

Abstract
Cardiac hypertrophy develops in response to a variety of cardiovascular stresses and results in activation of numerous signaling cascades and proteins. In the present study, we demonstrate that cytoglobin is a stress-responsive hemoprotein in the hypoxia-induced hypertrophic myocardium and it is transcriptionally regulated by calcineurin-dependent transcription factors. The cytoglobin transcript level is abundantly expressed in the adult heart and in response to hypoxia cytoglobin expression is markedly up-regulated within the hypoxia-induced hypertrophic heart. To define the molecular mechanism resulting in the induction of cytoglobin, we undertook a transcriptional analysis of the 5′ upstream regulatory region of the cytoglobin gene. Evolutionarily conserved binding elements for transcription factors HIF-1, AP-1, and NFAT are located within the upstream region of the cytoglobin gene. Transcriptional assays demonstrated that calcineurin activity modulates cytoglobin transcription. Increased calcineurin activity enhances the ability of NFAT and AP-1 to bind to the putative cytoglobin promoter, especially under hypoxic conditions. In addition, inhibition of calcineurin, NFAT, and/or AP-1 activities decreases endogenous cytoglobin transcript and protein levels. Thus, the regulation of cytoglobin transcription by calcineurin-dependent transcription factors suggests that cytoglobin may have a functional role in calcium-dependent events accompanying cardiac remodeling.
The development of ventricular hypertrophy is an adaptive response to a variety of cardiovascular stresses (e.g. hypoxia, ischemia, hypertension, pulmonary hypertension). Although hypertrophy serves to initially enhance cardiac output and normalize ventricular wall stress, the failure to alleviate the inciting stress results in this response becoming maladaptive, eventually leading to cardiac decompensation and heart failure. Ventricular hypertrophy has been implicated as an important predisposing risk factor for increased cardiovascular morbidity and mortality in humans (1). Alterations in the redox state and activation of various signaling transduction pathways and stress-responsive proteins are known to occur in ventricular hypertrophy and these either directly contribute to cardiac growth or are activated in response to hypertrophy (2, 3). Therefore, recent research efforts have focused on identifying signaling pathways and candidate proteins that may serve as therapeutic targets to ameliorate the development of ventricular hypertrophy.
Cytoglobin (Cygb)4 is a recently identified stress-responsive hemoprotein, evolutionarily conserved among human, rat, and mouse (4–7). The Cygb protein has a moderate degree of homology to myoglobin (Mb), and shares several key amino acid residues that are important for the structure and function of all hemoproteins (4, 5, 8, 9). However, unlike Mb, which is tissue-restricted to cardiomyocytes and oxidative skeletal myofibers, Cygb is more broadly expressed in adult tissues (4, 5, 9). In vitro studies indicate that Cygb is able to bind to oxygen (O2), nitric oxide (NO), and free radicals (5, 10). These studies also suggest that Cygb may be more important in scavenging free radicals than in modulating O2 metabolism.
Studies to date that have defined the functional role(s) for Cygb in mammalian tissues are limited and have not investigated the role of Cygb in cardiovascular biology (11, 12). The objective in the present study was to define changes in Cygb gene expression in response to myocardial hypoxia and to investigate the regulatory mechanisms that govern changes in its gene expression profile. Identifying the regulatory mechanisms that modulate Cygb expression will provide important biological insight into its cellular function.
The regulatory region of the Cygb gene has not been fully investigated. However, in the case of Mb, it has been well established that the 2-kb upstream region of the Mb gene contains the essential motifs that regulate its expression (13). Specifically, there are two evolutionarily conserved motifs in the Mb promoter that are recognized by nuclear factor of activated T cell (NFAT), a substrate for calcineurin, and it has been clearly demonstrated that these two motifs directly regulate Mb transcription (13–17). Based upon the close homology of Cygb to Mb, we hypothesized that Cygb transcription is regulated by calcineurin-dependent transcription factors (NFAT and/or activator protein-1 (AP-1)). Given its response to hypoxia, we further hypothesized that Cygb expression is regulated by a hypoxia-responsive transcription factor. Therefore, in addition to NFAT and AP-1, we evaluated whether the transcription factor, hypoxia-inducible factor-1 (HIF-1), a heterodimer composed of HIF-1α and HIF-1β subunits, also regulates Cygb gene expression.
In this study, we demonstrate that Cygb transcription is increased in the hypoxia-induced hypertrophic myocardium. We present evidence that hypoxia increases binding of the transcription factors NFAT and AP-1 to the proximal promoter region of the Cygb gene. Furthermore, activation of both of these transcriptional elements is dependent on the activity of the phosphatase, calcineurin. Thus, our study enhances our overall understanding of the potential functional role for Cygb and supports the concept that Cygb is a stress-responsive hemoprotein that is sensitive to hypoxic stress.
EXPERIMENTAL PROCEDURES
Animals and Tissue Processing—Adult C57BL/6 mice (2–4 months of age) were handled in accordance with National Institutes of Health and the University of Texas Southwestern Medical Center's (UTSW) Institutional Guidelines. Transgenic mice overexpressing either constitutively active calcineurin (CnA*) or regulator of calcineurin-1 (Rcan1) within the heart were generated utilizing the α-MHC promoter to specifically direct expression to the cardiomyocyte as previously described (18, 19). Hearts were harvested from control and experimental C57BL/6 adult male mice and processed for morphological analyses and Western blot analysis as described previously (7, 20).
Hypoxia Chamber—An in vivo plexiglass hypoxia chamber (87 × 42 × 45 cm) was engineered to maintain a constant hypoxic environment of 10% oxygen (O2) and was monitored continuously for oxygen and carbon dioxide concentrations, temperature, and humidity. A detailed description of the hypoxia chamber has been published previously (20). In addition, an in vitro hypoxia chamber (Billups-Rothenberg Inc.) was designed to expose C2C12 myoblasts to hypoxic conditions (1% O2, 5% CO2, 94% N2) for varying lengths of time. This airtight modular chamber is 30 cm in diameter with inflow and outflow valves that maintain a constant hypoxic environment (1% O2). The humidity within the chamber was maintained constant during the duration of a hypoxic experiment by placing a Petri dish filled with 15 ml of sterile water within the chamber.
RNA Isolation and Quantitative Reverse Transcription-PCR—Total RNA was isolated from normoxic and hypoxic hearts utilizing the TriPure isolation kit (Roche Diagnostics). Reverse transcription (RT) was performed using Superscript™ II RNase H-reverse transcriptase (Invitrogen) to obtain cDNA as described previously (7, 21). Quantitative reverse transcription polymerase chain reaction (qRT-PCR) was performed as described previously, and Cygb expression was normalized to 18s ribosome (18s-RIB) expression (21). The primer sets used to assess Cygb and 18s-RIB expression levels in these experiments were as follows: Cygb forward primer, 5′-CCAACTGCGAGGAC-3′; Cygb reverse primer, 5′-AGTTCACAAAGAAC-3′; 18s-RIB forward primer, 5′-TGCCACTGGCTGGT-3′; and 18s-RIB reverse primer, 5′-TTCACCACTTCTCT-3′.
Northern Blot Analysis—The Strip-EZ DNA probe synthesis kit (Ambion) was utilized to construct the Cygb radioactive probe. Twenty-five nanograms of linearized Cygb cDNA and 5 μl of [α-32P]dATP (1000 Ci/mmol; Amersham Biosciences) were used to produce the radioactive Cygb cDNA probe. A commercially purchased Northern blot membrane (MTN blot; BD Sciences) containing full-length transcripts from various adult murine tissues was incubated with the Cygb radioactive probe in Ultrahyb solution (Ambion) at 42 °C overnight. Following rinsing, the hybridized membrane was exposed to radiographic film at –80 °C for various lengths of time (6–48 h). β-Actin cDNA was used as a control probe to ensure equal loading of the lanes with RNA.
