purchased from ChemBridge, Specs (Delft, Netherlands), or Otava Chemicals (Kiev, Ukraine).
Enzyme Isolation—GGT was isolated from human kidney (National Disease Research Interchange, Philadelphia, PA), male Sprague-Dawley rat kidney and female BALB/c mouse kidney. Tissue was homogenized in 4 volumes of 25 mm Tris, pH 7.5, containing 0.33 m sucrose, 0.2 mm EDTA, 1 μm leupeptin, and 1.4 μg/ml aprotinin. A 9000 × g supernatant was spun at 100,000 × g for 1 h. The microsomal pellet was homogenized in 25 mm Tris, pH 7.35, 0.5% Triton X-100, 1 μm leupeptin, 1.4 μg/ml aprotinin and then centrifuged again at 100,000 × g for 1 h. The supernatant was assayed for GGT activity, aliquoted, and stored at –80 °C until further use. All solutions were maintained at 4 °C throughout the isolation. The specific activities of GGT were 3.4, 7.4, and 1.5 units/mg of protein for human, rat, and mouse preparations, respectively. Prior to use in the GGT assay, the enzyme was diluted in phosphate-buffered saline containing 0.025% Triton X-100, and 0.19 milliunits of enzyme were used per assay unless otherwise indicated.
Cloning of Human GGT and Expression in Pichia pastoris—The soluble domain of human GGT (amino acids 28–569) was amplified by PCR. The forward primer introduced an EcoRI restriction site and a tobacco etch virus protease-cleavable N-terminal His6 tag, whereas the reverse primer introduced a NotI restriction site after the stop codon of genomic GGT. The PCR product was digested with EcoRI and NotI and inserted into the corresponding sites of pPICZαA (Invitrogen), generating plasmid pMW-102. This construct was amplified in Escherichia coli DH5α cells. The fidelity of the recombinant open reading frame was verified by sequencing (the Oklahoma Medical Research Foundation, DNA Sequencing Core Facility, Oklahoma City, OK). The SacI-linearized plasmid, pMW-102, was purified and transformed into wild-type P. pastoris strain X-33, selected, and induced as recommended by Invitrogen. Secreted, recombinant, His6-tagged, human GGT was collected from the media with a nickel-nitrilotriacetic acid column. The hexahistidine tag was cleaved with His6-tagged tobacco etch virus protease. Tobacco etch virus protease and the cleaved histidine tag were separated from human GGT by collecting the flow-through from a second nickel-nitrilotriacetic acid column purification. The specific activity of the purified enzyme was 397.8 units/mg. Prior to use in the GGT assay, the enzyme was diluted in phosphate-buffered saline, and 2.8 milliunits of enzyme were used per assay.
Kinetic Studies—The assay buffer contained: 100 mm Na2HPO4, pH 7.4, with 3.2 mm KCl, 1.8 mm KH2PO4, and 27.5 mm NaCl. The concentrations of the substrate GpNA and the acceptor glygly were varied as indicated. Purified GGT was added to initiate the assay. The reaction was monitored as described for the high throughput screening.
Cytotoxicity Assays—786-O (ATCC CRL-1932), a human renal cell adenocarcinoma line, was plated in Dulbecco's modified Eagle's medium (DMEM) supplemented with 5% fetal bovine serum and penicillin/streptomycin (50 units/ml, 50 μg/ml) at 103 cells/well in 96-well plates. The next day, the medium was changed to fresh DMEM containing fetal bovine serum, antibiotics, and the test compound. Equivalent concentrations of DMSO were added to control wells. Cell viability was determined by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay 72 h after the addition of the test compound (20).
Cell Lines—LLC-PK1 (ATCC CRL-1392), a pig kidney cell line, NRK-52E (ATCC CRL-1571), a normal rat kidney cell line, LLC-MK2 (ATCC CCL-7), a normal kidney monkey cell line, and 786-O cells were grown in complete DMEM supplemented with 5% fetal bovine serum (Hyclone, Logan, UT), and penicillin/streptomycin (50 units/ml, 50 μg/ml) (Invitrogen). NIH/3T3 cells transfected with cDNA encoding human GGT were described previously and were grown in DMEM/F12 containing 200 μg/ml G418 (6). HK-2 (ATCC CRL-2190), a human cell line derived from immortalized normal proximal tubule cells, were grown in keratinocyte serum-free medium from ATCC (Manassas, VA). The cells were trypsinized off the plates and suspended in phosphate-buffered saline for the GGT assay.
Glutathione Degradation—Incubation mixture contained 1 mm glutathione, 40 mm glygly, and inhibitor in the GGT assay buffer. Reactions were initiated with the addition of 1.9 milliunits of GGT and incubated at 37 °C. The final volume of the reaction was 100 μl. Aliquots of the reaction mixture were removed at the specified time points, immediately acidified with an equal volume of cold 4.31% 5-sulfosalicylic acid, and maintained at 4 °C until the glutathione concentration was determined by the method of Tietze (21).
Data Analysis—Initial velocity data were first analyzed graphically using double reciprocal plots of initial velocities versus substrate concentration and suitable secondary plots. Data were then fitted using the appropriate equation and the Marquardt-Levenberg algorithm supplied with the EnzFitter program from BIOSOFT (Cambridge, UK). Kinetic parameters and their corresponding standard errors were estimated using a simple weighting method.
Data for competitive and uncompetitive inhibition were fitted using Equations 1 and 2.
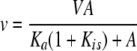 |
(Eq. 1) |
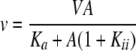 |
(Eq. 2) |
In Equations 1 and 2, v and V are the measured initial rate and maximum rate, respectively, Ka is the Michaelis constant for the varied substrate, and Kis and Kii are slope and intercept inhibition constants.
The LD50 and 95% confidence intervals of the test compounds in cell lines were calculated with a Prism log (inhibitor) versus normalized response (variable slope) curve fit (Prism, GraphPad Software Inc., San Diego, CA). A two-tailed t test was used to detect significant differences between OU749 inhibition of human GGT and inhibition of GGT from other species.
RESULTS
Screening Chemical Library for Inhibitors of GGT—A 10,000-compound chemical diversity library from ChemBridge Corp. was screened for inhibitors of GGT activity. Compounds in the DIVERSet library were chosen for maximum diversity of chemical structure. All compounds satisfied several physiochemical properties important for drug discovery: molecular weight ≤ 500, clogP ≤ 5, tPSA ≤ 100, rotatable bonds ≤ 8, hydrogen bond acceptors ≤ 10, and hydrogen bond donors ≤ 5 (22).
The initial screen identified 16 hits. When retested, 12 of the compounds were only weak inhibitors and not pursued further. Three of the remaining compounds were derivatives of isoindole-1,3-dione. Additional evaluation of this group of compounds revealed low solubility in aqueous solution and severe toxicity in a cell-based assay. These compounds were not pursued further. The last of the initial hits was a novel inhibitor of GGT. The inhibitor, N-[5-(4-methoxybenzyl)-1,3,4-thiadiazol-2-yl]benzenesulfonamide, was designated OU749. The structure of OU749 reveals that it is not a glutamine analogue (Fig. 2). It inhibited GGT isolated from human kidney in a dose-dependent manner (Fig. 2).
FIGURE 2.

Inhibition of GGT by OU749. Left and right panel, structure of OU749 (left panel) and substrate velocity curves (right panel) for the inhibition of human kidney GGT by 0 μm (▪), 15.2 μm (▵), 31.3 μm (▿), 62.5 μm (⋄), and 125 μm (○) OU749 in the presence of 40 mm glygly. Data shown are the average of triplicate values ± S.D. (For many points, the error bars are smaller than the symbol.)