Western Blot Analysis—Protein from whole heart or cell lysates was isolated from control and experimental groups. Western blot analysis was performed as published previously (7, 19, 20). Western blot analyses of nuclear and cytosolic extracts from cells and murine hearts were also processed as described previously (22). Polyclonal rabbit anti-Cygb serum was designed and engineered by the Antibody Core Facility at the University of Texas Southwestern Medical Center. This polyclonal rabbit anti-Cygb serum (1:100 dilution) was used as the primary antiserum, which was detected using a horseradish peroxidase-conjugated secondary anti-serum. Western blot analyses to assess CnA, c-Jun, and c-Fos protein expression levels were performed using the respective antisera (CnA 1:1000, Sigma; c-Jun 1:200 and c-Fos 1:200, Santa Cruz Biotechnology). Antisera to lamin A/C and Sp1 were used to ensure the purity of the nuclear extracts (lamin A/C 1:200 and Sp1 1:200; Santa Cruz Biotechnology.). The mouse monoclonal α-tubulin antibody (1:3,000; Sigma) was used as a standard to ensure equal protein loading of the lanes. Band intensity was quantitated using a computerized digital analysis program (ImageJ 1.24; Scion Corp.).
Plasmid Construction and Luciferase Reporter Assay—Segments of the 5′ upstream region of the Cygb gene were cloned into the pGL3-Basic vector (Promega) and inserted upstream of the luciferase reporter. NFAT and AP-1 responsive elements (NRE and ARE) within the mouse Cygb promoter region were disrupted using PCR-based site-directed mutagenesis as described previously (14, 17). CMV-driven expression vectors were generously provided by Drs. Eric Olson (UTSW; CnA*, NFATc1-GFP, and Rcan1), Richard Bruick (UTSW; constitutively active hypoxia-inducible factor 1α (HIF-1*) and hypoxia response element fused to a luciferase reporter (HRE-Luc)), and Michael Birrer (NCI, National Institutes of Health; c-Fos, and c-Jun) (18, 19, 23, 24).
Because transient transfection of plasmids into primary cardiomyocytes is inefficient, with low transfection rates, we utilized C2C12 cells as a muscle cell line to undertake our transcriptional analysis. C2C12 myoblasts were cultured in growth media as described previously (14–19). Utilizing Lipofectamine (Invitrogen), each well of a 6-well plate was co-transfected with the Cygb promoter-reporter plasmid (0.5 μg) and an expression plasmid driven by a CMV promoter (CnA*, HIF-1*, Rcan1, NFATc1-GFP, and/or c-Fos/c-Jun) or empty vector (pCl-NEO; 0.5 μg), along with a CMV-LacZ plasmid (0.5 μg) as an internal control for transfection efficiency. In experiments designed to assess the effect of inhibiting calcineurin activity on Cygb transcription, the cells were either co-transfected with a CMV-driven Rcan1 expression plasmid or treated with cyclosporin A (CsA, 10 μm; Calbiochem). After transfection of the respective plasmids, the cells were cultured under either normoxic (21% O2, 5% CO2, 74% N2) or hypoxic (1% O2, 5% CO2, 94% N2) conditions. After a 16-h exposure to either normoxia or hypoxia, the C2C12 cells were harvested. Luciferase and β-galactosidase assays of whole cell extracts were conducted as described previously (14–19). Finally, to demonstrate that the cells were exposed to hypoxic conditions, HRE-Luc was transfected into C2C12 cells with or without the HIF-1* plasmid, and luciferase activity was assessed (data not shown).
Quantification of NFAT Nuclear Translocation—C2C12 cells were transiently transfected with an NFATc1-GFP expression plasmid in 6-well culture plates. Utilizing a Zeiss Axioplan 2iE photomicroscope equipped with epifluorescence, subcellular localization of NFATc1-GFP fluorescence was monitored in response to hypoxia (1% O2 for 16 h) and in the presence of CnA*. Each well was divided into four quadrants, and photomicrography of each quadrant was achieved using this microscope and a Zeiss Axiocam digital charge-coupled device grayscale camera interfaced with a Macintosh G4 computer. Images were captured using Openlab 4.0.1 acquisition and analysis software (Improvision Inc.). In each quadrant, the number of GFP-positive nuclei was divided by the total number of GFP-positive cells. This value represented the percentage of cells in that quadrant in which NFAT had translocated into the nucleus. The nuclei were identified by Hoechst staining. The final quantification for each well was the average value obtained from the four quadrants, and the experiment was repeated in triplicate.
Electrophoretic Mobility Shift Assay—To discern the authenticity of the AP-1 binding site within the Cygb promoter region, an electrophoretic mobility shift assay (EMSA) was undertaken with double-stranded oligonucleotides corresponding to the conserved ARE site at –432. The assay was performed as described previously (18, 22, 25). In brief, cell lysates were harvested from normoxic and hypoxic C2C12 myoblasts and cells overexpressing CnA* with and without CsA. Nuclear and cytosolic extracts were prepared as described (22, 25). The sequences for the probes were as follows: wild-type (WT) AP-1 probe, 5′-CTTCTCCGCGTGACCCCCTGATCCT-3′; mutated AP-1 probe, 5′-CTTCTCCGCGAAAACCCCTGATCCT-3′. The 32P-labeled probe was mixed with 10 μg of nuclear protein extract in 2 μl of 10× binding buffer (20 mm HEPES, 4 mm EDTA, 4 mm dithiothreitol, and 50% glycerol). To this mixture was added 2 μl of 10% Nonidet P-40 and 1 μl of 1 μg/μl poly(dI:dC). The final volume was 20 μl, and the reaction mixture was incubated on ice for 20 min. The reaction products were resolved on a 5% nondenaturing polyacrylamide gel. The gel was dried under vacuum at 80 °C for 1 h and then exposed to radiographic film. Cold competition was carried out in the presence of a 10× molar excess of unlabeled probe, and for the supershift reactions, the nuclear extracts were incubated with 1 μg of polyclonal rabbit anti-c-Jun and anti-c-Fos sera (Santa Cruz Biotechnology Inc.) for 30 min prior to initiation of the binding reaction.
Chromatin Immunoprecipitation Assay—Cell lysates were harvested from normoxic and hypoxic C2C12 myoblasts and C2C12 cells overexpressing CnA*. Utilizing the ChIP Kit-M (Progeneron), a chromatin immunoprecipitation (ChIP) assay was undertaken to evaluate the physical interaction among AP-1, NFAT, and the endogenous 5′ upstream fragment of the putative Cygb promoter region within the experimental groups. Immunoprecipitation of the AP-1 complex with the endogenous Cygb promoter fragment was performed using polyclonal rabbit anti-c-Jun and anti-c-Fos sera (Santa Cruz Biotechnology Inc.), and immunoprecipitation of NFAT with the Cygb promoter was performed using the polyclonal rabbit anti-NFATc1 serum (Santa Cruz Biotechnology Inc.).
Quantitative RT-PCR was utilized to assess the -fold enrichment of the immunoprecipitated protein-DNA complex for AP-1 and NFAT. Primers were designed to span the 5′ upstream region of the Cygb gene that included the AP-1 binding motif but did not include either the HRE or the NRE. The amplicon produced was 224 bp in length. The primer set used in the ChIP assay was as follows: forward primer, 5′-CACACGTCGAGAGCTGAGAC-3′; reverse primer, 5′-TTGGCAAGGTGAAGGTTAGG-3′.
Quantitative RT-PCR was performed using SYBR Green PCR Master Mix (Applied Biosystems) on an ABI Prism 7000 sequence detection System (Applied Biosystems) as described previously (21). Signals were normalized to control samples processed from mock immunoprecipitation without the AP-1 or NFAT antibodies. Normalization was performed according to the ΔΔCt method (21, 26). In addition, control primers (forward primer, 5′-GCTATGAGGAATGGCTGCAT-3′; reverse primer, 5′-CTGAGCAGGTCACAACAGGC-3′), which amplified a 200-bp noncoding region on murine chromosome 13, was used as a control reaction to address the specificity of the immunoprecipitated protein-DNA complex for AP-1 or NFAT.