Kinetic Analysis of Inhibition of GGT by OU749—To determine the mechanism of OU749 inhibition of human kidney GGT, inhibition patterns were obtained varying each of the substrates with the second maintained at a fixed concentration. With GpNA (the first substrate) varied, glygly was maintained at 40 mm (Kglygly is 11.4 ± 1.2 mm), whereas GpNA was maintained at 3 mm (KGpNA is 1.07 ± 0.04 mm) when glygly was varied. Inhibition by OU749 was uncompetitive with respect to GpNA, indicating that it binds to the covalent E-γ-glutamyl complex, the F form of the enzyme (Fig. 3A). The Ki value obtained was 73.8 ± 2.5 μm. Inhibition by OU749 was competitive with respect to glygly, indicating that OU749 was occupying the acceptor site (Fig. 3B). A Ki of 17.6 ± 3.8 μm was obtained. The Ki of 17.6 μm is the intrinsic value (23). The value obtained varying GpNA at a fixed glygly concentration must be the same once corrected for the concentration of fixed reactant. Given appKi = Ki(1 + [glygly]/Kglygly), the calculated value of Ki is 16.5 μm. A similar analysis of OU749 inhibition, carried out with a highly purified preparation of human GGT expressed in yeast, yielded similar results (Fig. 3, C and D). The appKi of OU749 as a competitive inhibitor of glygly was 25 ± 2 μm, whereas that obtained varying GpNA was 58.2 ± 1.6 μm. A fit of the data displayed in Fig. 3, A–D, to the equation for noncompetitive inhibition (v = VA/[Ka(1 + I/Kis) + A(1 + I/Kii)]) does give a finite but very high Ki (>200 μm) binding to the free form of the enzyme (E form). This is not surprising because the site for glygly, and thus OU749, must be present in the E form. However, the much weaker binding, >10-fold, is consistent with synergy of binding of OU749 in the presence of the γ-glutamyl moiety.
FIGURE 3.

Kinetic Analysis of GGT inhibition by OU749. A–D, double reciprocal plots of the initial velocities of human kidney GGT (A and B) or human GGT transfected into and isolated from P. pastoris (C and D) in the presence of 40 mm glygly (A and C) or 3 mm GpNA (B and D) with 0 μm (▪), 15.2 μm (▴), 31.3 μm (▵), 62.5 μm (⋄), and 125 μm (○) OU749. Data shown are average of triplicate values ± S.D. (For many points, the error bars are smaller than the symbol.)
Structure Activity Studies—A series of structural analogues of OU749 was evaluated as inhibitors of GGT. The appKi for each compound, obtained by varying GpNA, was determined experimentally with GGT isolated from human kidney. Compounds that had no inhibitory activity aided in defining elements that were essential for inhibition (Fig. 4). The data revealed that inhibition of GGT activity required the benzene ring attached to the sulfonamide group (Fig. 4, compound 1). The addition of a spacer carbon atom between the sulfur and the benzene ring eliminated inhibitory activity (Fig. 4, compound 2). Removing the carbon spacer atom between the thiadiazole ring and the end ring (Fig. 4, compound 3) or substitution of the thiadiazole ring with more complex structures eliminated inhibitory activity (Fig. 4, compounds 4 and 5). A core structure containing all the essential elements is shown in Fig. 5. The Ki for the unsubstituted core structure is 99.7 ± 3.1 μm. Inhibition is enhanced by the substitution of either the X or the Y position with chlorine (Fig. 5, compounds 2 and 4). Substitution of both the X and the Y position with chlorine provides the most potent inhibitor with an αKi of 28.7 ± 1.0 μm (Fig. 5, compound 1). The addition of a methoxy group at the Y position of the core compound yields our lead compound OU749, which is the third most potent inhibitor of the analogues tested (Fig. 5, compound 3). Substitution of the X position with either a methoxy compound or a nitroso group weakened the inhibition (Fig. 5, compounds 5 and 7–9). The addition of an acetamide group at the X position eliminates inhibition (Fig. 5, compound 10).
FIGURE 4.

Structural analogues of OU749 that do not inhibit GGT activity. 1, compound 1; 2, compound 2; 3, compound 3; 4, compound 4; 5, compound 5.
FIGURE 5.

Inhibition of GGT by structural analogues of OU749.
Toxicity of OU749—The glutamine analogues that inhibit GGT activity are toxic to dividing cells. We evaluated the toxicity of OU749 and several of its structural analogues toward cells in log growth using 786-O cells, a human renal tumor cell line. Cells were grown in the presence of the test compounds for 3 days. As shown in Table 1, the glutamine analogue, acivicin, had an LD50 of 0.81 μm. All four of the compounds tested (Fig. 5, compounds 1–3 and 6) were at least an order of magnitude less toxic than acivicin. OU749 (compound 3) was more than 150-fold less toxic than acivicin. These data emphasize the reduced toxicity of GGT inhibitors that are not glutamine analogues.
TABLE 1.
Toxicity of GGT inhibitors toward dividing 786-O cells
Although our structure activity analysis revealed two compounds that were more potent inhibitors of GGT than OU749 (Fig. 5, compounds 1 and 2), these compounds were 17–20-fold more toxic than OU749, making them less promising as candidates for further development for clinical use. Therefore OU749 continued as our lead compound in further characterization studies of inhibition by this class of compounds.
Species Specificity of GGT Inhibition by OU749—OU749 inhibits GGT isolated from human kidney in a dose-dependent manner. However, OU749 is 7-fold less potent as an inhibitor of GGT isolated from rat kidney and 10-fold less potent inhibiting GGT from mouse kidney (Fig. 6A). The species specificity of GGT inhibition by OU749 was further evaluated with cells lines from five different species (Fig. 6B). OU749 showed dose-dependent inhibition of GGT in the two human kidney cell lines, 786-O a renal tumor cell line and HK-2, an immortalized renal proximal tubule cell line. Inhibition of GGT in the human cell lines was of similar potency to the inhibition of GGT isolated from human kidney. GGT in the rat kidney cell line NRK-52E was only weakly inhibited by OU749, equivalent to the weak inhibition of GGT isolated from rat kidney. GGT in the pig kidney cell line LLC-PK1 was not inhibited by OU749. GGT in the monkey kidney cell line LLC-MK2 was only weakly inhibited by OU749, similar to the inhibition of GGT in rat kidney cells.
FIGURE 6.

Species-specific inhibition of GGT by OU749. A, inhibition of GGT from human kidney (•), rat kidney (▵), and mouse kidney (□) by OU749. Inhibition of human GGT was significantly more potent than inhibition of rat or mouse GGT (p = 0.03). B, inhibition of GGT in 786-O human renal adenocarcinoma cells (•), HK-2 normal human kidney-derived cells (▴), and NIH/3T3 mouse fibroblast transfected with human GGT cDNA (♦), LLC-MK2 monkey kidney cells(○), NRK-52E rat kidney cells (▵), and LLC-PK1 pig kidney (▿) cells. There was no significant difference in inhibition among the cell lines expressing human GGT, but OU749 inhibition of GGT in human cell lines was significantly more potent than inhibition of GGT in monkey, rat, or pig cell lines (p < 0.04).
GGT is heavily glycosylated (24). To determine whether the sensitivity of GGT to inhibition by OU749 was determined by the primary structure or by post-translational modifications such as glycosylation, we tested the sensitivity of human GGT expressed in mouse NIH/3T3 fibroblasts to inhibition by OU749 (Fig. 6B). The data revealed that human GGT expressed in mouse cells was inhibited by OU749 to the same extent as human GGT expressed in other human cells. Therefore the sensitivity of GGT to inhibition by OU749 was determined by the peptide sequence rather than species-specific post-translational modifications. Data in Fig. 2 further confirm that the primary structure rather than post-translational modifications determine the degree of inhibition of GGT by OU749. GGT isolated from human kidney (Fig. 2, A and B) is inhibited by OU749 to the same extent as human GGT expressed in yeast (Fig. 2, C and D).