Dominant Negative Constructs and Assessment of Endogenous Cytoglobin Transcript Levels—A dominant negative plasmid recognizing the hypoxia-responsive element (dnHIF-1) was a gift from Dr. Joseph Garcia (UTSW). In addition, dominant negative constructs for NFAT (dnNFAT) and c-Jun (TAM67) were generously provided by Drs. Leon DeWindt (University Medical Center Utrecht), and Michael Birrer (NCI, National Institutes of Health), respectively. These CMV-driven expression vectors (dnHIF-1, dnNFAT, TAM67) have previously been reported to efficiently inhibit the activity of all HIF isoforms, all NFAT isoforms, and AP-1, respectively (24, 27–31). Transient transfection of these constructs (0.5 μg), individually and in combination, into C2C12 myoblasts was undertaken as described above. After exposure to 16 h of normoxia or hypoxia, the cells were harvested, RNA extracted, and cDNA made as described previously (7, 21). Assessment of endogenous Cygb transcript levels in these cell extracts was performed by qRT-PCR as described above and in previous reports (21).
Statistical Analysis—Data were reported as the means ± S.E. Statistical significance was assessed by performing a two-tailed Student's t test or one-way analysis of variance between groups with a Bonferroni's post-test analysis. A p value of less than 0.05 was considered significant.
RESULTS
Induction of Cytoglobin Expression in the Hypoxic Heart
Northern blot analysis demonstrated that Cygb transcript is expressed in a variety of adult tissues and is most abundant in the whole heart and brain (Fig. 1A). To determine whether the expression of Cygb changes in response to cardiac hypoxia, WT mice were exposed to chronic hypoxia (10% O2 for 2 weeks). We have shown previously that this in vivo model of chronic hypoxia produces a significant polycythemic response and the induction of angiogenesis within the myocardium, indicative of an adaptive response to hypoxia (20). Exposure to such a stress resulted in the development of significant right and left ventricular hypertrophy in the hypoxic heart compared with the normoxic heart (Fig. 1, B and C). The magnitude of the increase in the heart-to-body weight ratio in hypoxic hearts compared with normoxic hearts was 1.6. Despite the development of significant amount of hypertrophy, we did not observe any fibrosis in the hypoxia-induced hypertrophic heart (data not shown). Quantitative RT-PCR and Western blot analysis revealed a significant increase in Cygb transcript and protein levels in the hypoxic heart (Fig. 1, D–F). Specifically, the magnitude of the increase in Cygb transcript and protein levels in hypoxic hearts compared with normoxic hearts was 6.0 and 3.8, respectively (Fig. 1, D–F). This degree of increase in the protein level is similar to that found in our previous report of increased neuronal Cygb expression within the hypoxic brain (7). Finally, cultured C2C12 myoblasts exposed to hypoxia (1% O2 for 16 h) also demonstrated increased Cygb protein levels (Fig. 1G).
FIGURE 1.

Enhanced cytoglobin expression in adult hypoxic hearts. A, Northern blot analysis of total RNA isolated from various adult tissues revealed Cygb expression is highest in the adult heart and brain. β-Actin was used as a RNA loading control. B, adult WT male mice were exposed to chronic hypoxia (10% O2) for 2 weeks. Histological (hematoxylin/eosin staining) analysis of normoxic and hypoxic hearts demonstrated a significant amount of right and left ventricular hypertrophy in the hypoxic heart. C, hypoxic hearts have a 1.6-fold increase in the heart-to-body weight (Hrt/BW) ratio compared with normoxic hearts. D, quantitative RT-PCR analysis revealed a 6.0-fold increase in Cygb mRNA levels in hypoxic hearts compared with normoxic hearts. The 18s-RIB sequence was amplified as a loading control in this analysis. E, Western blot analysis showed a marked increase in Cygb protein levels in hypoxic hearts. F, quantification of Cygb protein levels depicted in the Western blot in panel E revealed a 3.8-fold increase in Cygb levels in hypoxic hearts compared with normoxic hearts. G, Western blot analysis revealed a marked increase in Cygb protein levels in hypoxic C2C12 cells (1% O2 for 16 h) compared with normoxic cells. α-Tubulin (α-Tub) was used as a loading control in the Western blots. (*, p < 0.005 hypoxic versus normoxic hearts; n = 3–5 in each group). Bar = 1 mm.
Transcriptional Regulation of Cytoglobin Expression
Evolutionarily Conserved Response Elements within the Cytoglobin Promoter—Having demonstrated an increase in both Cygb transcript and protein levels within the hypoxic myocardium, a series of experiments were designed to determine the mechanisms that control Cygb transcription in response to hypoxia. Initially, a data-base analysis utilizing rVISTA, a Web-based program to assess for potential regulatory elements of a gene, was undertaken to identify evolutionarily conserved motifs within the 5′ upstream region of the human, rat, and mouse Cygb gene (32). Our analysis identified two putative Cygb promoter regions (4.6 and 0.6 kb) upstream of the 5′-end of the Cygb gene that contained evolutionarily conserved consensus sites recognized by the transcription factors HIF-1, AP-1, and NFAT (Fig. 2A). These transcription factors are known to mediate transcriptional activation of a variety of genes in response to hypoxia, hypertrophy, and oxidative stress (33–36). In addition, both NFAT and AP-1 are known substrates for the calcium-calmodulin-activated phosphatase, calcineurin (37–39). To assess the role of these transcription factors in regulating Cygb transcription, we fused these two upstream fragments to a luciferase reporter and undertook transcriptional assays. Because transient transfection of either the 4.6-kb upstream fragment of the Cygb gene-luciferase reporter (4.6-kb Cygb-Luc) or the 0.6-kb upstream fragment of the Cygb gene-luciferase reporter (0.6-kb Cygb-Luc) into C2C12 myoblasts revealed similar transcriptional activation patterns, we focused our analysis on the 0.6-kb Cygb promoter region (Fig. 2, B–D).
FIGURE 2.

Calcineurin regulates the transcriptional activity of the 5′ upstream region of the cytoglobin gene. A, depiction of two regions (4.6- and 0.6-kb fragments) upstream of the 5′-end of the Cygb gene illustrates the evolutionarily conserved motifs for HIF-1, AP-1, and NFAT located within the first 600 base pairs. B, the transcriptional activity of the 4.6-kb Cygb-Luc plasmid was assessed in C2C12 myoblasts. Co-transfection with a CnA* expression plasmid resulted in a 39.3 ± 3.0-fold induction of transcriptional activity under hypoxic conditions. This response was significantly blunted by inhibitors of CnA* (Rcan1 or CsA). Co-transfection with a HIF* expression plasmid increased both basal and CnA*-stimulated transcription. C, inhibition of endogenous calcineurin activity (by overexpressing Rcan1 or exposure to CsA) in C2C12 cells resulted in complete attenuation of transcriptional activation of the 4.6-kb Cygb-Luc reporter under both normoxic and hypoxic conditions. D, transcriptional assays of the 0.6-kb Cygb-Luc plasmid transfected into C2C12 cells revealed a pattern of activation similar to the 4.6-kb Cygb-Luc reporter. Co-transfection with a CnA* expression plasmid resulted in a 16.8 ± 2.6-fold induction of transcriptional activity under hypoxic conditions. This response was also minimally augmented with HIF-1* but was significantly blunted by inhibitors of calcineurin (Rcan1 and CsA). *, p < 0.005 normoxic experimental cells versus normoxic control cells transfected with only the promoter reporter plasmid; **, p < 0.005 hypoxic experimental cells versus normoxic control cells transfected with only the promoter reporter plasmid; #, p < 0.01 normoxic experimental cells with inhibited calcineurin activity versus normoxic cells overexpressing CnA*; ##, p < 0.01 hypoxic experimental cells with inhibited calcineurin activity versus hypoxic cells overexpressing CnA* (n = 9 in each group).