OU749 Inhibits the Cleavage of Glutathione by GGT—The standard assay for GGT activity monitors the release of p-nitroaniline from the first substrate, γ-glutamyl-p-nitroanilide. To confirm that OU749 inhibits the cleavage of glutathione, the primary physiologic substrate of GGT, we monitored the breakdown of glutathione by human kidney GGT in the presence of the inhibitor. OU749 inhibited the cleavage of glutathione in a dose-dependent manner (Fig. 7A). OU749 also inhibited the cleavage of oxidized glutathione by human GGT (data not shown). To determine whether the species specificity of inhibition of GGT by OU749 was also relevant to the physiologic substrate, OU749 was evaluated for its ability to inhibit the degradation of glutathione by rat GGT. Rat GGT cleaved glutathione in a time-dependent fashion, as had been observed for human GGT (Fig. 7B). OU749 was unable to inhibit degradation of glutathione by GGT from rat kidney (Fig. 7B). These data corroborate the data regarding the species specificity of OU749 obtained using the synthetic substrate, GpNA.
FIGURE 7.

Species-specific inhibition of glutathione degradation by OU749. A and B, cleavage of glutathione by human GGT (A) or rat GGT (B) in the presence of 0 μm (▪), 62.5 μm (⋄), 125 μm (○), 250 μm (*), or 500 μm (□) OU749. Reactions contained 1.9 milliunits of GGT, 40 mm glygly, and 1 mm glutathione. OU749 inhibited glutathione breakdown by human GGT but did not inhibit glutathione breakdown by rat GGT.
DISCUSSION
We have identified a novel class of GGT inhibitors that are not glutamine analogues. Kinetic studies of the lead compound OU749 revealed that the mechanism of inhibition was uncompetitive relative to the γ-glutamyl substrate, indicating that the inhibitor bound the enzyme-substrate complex. In contrast to competitive inhibitors, which lose potency as substrate concentration builds, uncompetitive inhibitors become more potent as the substrate concentration rises in an inhibited open system. Data from the GGT knock-out mice show that in the absence of GGT activity, glutathione levels decrease in tissues, but the concentration of glutathione in the serum rises more than 6-fold, likely due to the inability of cells to cleave glutathione and recover the amino acids (11). In addition, the glutathione concentration in the urine increases more than 2400-fold as the glutathione transits the proximal tubules intact in the absence of GGT. Westley and Westley (25) have argued that uncompetitive inhibitors are superior to competitive inhibitors for instituting change in open systems such as those found in vivo where substrate concentrations rise with enzyme inhibition. Many uncompetitive inhibitors function by locking the enzyme-substrate complex in form after initial product release but before conversion of the enzyme back to the native form. In GGT, the enzyme-substrate complex consists of the glutamyl group covalently bound to the enzyme following release of the remainder of the γ-glutamyl substrate. GGT from E. coli has been crystallized (26). Analysis of the crystal structure of E. coli GGT and the γ-glutamyl-enzyme intermediate revealed that the N-terminal threonine of the small subunit is the catalytic nucleophile in the enzymatic reaction (27). This threonine is conserved in human, rat, mouse, and pig. Site-directed mutagenesis of human GGT has identified four amino acids that are essential to GGT activity (one in the large subunit, Arg 107, and three in the small subunit, Asp-423, Ser-451, and Ser-452) (28–30). All four of these amino acids are identical in human, rat, mouse, and pig.
Kinetic studies further revealed that while binding the enzyme-γ-glutamyl complex, OU749 occupies the acceptor site. The acceptor site is not well defined. To date, only the crystal structures of GGT isolated from E. coli and Helicobacter pylori have been published (27, 31, 32). No information on mutational analysis that alters the acceptor site has been published. Our finding that inhibition by OU749 is species-specific with high affinity for human, lower affinity for monkey, rat, and mouse, and no inhibition of pig GGT may aid in the delineation of the acceptor site. The sequence of GGT from human, rat, mouse, and pig have been published. Alignment of these sequences is included in the supplemental data. However, no pattern is apparent in the sequence alignment that would provide insight into the amino acids or regions of the polypeptide sequence that are critical to inhibitor binding. Alignment of the GGT peptide sequences does reveal that one of the seven potential N-glycosylation sites in human GGT (Asn-266) is not present in rat, mouse, or pig. Modeling the mammalian sequences based on the crystal structure of the bacterial enzyme does not highlight any of the amino acids as important in inhibitor binding. There are no other reports of species-specific inhibitors of GGT, although the effect of sulfhydral reagents on GGT activity appears to have some species specificity (33).
Structural alterations of OU749 increased the inhibitory activity of the compound but not without accompanying increases in toxicity. The in vitro toxicity profile of OU749 is favorable. In a dividing cell model, OU749 is 150-fold less toxic than acivicin, which was abandoned after phase I clinical trials due to toxicity.
In addition to its role in cancer therapy, GGT also plays a critical role in drug metabolism due to the ubiquitous presence of glutathione. GGT is an essential enzyme in the formation of mercapturic acids in the kidney and initiates the activation of halogenated alkenes and other drugs to potent kidney toxins through this pathway (34, 35). Cisplatin has been shown to be bioactivated to a renal toxin through the mercapturic acid pathway (36, 37). Inhibition of GGT during cisplatin-based chemotherapy would not only sensitize the tumors to the therapy, it would also block the kidney toxicity of cisplatin. Additional clinical conditions for which a GGT inhibitor may have therapeutic benefit include cardiovascular disease and asthma as nitric oxide is transported in the blood as a glutathione conjugate and requires GGT activity for its release (38, 39). Finally, GGT is one of two enzymes that metabolize leukotriene C4 to leukotriene D4, a mediator of inflammation common to many diseases (40).
Previous clinical studies have attempted to overcome drug resistance in tumors by inhibiting glutathione synthesis with buthionine sulfoximine (41), an inhibitor of the rate-limiting enzyme in the synthesis of glutathione. However, there was no depletion of cysteine in the body with this protocol. The sulfur of cysteine, the active nucleophilic group of the glutathione molecule, binds and inactivates reactive, electrophilic compounds. Free cysteine will also react with, and inactivate, chemotherapy drugs (42). Inhibition of GGT both reduces intracellular glutathione and depletes cysteine levels, increasing the sensitivity of the tumor to the drug. Studies in mice have shown that inhibiting GGT for as little as 2 h selectively lowers the intracellular cysteine concentration in GGT-positive tumors (3). Mena et al. (4) used acivicin to deplete tumor glutathione in combination with aggressive therapy and achieved complete cure of metastatic melanoma to the liver in 90% of test animals. Development of less toxic GGT inhibitors, such as OU749, holds great promise for enhanced cancer therapy.
Supplementary Material
Acknowledgments
We gratefully acknowledge the assistance of Terri McHugh, Department of Cell Biology, OUHSC for assistance with the enzyme assays and Kostyantyn Bobyk in the Department of Chemistry and Biochemistry, University of Oklahoma for assistance with enzyme kinetics calculations and preparing the graphs for Fig. 3.
This work was supported, in whole or in part, by National Institutes of Health Grant RO1CA57530 from the NCI (to M. H. H.) and by Grant HR03-044 from the Oklahoma Center for the Advancement of Science and Technology (to M. H. H.). This work was also supported by the Grayce B. Kerr endowment to the University of Oklahoma to support the research of P. F. C.
The on-line version of this article (available at http://www.jbc.org) contains a supplemental figure.
Footnotes
The abbreviations used are: GGT, γ-glutamyl transpeptidase; GpNA, l-glutamic acid γ-4-nitroanilide HCl; glygly, glycyl-glycine; DMSO, dimethyl sulfoxide; DMEM, Dulbecco's modified Eagle's medium.