Calcineurin Activity Modulates Cytoglobin Transcription—The evolutionarily conserved consensus sequences for HIF-1, AP-1, and NFAT within the 0.6-kb Cygb promoter region are illustrated in Fig. 2A. Unlike the HRE and ARE consensus sites, the NRE site is located on the noncoding strand (3′ to 5′ arm) of the DNA sequence in the upstream region of the Cygb gene. In C2C12 myoblasts transiently transfected with the 0.6-kb Cygb-Luc reporter plasmid, hypoxia (1% O2 for 16 h) alone activated the reporter plasmid by 2-fold (Fig. 2D). Co-transfection of the 0.6-kb Cygb-Luc plasmid along with a plasmid overexpressing CnA* resulted in a greater increase in reporter activity under both normoxic and hypoxic conditions compared with activation of the 0.6-kb Cygb-Luc plasmid alone under normoxic conditions (Fig. 2D). In contrast, co-transfection of the 0.6-kb Cygb-Luc plasmid along with a plasmid overexpressing HIF-1* resulted in minimal, but significant, activation of the 0.6-kb Cygb regulatory region in the absence of CnA* (Fig. 2D). Finally, overexpression of both CnA* and HIF-1* resulted in only a 1.7 (normoxia)- and 1.4 (hypoxia)-fold increase in Cygb transcription as compared with CnA* overexpression alone (Fig. 2D). These observations suggest that calcineurin (either via NFAT and/or AP-1 activation) is more potent, as compared with HIF-1, in inducing Cygb transcription under both normoxic and hypoxic conditions. To further examine calcineurin-mediated transcriptional regulation of the Cygb gene, we co-transfected C2C12 cells with either a CMV-Rcan1 expression vector (inhibits calcineurin activity) with the 0.6-kb Cygb-Luc and CnA* plasmids or alternatively exposed the transfected cells to CsA, a phamacological inhibitor of calcineurin (18, 40). As depicted in Fig. 2D, inhibition of calcineurin activity resulted in a 60% (CsA exposure) to 75% (Rcan1 overexpression) reduction of transcriptional activity under both normoxic and hypoxic conditions (Fig. 2D). Furthermore, inhibition of calcineurin activity blunted the reporter activity in response to hypoxia compared with normoxia with the 0.6-kb Cygb-Luc plasmid (Fig. 2D). Likewise, inhibition of endogenous calcineurin activity attenuated any significant increase in reporter activity with the 4.6-kb Cygb-Luc plasmid under hypoxic conditions (Fig. 2C). Finally, in vitro studies demonstrated increased Cygb protein expression in C2C12 cells overexpressing CnA* as compared with control C2C12 cells (Fig. 3A). Results from these experiments further support the hypothesis that calcineurin plays a critical role in the transcriptional regulation of Cygb under both normoxic and hypoxic conditions.
FIGURE 3.

Calcineurin activity increases cytoglobin levels in C2C12 cells and transgenic murine hearts. A, under normoxic conditions, Western blot analysis demonstrated an increase in Cygb protein levels in C2C12 myoblasts transfected with a CnA* expression plasmid compared with control C2C12 cells. B and C, Western blot analysis with the corresponding quantification demonstrated enhanced Cygb protein levels in transgenic murine hearts carrying a cardiac-specific CnA* transgene (*, p < 0.05 TgCnA* versus WT hearts; n = 3 in each group). D and E, conversely, in murine hearts with a cardiac-specific Rcan1 transgene, Western blot analysis demonstrated decreased Cygb protein levels (*, p < 0.05 TgRcan1 versus WT hearts; n = 3 in each group). F, echocardiography in unanesthetized mice revealed depressed left ventricular systolic function in 2-month-old transgenic mice compared with 2-month-old WT littermate mice (*, p < 0.001 2-month-old TgCnA* or TgRcan1 mice versus 2-month-old WT mice; n = 3 in each group). α-Tubulin (α-Tub) was used as a loading control in the Western blots. TgCnA*, transgenic mice with cardiac-specific overexpression of CnA*; TgRcan1, transgenic mice with cardiac-specific overexpression of Rcan1.
Cytoglobin Expression in Transgenic Mice with Altered Calcineurin Activity—Extending our in vitro analysis of the Cygb promoter, we utilized transgenic mouse models with cardiac-specific overexpression of activated calcineurin (TgCnA*) or Rcan1 (TgRcan1) to assess whether calcineurin activity alters Cygb protein levels in vivo (18, 19). Western blot analysis revealed a significant increase in Cygb protein levels in TgCnA* hearts (Fig. 3, B and C), whereas a significant reduction in Cygb levels was found in TgRcan1 hearts (Fig. 3, D and E).
To determine whether the increase in Cygb protein in TgCnA* mice was a direct consequence of calcineurin activity or secondary to the development of heart failure in these mice, we assessed left ventricular systolic function by echocardiography in these transgenic mice. At 1 month of age, TgCnA* mice had fractional shortening similar to that in WT or TgRcan1 mice (Fig. 3F). By 2 months of age, TgCnA* mice developed a significant reduction in cardiac systolic function compared with WT mice (Fig. 3F). We concluded from these studies that increased Cygb expression in 1-month-old TgCnA* mice and decreased Cygb expression in 1-month-old TgRcan1 mice were due to changes in calcineurin activity and were not a secondary consequence of decreased cardiac function.
Activation of the Calcineurin/NFAT Pathway under Hypoxia and Regulation of Cytoglobin Transcription—Although calcineurin signaling activates both the transcription factors, NFAT and AP-1, it is the calcineurin/NFAT pathway that has been more extensively examined as compared with the calcineurin/AP-1 pathway (37, 41, 42). Therefore, a series of experiments were undertaken to assess the activation of NFAT in response to hypoxia. C2C12 myoblasts were transiently transfected with NFATc1-GFP or CnA* expression plasmids under varying O2 tension levels. By monitoring NFATc1-GFP fluorescence, we demonstrated that NFATc1 translocates to the nucleus under hypoxic conditions, suggesting that hypoxia can activate calcineurin and thus increase nuclear occupancy of NFAT (Fig. 4, A and B). As a control for nuclear translocation, the NFATc1-GFP plasmid was co-transfected with the CnA* expression plasmid into C2C12 cells under normoxic conditions. In these experiments we observed a significant increase in the number of NFATc1-GFP-positive nuclei compared with normoxic NFATc1-GFP-overexpressing cells (Fig. 4, A and B).
FIGURE 4.

Increased NFAT occupancy in hypoxic nuclei and the significance of the conserved NFAT response element. A, C2C12 cells transiently transfected with plasmids overexpressing NFATc1-GFP and/or CnA* were exposed to normoxic or hypoxic conditions. Using fluorescent microscopy, the data indicate NFATc1-GFP translocated from the cytosol to the nucleus under conditions of hypoxia and/or in the presence of CnA*. B, quantification of the degree of NFAT translocation revealed a significant increase in NFAT within the nuclei of hypoxic cells transfected with NFATc1-GFP (18.2 ± 2.3-fold) as well as normoxic cells co-transfected with NFATc1-GFP and CnA* expression plasmids (34.6 ± 2.1-fold) compared with normoxic cells overexpressing NFATc1-GFP. (*, p < 0.005 hypoxic or normoxic+CnA* cells versus normoxic control cells; n = 3 in each group). C, deletion of the NFAT site within the 0.6-kb upstream region of the Cygb gene failed to blunt luciferase activity of the mutated 0.6-kb Cygb-Luc reporter. (*, p < 0.005 normoxic cells transfected with the ΔNFAT 0.6-kb Cygb-Luc plasmid versus normoxic cells transfected with the WT 0.6-kb Cygb-Luc plasmid; **, p < 0.005 hypoxic cells transfected with the ΔNFAT 0.6-kb Cygb-Luc plasmid versus normoxic cells transfected with the WT 0.6-kb Cygb-Luc plasmid; n = 9 in each group). ΔNFAT, deletion of the conserved NFAT motif within the 0.6-kb Cygb promoter region. Bar = 20 μm.