References
- 1.Benlloch, M., Ortega, A., Ferrer, P., Segarra, R., Obrador, E., Asensi, M., Carretero, J., and Estrela, J. M. (2005) J. Biol. Chem. 280 6950–6959 [DOI] [PubMed] [Google Scholar]
- 2.Ahmad, S., Okine, L., Wood, R., Aljian, J., and Vistica, D. T. (1987) J. Cell. Physiol. 131 240–246 [DOI] [PubMed] [Google Scholar]
- 3.Ruoso, P., and Hedley, D. W. (2004) Cancer Chemother. Pharmacol. 54 49–56 [DOI] [PubMed] [Google Scholar]
- 4.Mena, S., Benlloch, M., Ortega, A., Carretero, J., Obrador, E., Asensi, M., Petschen, I., Brown, B. D., and Estrela, J. M. (2007) Clin. Cancer Res. 13 2658–2666 [DOI] [PubMed] [Google Scholar]
- 5.Lee, C. Y., Wey, S. P., Liao, M. H., Hsu, W. L., Wu, H. Y., and Jan, T. R. (2008) Int. Immunopharmacol. 8 732–740 [DOI] [PubMed] [Google Scholar]
- 6.Hanigan, M. H., and Ricketts, W. A. (1993) Biochemistry 32 6302–6306 [DOI] [PubMed] [Google Scholar]
- 7.Hanigan, M. H., Frierson, H. F., Jr., Swanson, P. E., and De Young, B. R. (1999) Hum. Pathol. 30 300–305 [DOI] [PubMed] [Google Scholar]
- 8.Hanigan, M. H., Gallagher, B. C., Townsend, D. M., and Gabarra, V. (1999) Carcinogenesis 20 553–559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ahluwalia, G. S., Grem, J. L., Hao, Z., and Cooney, D. A. (1990) Pharmacol. Ther. 46 243–271 [DOI] [PubMed] [Google Scholar]
- 10.Lyons, S. D., Sant, M. E., and Christopherson, R. I. (1990) J. Biol. Chem. 265 11377–11381 [PubMed] [Google Scholar]
- 11.Lieberman, M. W., Wiseman, A. L., Shi, Z., Carter, B. Z., Barrios, R., Ou, C., Chevez-Barrios, P., Wand, Y., Habib, G. M., Goodman, J. C., Huang, S. L., Lebovitz, R. M., and Matzuk, M. M. (1996) Proc. Natl. Acad. Sci. U. S. A. 93 7923–7926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Folk, J. E. (1969) J. Biol. Chem. 244 3707–3713 [PubMed] [Google Scholar]
- 13.Tate, S. S., and Meister, A. (1978) Proc. Natl. Acad. Sci. U. S. A. 75 4806–4809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lherbet, C., Gravel, C., and Keillor, J. W. (2004) Bioorg. Med. Chem. Lett. 14 3451–3455 [DOI] [PubMed] [Google Scholar]
- 15.Lherbet, C., and Keillor, J. W. (2004) Org. Biomol. Chem. 2 238–245 [DOI] [PubMed] [Google Scholar]
- 16.Han, L., Hiratake, J., Kamiyama, A., and Sakata, K. (2007) Biochemistry 46 1432–1447 [DOI] [PubMed] [Google Scholar]
- 17.Han, L., Hiratake, J., Tachi, N., Suzuki, H., Kumagai, H., and Sakata, K. (2006) Bioorg. Med. Chem. 14 6043–6054 [DOI] [PubMed] [Google Scholar]
- 18.Taylor, S. A., Crowley, J., Pollocck, T. W., Eyre, H. J., Jaeckle, C., Hynes, H. E., and Stephens, R. L. (1991) J. Clin. Oncol. 9 1476–1479 [DOI] [PubMed] [Google Scholar]
- 19.Orlowski, M., and Meister, A. (1963) Biochim. Biophys. Acta 73 679–681 [DOI] [PubMed] [Google Scholar]
- 20.Hansen, M. B., Nielsen, S. E., and Berg, K. (1989) J. Immunol. Methods 119 203–210 [DOI] [PubMed] [Google Scholar]
- 21.Tietze, F. (1969) Anal. Biochem. 27 502–522 [DOI] [PubMed] [Google Scholar]