Subsequently, we undertook deletional studies to investigate the role of NFAT in the regulation of Cygb transcription. We found that a reporter construct in which the conserved NRE was deleted (ΔNFAT 0.6-kb Cygb-Luc) was still activated by hypoxia and/or co-transfection with the CnA* expression plasmid (Fig. 4C). These data suggest that this conserved NFAT binding site is not solely responsible for the regulation of Cygb expression in response to hypoxia and/or calcineurin. Therefore, we initiated experiments to examine the role of the conserved AP-1 binding site in transactivating Cygb expression.
The Calcineurin/AP-1 Pathway Regulates Cytoglobin Transcription—A 100-bp fragment containing the conserved ARE was cloned into a luciferase reporter construct (0.1-kb Cygb-Luc; Fig. 5A). Transient transfection assays in C2C12 myoblasts indicated that this promoter fragment responded to both hypoxia and calcineurin overexpression, despite the absence of the conserved HIF-1 and NFAT binding sites (Fig. 5B). Inhibition of endogenous calcineurin activity with either overexpression of Rcan1 or exposure to CsA resulted in a blunted response of the 0.1-kb Cygb-Luc promoter reporter plasmid to hypoxia (Fig. 5C). As AP-1 is a heterodimer formed by the dimerization of c-Jun and c-Fos, we co-transfected the 0.1-kb Cygb-Luc plasmid with c-Jun/c-Fos expression plasmids into C2C12 cells (43, 44). This resulted in a 26 (normoxia)- and 39 (hypoxia)-fold increase in luciferase activity compared with the 0.1-kb Cygb-Luc plasmid alone under normoxic conditions (Fig. 5D). Furthermore, deletion or site-directed mutagenesis of the AP-1 site resulted in a loss of response to hypoxia and/or calcineurin (Fig. 6). Taken together, these results support the hypothesis that AP-1 is involved in calcineurin-dependent transactivation of Cygb gene expression in response to hypoxia.
FIGURE 5.
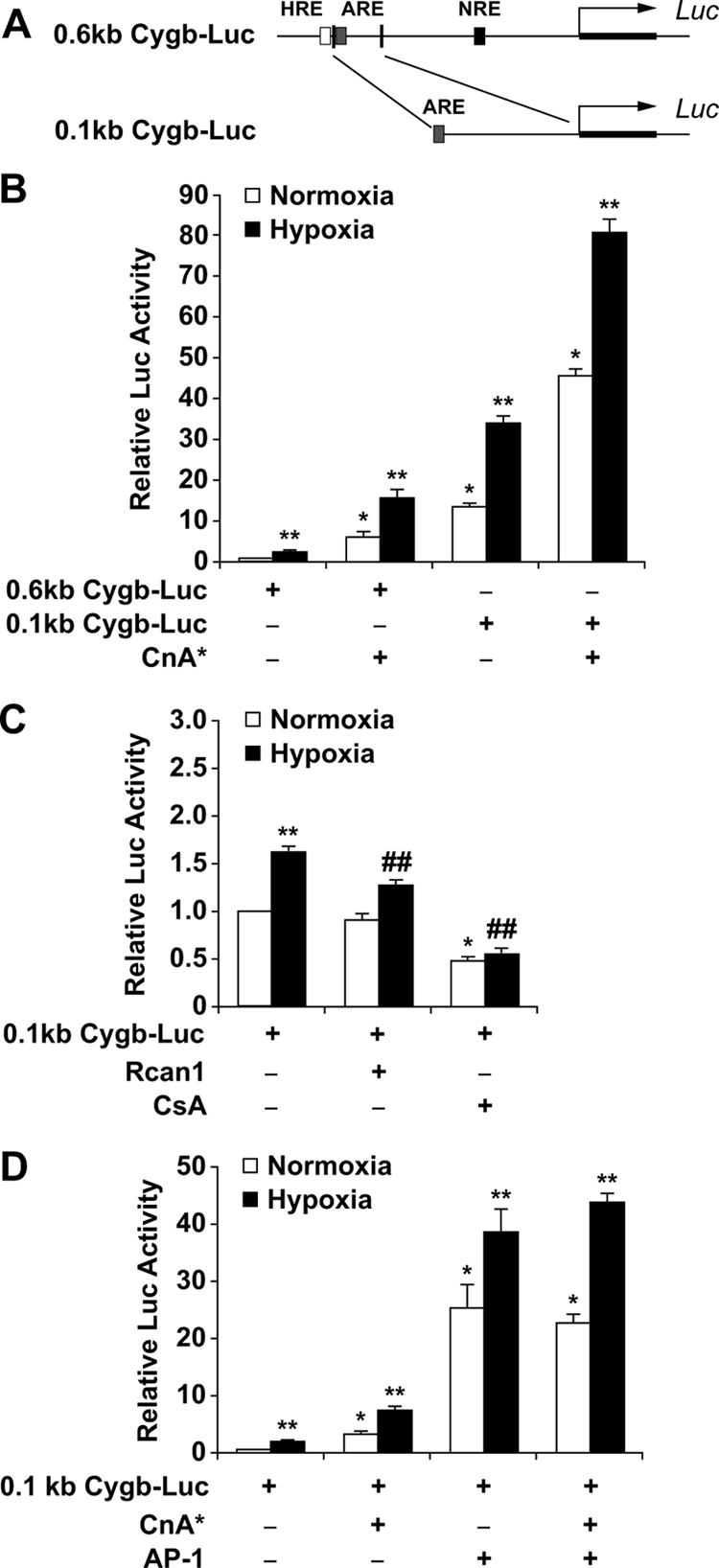
Induction of cytoglobin transcriptional activity by AP-1. A, illustration depicting the location of the 0.1-kb Cygb promoter fragment within the 0.6-kb Cygb promoter region. This smaller DNA fragment contains the evolutionarily conserved motif for AP-1 but not HIF-1 or NFAT. B, the transcriptional activity of the 0.1-kb upstream fragment of the Cygb gene was compared with the activity of the 0.6-kb Cygb-Luc plasmid in C2C12 myoblasts. Under hypoxic conditions, overexpression of CnA* resulted in a 81.7 ± 2.9-fold induction of transcriptional activity of the 0.1-kb Cygb-Luc plasmid compared with a 15.7 ± 2.0-fold induction of transcriptional activity of the 0.6-kb Cygb-Luc plasmid. C, inhibition of endogenous calcineurin activity (by overexpressing Rcan1 or exposure to CsA) in C2C12 cells resulted in attenuation of transcriptional activation of the 0.1-kb Cygb-Luc reporter under both normoxic and hypoxic conditions. D, co-transfection of AP-1 expression plasmids along with the 0.1-kb Cygb-Luc plasmid resulted in a 39 ± 3.9-fold induction of transcriptional activity under hypoxic conditions. *, p < 0.005 normoxic experimental cells versus normoxic control cells transfected with only the promoter reporter plasmid; **, p < 0.005 hypoxic experimental cells versus normoxic control cells transfected with only the promoter reporter plasmid; ##, p < 0.01 hypoxic experimental cells with inhibited calcineurin activity versus hypoxic cells transfected with only the promoter reporter plasmid (n = 9 in each group).
FIGURE 6.

Site-directed mutagenesis of the AP-1 site blunts transcriptional activation of cytoglobin in response to activated calcineurin and hypoxia. The ARE within the 0.1-kb upstream fragment of the Cygb gene was either deleted or mutated. Deletion (Δ) or mutation (Mut) of the ARE suppressed the transcriptional activation of Cygb, especially under hypoxic conditions and/or in the presence of CnA*. *, p < 0.005 normoxic CnA*-overexpressing cells versus normoxic control cells transfected with only the promoter reporter plasmid; **, p < 0.005 hypoxic cells versus normoxic control cells transfected with only the promoter reporter plasmid; #, p < 0.01 normoxic CnA*-overexpressing cells transfected with the deleted/mutated AP-1 Cygb-Luc plasmid versus normoxic CnA*-overexpressing cells transfected with the WT 0.1-kb Cygb-Luc plasmid; ##, p < 0.01 hypoxic CnA*-overexpressing cells transfected with the deleted/mutated AP-1 Cygb-Luc plasmid versus hypoxic CnA*-overexpressing cells transfected with the WT 0.1-kb Cygb-Luc plasmid (n = 9 in each group).
To assess the effect of modulating calcineurin signaling upon endogenous AP-1, we assessed c-Jun and c-Fos (components of the AP-1 heterodimer) protein levels in C2C12 myoblasts overexpressing CnA* and in transgenic hearts with altered calcineurin activity (TgCnA* or TgRcan1 mice). Western blot analysis of C2C12 cells revealed a significant increase in c-Jun and c-Fos protein levels in the nuclei of both normoxic and hypoxic cells transiently transfected with the CnA* expression plasmid (Fig. 7, A–C). In addition, there was also a significant increase of both c-Jun and c-Fos protein levels in the nuclear extracts of TgCnA* hearts compared with the nuclear extracts from TgRcan1 or WT hearts (Fig. 7, D–F). Thus, increased calcineurin activity is associated with increased nuclear localization of c-Jun and c-Fos proteins.
FIGURE 7.

Modulation of c-Jun and c-Fos protein levels in C2C12 cells and transgenic murine hearts with varying calcineurin activity. A, representative Western blot analysis demonstrated an increase in c-Jun and c-Fos protein levels in nuclear extracts from CnA*-overexpressing C2C12 myoblasts compared with control C2C12 cells. Hypoxia further enhances this up-regulation in c-Jun and c-Fos protein levels. B and C, quantification of c-Jun and c-Fos protein levels in cytosolic and nuclear extracts from cells that revealed increased protein levels within the nuclei of hypoxic and/or CnA*-overexpressing cells. *, p < 0.05 normoxic experimental cytosolic/nuclear extracts versus normoxic control cytosolic/nuclear extracts; **, p < 0.05 hypoxic experimental cytosolic/nuclear extracts versus normoxic control cytosolic/nuclear extracts (n = 3 in each group). D, representative Western blot analysis demonstrated increased c-Jun and c-Fos protein levels in nuclear extracts from transgenic murine hearts that overexpress CnA*. Conversely, in murine hearts that overexpress Rcan-1, Western blot analysis demonstrated decreased c-Jun and c-Fos protein levels within the nucleus. Lamin A/C and Sp1 were used as controls to assess the purity of the nuclear extracts, whereas α-tubulin (α-Tub) was used as a loading control in the Western blots. E and F, quantification of c-Jun and c-Fos protein levels in cytosolic and nuclear extracts from transgenic mice revealed increased protein levels within the nuclei of CnA*-overexpressing cardiomyocytes. *, p < 0.05 TgCnA* versus WT (n = 3 in each group). TgCnA*, transgenic mice with cardiac-specific overexpression of CnA*; TgRcan1, transgenic mice with cardiac-specific overexpression of Rcan1.
To directly verify that AP-1 binds to the DNA sequence in the upstream fragment of the Cygb gene, we undertook both EMSA and ChIP assays. Utilizing a WT AP-1 probe containing the ARE sequence in the 0.1-kb Cygb fragment, we demonstrated the formation of a specific protein-DNA complex in EMSA of nuclear extracts of control C2C12 cells (Fig. 8, A and B). This complex formation was enhanced in hypoxic cells overexpressing CnA* and attenuated with the addition of CsA (Fig. 8B). We used three methods to verify that our EMSA result was actually an AP-1-DNA complex. First, the complex formation was lost when we utilized a probe with the AP-1 site mutated (Fig. 8, A and C). Second, the addition of c-Jun and c-Fos antibodies disrupted the complex formation (Fig. 8A). Third, the use of a cold (nonradioactive) oligo probe containing the consensus sequence for the AP-1 binding site competed away the binding of the radioactive WT AP-1 probe from binding to the endogenous DNA under both normoxic and hypoxic conditions (Fig. 8A). Thus, the EMSA data provided in vitro evidence that AP-1 binding increases in response to hypoxia and/or calcineurin activation. However, inhibition of calcineurin activity attenuates the DNA binding affinity of AP-1 for the endogenous Cygb promoter (Fig. 8, A and B).
FIGURE 8.

Calcineurin enhances AP-1 binding to the endogenous cytoglobin promoter. A–C, utilizing EMSA, AP-1 was demonstrated to bind to the endogenous Cygb promoter. The binding affinity of AP-1 to DNA was enhanced in the presence of CnA* and in response to hypoxia. Inhibition of calcineurin activity in cells overexpressing CnA* attenuated the binding affinity of AP-1 to DNA. The use of a mutated AP-1 probe (Mut. AP-1), cold competition (Cold Comp.) with a nonradioactive WT AP-1 probe, incubating the cell lysates with c-Jun and c-Fos antibodies (Ab), and exposure of the cells to CsA markedly reduced the ability of AP-1 to bind to the endogenous Cygb promoter. D, a ChIP assay provided in vivo data that AP-1 was able to bind to the endogenous Cygb promoter. Utilizing qRT-PCR there was an increase in fold enrichment in the amount of AP-1 bound to the endogenous Cygb promoter region in response to hypoxia and a further increase in the presence of CnA* compared with that found under normoxic conditions. Similar to AP-1, an increase in fold enrichment of NFAT bound to the immunoprecipitated protein-DNA complex was noted in response to hypoxia and in the presence of CnA*. Use of control primers targeting a noncoding region significantly away from the Cygb gene did not produce an amplicon from the immunoprecipitated protein-DNA complex (data not shown). *, p < 0.05 hypoxic or CnA*-overexpressing cells versus normoxic control cells (n = 3 in each group).
A ChIP assay was undertaken to provide further in vivo evidence that AP-1 binds to the endogenous Cygb promoter region in a calcineurin-dependent manner. The ChIP assay was performed with primers flanking the ARE region within the upstream Cygb promoter region. C2C12 myoblasts and cells transiently transfected with the CnA* expression plasmid were exposed to normoxic or hypoxic conditions for 16 h. Cross-linked protein-DNA complexes were precipitated with both c-Jun and c-Fos antibodies and then tested for the presence of the Cygb promoter using ChIP primers flanking the ARE region. Utilizing qRT-PCR, we demonstrated that there was a significant increase in the amount of AP-1 bound to the endogenous Cygb upstream fragment under hypoxic conditions and an even further increase following overexpression of CnA* (Fig. 8D). No significant qRT-PCR product was generated when control primers, targeted to a noncoding region within murine chromosome 13, were used to produce an amplicon from the immunoprecipitated protein-DNA complex (data not shown). Thus, both the EMSA and ChIP assays demonstrated that AP-1 is capable of binding to the endogenous 5′ upstream region of the Cygb gene in a calcineurin-dependent manner under both normoxic and hypoxic conditions.
In parallel experiments, using a NFATc1 antibody in the ChIP assay we also found an increase in binding of NFAT to the Cygb promoter in response to both hypoxia and activated calcineurin (Fig. 8D). Thus, taken together our data indicate that hypoxia results in a calcineurin-dependent increase in Cygb levels through a mechanism involving increased occupancy by both NFAT and AP-1 onto the proximal promoter region of the Cygb gene.