- 22.Lipinski, C. A., Lombardo, F., Dominy, B. W., and Feeney, P. J. (2001) Adv. Drug Delivery Rev. 46 3–26 [DOI] [PubMed] [Google Scholar]
- 23.Cook, P. F., and Cleland, W. W. (2007) Enzyme Kinetics and Mechanisms, Garland Press, London
- 24.Yamashita, K., Hitoi, A., Matsuda, Y., Miura, T., Katunuma, N., and Kobata, A. (1986) J. Biochem. (Tokyo) 99 55–62 [DOI] [PubMed] [Google Scholar]
- 25.Westley, A. M., and Westley, J. (1996) J. Biol. Chem. 271 5347–5352 [DOI] [PubMed] [Google Scholar]
- 26.Kumagai, H., Nohara, S., Suzuki, H., Hashimoto, W., Yamamoto, K., Sakai, H., Sakabe, K., Fukuyama, K., and Sakabe, N. (1993) J. Mol. Biol. 234 1259–1262 [DOI] [PubMed] [Google Scholar]
- 27.Okada, T., Suzuki, H., Wada, K., Kumagai, H., and Fukuyama, K. (2006) Proc. Natl. Acad. Sci. U. S. A. 103 6471–6476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ikeda, Y., Fujii, J., Anderson, M. E., Taniguchi, N., and Meister, A. (1995) J. Biol. Chem. 270 22223–22228 [DOI] [PubMed] [Google Scholar]
- 29.Ikeda, Y., Fujii, J., Taniguchi, N., and Meister, A. (1995) J. Biol. Chem. 270 12471–12475 [PubMed] [Google Scholar]
- 30.Ikeda, Y., Fujii, J., and Taniguchi, N. (1993) J. Biol. Chem. 268 3980–3985 [PubMed] [Google Scholar]
- 31.Morrow, A. L., Williams, K., Sand, A., Boanca, G., and Barycki, J. J. (2007) Biochemistry 46 13407–13414 [DOI] [PubMed] [Google Scholar]
- 32.Boanca, G., Sand, A., Okada, T., Suzuki, H., Kumagai, H., Fukuyama, K., and Barycki, J. J. (2007) J. Biol. Chem. 282 534–541 [DOI] [PubMed] [Google Scholar]
- 33.Allison, R. D. (1985) Methods Enzymol. 113 419–437 [DOI] [PubMed] [Google Scholar]
- 34.Anders, M. W., and Dekant, W. (1998) Annu. Rev. Pharmacol. Toxicol. 38 501–537 [DOI] [PubMed] [Google Scholar]
- 35.Bai, F., Jones, D. C., Lau, S. S., and Monks, T. J. (2001) Chem. Res. Toxicol. 14 863–870 [DOI] [PubMed] [Google Scholar]
- 36.Hanigan, M. H., Gallagher, B. C., Taylor, P. T. Jr., and Large, M. K. (1994) Cancer Res. 54 5925–5929 [PubMed] [Google Scholar]
- 37.Hanigan, M. H., Lykissa, E. D., Townsend, D. M., Ou, C. N., Barrios, R., and Lieberman, M. W. (2001) Am. J. Pathol. 159 1889–1894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Henson, S. E., Nichols, T. C., Holers, V. M., and Karp, D. R. (1999) J. Immunol. 163 1845–1852 [PubMed] [Google Scholar]
- 39.Lipton, A. J., Johnson, M. A., Macdonald, T., Lieberman, M. W., Gozal, D., and Gaston, B. (2001) Nature 413 171–174 [DOI] [PubMed] [Google Scholar]
- 40.Carter, B. Z., Wiseman, A. L., Orkiszewski, R., Ballard, K. D., Ou, C. N., and Lieberman, M. W. (1997) J. Biol. Chem. 272 12305–12310 [DOI] [PubMed] [Google Scholar]
- 41.O'Dwyer, P. J., Hamilton, T. C., Young, R. C., LaCreta, F. P., Carp, N., Tew, K. D., Padavic, K., Comis, L., and Ozols, R. F. (1992) J. Natl. Cancer Inst. 84 264–267 [DOI] [PubMed] [Google Scholar]
- 42.Bose, R. N., Ghosh, S. K., and Moghaddas, S. (1997) J. Inorg. Biochem. 65 199–205 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.