Inhibition of Endogenous HIF-1 and Calcineurin-dependent Transcription Factors on Endogenous Cytoglobin Transcript Levels—To assess the effects of HIF-1 and the calcineurin-dependent transcription factors on endogenous Cygb gene expression under both normoxic and hypoxic conditions, dominant negative constructs previously demonstrated to inhibit the activity of HIF-1, calcineurin, NFAT, and AP-1 were utilized (19, 24, 27–31, 40, 45). Inhibition of calcineurin activity by either CsA (pharmacologic inhibition) or Rcan1 overexpression (endogenous inhibition) resulted in a marked decrease in endogenous Cygb transcript levels under both normoxic and hypoxic conditions (Fig. 9A). In addition, the endogenous inhibition of calcineurin activity by Rcan1 resulted in loss of the hypoxic response in regards to induction of Cygb transcription. Although CsA-treated C2C12 cells resulted in decreased Cygb transcript levels, there was still a 1.8-fold increase in Cygb levels in cells treated under hypoxic rather than normoxic conditions (Fig. 9A). This observation was likely because of the inefficient pharmacologic inhibition of calcineurin by CsA.
FIGURE 9.

Inhibition of calcineurin-dependent transcription factor activities result in decreased endogenous cytoglobin transcript levels under hypoxia. A, pharmacologic (CsA) and endogenous (Rcan1) inhibition of calcineurin activity within C2C12 myoblasts resulted in a significant reduction in endogenous Cygb mRNA levels. B and C, transfection of a dominant negative HIF plasmid into C2C12 cells resulted in failure to augment endogenous Cygb transcript levels under hypoxia. However, inhibiting both calcineurin and HIF-1 activities not only blunted the hypoxia-induced increase in Cygb mRNA levels but actually resulted in a marked decrease in Cygb transcript levels under both normoxic and hypoxic conditions. D and E, inhibition of NFAT and AP-1 activities within C2C12 myoblasts utilizing either pharmacologic agents (11R-VIVIT, SP60025, Tanshinone IIA) or dominant negative mutants of NFAT and AP-1 (dnNFAT, TAM67) resulted in a significant reduction in endogenous Cygb mRNA levels, especially under hypoxic conditions. Reduction in endogenous Cygb transcript levels under hypoxic conditions was even more pronounced with the simultaneous inhibition of calcineurin and AP-1 activities. *, < 0.05 normoxic experimental cells versus normoxic control cells; **, p < 0.05 hypoxic experimental cells versus normoxic control cells (n = 3 in each group). dnHIF-1, dominant negative mutant HIF construct; dnNFAT, dominant negative mutant NFAT construct; TAM67, dominant negative mutant AP-1 construct.
Transient transfection of dnHIF-1 into C2C12 myoblasts resulted in more than 60% inactivation of the hypoxia response element (data not shown), which is in agreement with a previous report demonstrating the ability of this dnHIF-1 to effectively inhibit HIF-1α activity (28). In support of the transcriptional assays, inhibition of HIF-1α activity in hypoxic C2C12 cells resulted in failure to increase endogenous Cygb transcript levels compared with Cygb levels in normoxic control cells (Fig. 9B). However, inhibiting both endogenous calcineurin and HIF-1α activities resulted in not only loss of hypoxic induction of Cygb mRNA levels but also a marked decrease in endogenous Cygb transcript levels compared with normoxic control cells (Fig. 9C). These data further support the hypothesis that calcineurin-dependent transcription factors have a greater role in regulating Cygb transcription, especially under low O2 tension, as compared with HIF-1.
Inhibition of NFAT activity can be accomplished by either 11R-VIVIT (pharmacologic inhibition) or dnNFAT (endogenous inhibition). As a positive control we demonstrated that dnNFAT inhibited 70% activation of the interleukin-2 promoter by calcineurin in C2C12 myoblasts; these data were in agreement with the efficiency of the dnNFAT construct in inhibiting NFAT activity within a cell (data not shown) (27). Despite the inhibition of NFAT activity with either 11R-VIVIT or the dnNFAT construct, some induction of Cygb transcription was still observed under hypoxia (Fig. 9, D and E). However, the dnNFAT-treated normoxic and hypoxic cells had a significant decrease in overall Cygb transcript levels as compared with the levels within normoxic control cells (Fig. 9E).
It has been reported previously that TAM67, a dominant negative mutant form of c-Jun, is able to effectively suppress the activity of the AP-1 complex (24, 29–31). With specific pharmacologic inhibitors (SP60025, Tanshinone IIA) or TAM67, inhibition of AP-1 activity resulted in attenuation of Cygb induction by hypoxia (Fig. 9, D and E). In fact, hypoxic C2C12 myoblasts transfected with TAM67 resulted in a significant decrease in endogenous Cygb transcript levels compared with normoxic control cells (Fig. 9E). In addition, there was minimal synergy noted in C2C12 cells co-transfected with both dnNFAT and TAM67 (Fig. 9E). Finally, inhibition of both calcineurin (with either CsA or Rcan1) and AP-1 (with TAM67) activities resulted in the greatest decrease in Cygb transcript levels in hypoxic cells compared with normoxic control cells (Fig. 9E). Collectively, data obtained from these experiments support the transcriptional, EMSA, and ChIP assay data and highlight the importance of calcineurin-dependent transcription factors in regulating Cygb transcription, especially under hypoxic conditions.
DISCUSSION
This study provides several key observations regarding the biology of Cygb and its transcriptional regulation in myocytes. First, Cygb mRNA and protein levels are robustly up-regulated in response to hypoxia in both in vitro and in vivo studies. Second, we identified a key region within the 5′ upstream segment of the Cygb gene that regulates the transcriptional response to hypoxia. Third, we present evidence that hypoxic activation of Cygb transcription is mediated by calcineurin activity. Although calcineurin increases binding of both NFAT and AP-1 to the putative Cygb promoter region, AP-1 may serve as the primary transactivator of Cygb transcription, especially under hypoxic conditions.
Enhanced Cytoglobin Expression within the Hypoxic Myocardium—Previous reports indicate that Cygb is expressed in a variety of adult tissues (4, 5, 9). In this study, we demonstrate that Cygb is not uniformly expressed in all adult tissues. As a hemoprotein capable of scavenging reactive oxygen species, the marked abundance of Cygb within the heart and the brain and its increased expression within these organs exposed to hypoxic stress suggest that Cygb may serve a cytoprotective role within these tissues by modulating the redox state of the cell. This hypothesis is supported by two recent in vitro studies suggesting that overexpression of Cygb protects a cell from oxidative damage and cell death (11, 12).
In our present study, we utilized a chronic hypoxia mouse model to demonstrate an increase in Cygb expression in response to hypoxic stress. Exposure to chronic hypoxia has been demonstrated to increase calcineurin activity and initiate ventricular remodeling via the development of cardiac hypertrophy (20, 46–48). Cardiomyocytes that undergo hypertrophy are metabolically active cells that have altered calcium homeostasis, redox state, and stress response signaling (2, 49–51). The observed increase in Cygb protein expression within the hypoxia-induced hypertrophic myocardium may serve a cytoprotective role by regulating the altered redox state of the myocytes.
Calcineurin Activity Modulates Cytoglobin Transcript and Protein Levels within the Myocyte—Calcineurin is an important calcium-calmodulin-activated phosphatase that regulates cardiac growth/remodeling, hypertrophy, apoptosis, and metabolism by participating in signaling pathways involved in the regulation of a variety of cardiac genes (18, 41, 52, 53). In response to an environmental stress (e.g. hypoxia, ischemia, pressure overload), the calcium-calmodulin complex activates calcineurin, enabling a myocyte to respond to the external stress signal by the ability of calcineurin to modify various transcription factors (e.g. AP-1, MEF2, and NFAT) and thus gene expression. Alternatively, calcineurin may interact directly with specific proteins involved with various stress signaling pathways. An example of the latter interaction is demonstrated by recent data revealing direct protein-protein interaction between calcineurin and Bcl-2 (an anti-apoptotic protein) under hypoxic conditions (54, 55). Although there is controversy as to whether calcineurin is involved in pro- or anti-apoptotic pathways, emerging data largely support an anti-apoptotic regulatory role for calcineurin under conditions of increased oxidative stress (53, 56, 57).
Data obtained from the in vitro transcriptional analysis of the Cygb promoter, the use of transgenic mouse models having varying degrees of calcineurin activity, and experiments involving the chemical and endogenous inhibition of calcineurin activity clearly demonstrate that calcineurin regulates the activation of Cygb transcription within the myocyte. In fact, the in vitro transcriptional assays involving the inhibition of calcineurin activity blunted the activation of Cygb transcription under both normoxic and hypoxic conditions. In addition, inhibition of calcineurin activity in TgRcan1 mice resulted in decreased Cygb protein expression in the heart, whereas cells transfected with Rcan1 had decreased endogenous Cygb transcript levels under normoxia and failed to augment Cygb mRNA levels upon exposure to hypoxia. Both the in vitro and in vivo data support the hypothesis that Cygb transcription is mediated predominately by the level of calcineurin activity. Thus, these new data along with recent in vitro studies indicating a cytoprotective role of Cygb under oxidative stress conditions suggest Cygb may serve as a calcineurin-dependent, pro-survival, stress-responsive hemoprotein regulating the redox state of the cell and cellular apoptosis (11, 12).
Calcineurin Enhances AP-1-mediated Cytoglobin Transcription—Data from the present study established that AP-1 plays an important role in the induction of Cygb transcription, especially under conditions associated with hypoxic stress. As a transcription factor complex, AP-1 is formed by the dimerization of various basic region-leucine zipper (bZIP) proteins belonging to the Jun (c-Jun, JunB, and JunD), Fos (c-Fos, FosB, Fra-1, and Fra-2), Maf (c-Maf, MafB, MafA, MafG/F/K, and Nrl), and ATF (ATF2, LRF1/ATF3, B-ATF, JDP1, and JDP3) families. Of these bZIP proteins, c-Jun is the most potent transcriptional activator. Within the AP-1 complex, c-Fos enhances the DNA binding affinity of c-Jun by forming a stable heterodimer with c-Jun (58, 59). AP-1 activity is induced by various triggers of oxidative stress (e.g. hypoxia, ischemia, and pressure overload) (44, 60–64). As a redox-sensitive transcription factor, AP-1 is believed to play an important role in cell survival, differentiation, growth, and apoptosis (58, 59, 62). In addition, AP-1 has been proposed to have a role in the development of cardiac hypertrophy (60, 64, 65).
A recent study identified the involvement of a calcineurin/AP-1 pathway in regulating growth factor-induced genes (37). Chen et al. (37) provide data demonstrating that calcineurin can directly dephosphorylate the serine 243 located on the C terminus of c-Jun. Dephosphorylation of the C terminus of c-Jun not only enhances the DNA binding affinity of AP-1 (via c-Jun) but also its interaction with other transcription factors (e.g. Sp1, ATF, SMAD, and NFAT) to regulate gene expression (37, 66).
In this study, we have provided data in support of the existence of a calcineurin/AP-1 pathway. There is increased c-Jun and c-Fos protein levels in the nuclei of normoxic cells overexpressing CnA* with further increase under hypoxia. Furthermore, transgenic mice with increased calcineurin activity within the heart have increased c-Jun and c-Fos protein levels within cardiac nuclei, whereas the opposite was found in hearts from TgRcan1 mice that had decreased calcineurin activity. Dominant negative experiments involving the inhibition of both calcineurin and AP-1 activities in hypoxic C2C12 cells resulted in a marked reduction in endogenous Cygb transcript levels. Finally, data are also provided supporting the DNA binding affinity of AP-1 to the Cygb promoter region is enhanced with increased calcineurin activity and this effect is further augmented under hypoxic conditions. Collectively, we conclude that the calcineurin/AP-1 pathway is important in the transactivation of Cygb transcription under hypoxic stress.
Role of Calcineurin/NFAT Pathway in Regulating Cytoglobin Transcription—Although calcineurin regulates gene transcription through the modification of various transcription factors (e.g. AP-1, MEF2, and NFAT), it is the calcineurin/NFAT pathway that has been best characterized, especially as it pertains to cardiac remodeling and the development of ventricular hypertrophy (33, 41, 42). NFAT regulates the transcription of a wide variety of genes involved in muscle growth, differentiation, and metabolism (e.g. acid-binding protein-2, glucagon, muscle regulatory protein 5, myoglobin, myosin heavy chain II, p21, and troponin-I) (15, 33, 38). Because the DNA binding activity of NFAT is weak and short-lived, many of the downstream gene targets of NFAT are regulated by the ability of NFAT to interact synergistically with other transcription factors (e.g. AP-1, GATA isoforms, and MEF-2) (38, 67–69). Specifically, the interaction between NFAT and AP-1 in regulating the transcription of a variety of genes has been well established (15, 18, 69, 70).
In the present study, we have demonstrated that hypoxia results in a marked increase in calcineurin activity as is evident by a significant increase in NFAT occupancy in the nucleus of hypoxic cells. In addition, the ChIP assay revealed that the immunoprecipitated protein-DNA complex is enriched with NFAT, especially in the presence of activated calcineurin and/or hypoxia. Collectively, these data suggest that perhaps there exists an interaction between NFAT and AP-1 in regulating Cygb transcription. Because the deletion of the conserved NRE binding site, as well as the dominant negative experiments involving inhibition of NFAT activity, indicates that NFAT is not solely responsible for Cygb transcription, NFAT may interact with AP-1 in one of two ways. First, both NFAT and AP-1 may directly bind to the Cygb promoter with synergistic interaction between the two transcription factors to modulate Cygb transcription. Secondly, NFAT may not directly bind to the Cygb promoter but rather may complex with AP-1. Subsequently, the NFAT-AP-1 complex can bind directly to the Cygb promoter region, thus regulating Cygb transcription. Future studies will explore the interaction between NFAT and AP-1 in regulating Cygb transcription.
Role of HIF-1 as a Transactivator of Cytoglobin Transcription—An hypoxic insult to the myocardium may result in myocardial necrosis, initiation of apoptosis, and/or development of compensatory cardiac hypertrophy. The myocardium responds to reduced O2 availability by activating a variety of cardioprotective signaling cascades. The transcription factor HIF-1 is believed to be the master regulator of cellular O2 homeostasis and serves a vital role within the adaptive response of the myocyte to hypoxic stress by regulating the transcription of a variety of hypoxia-responsive genes (34, 71, 72). Although Cygb is a hypoxia-responsive hemoprotein, our data indicate HIF-1 is a relatively weak direct transcriptional activator of Cygb expression. Wystub et al. (73) previously reported that Cygb may be regulated by HIF-1 based on a bioinformatics analysis of a putative human Cygb promoter region. Indeed, we observed an approximately 2-fold induction of Cygb transcription with either HIF-1* or under hypoxic conditions, which is in agreement with the results recently obtained by Guo et al. (74). However, our in vitro transcriptional assays and dominant negative experiments involving dnHIF-1 suggest that the direct role of HIF-1 is not as significant as the contribution of calcineurin-dependent transcription factors (AP-1 and NFAT) in regulating Cygb transcription. However, HIF-1 may have an indirect role in regulating Cygb transcription. It has been demonstrated recently that phosphorylation of the N terminus of c-Jun is dependent on HIF-1α (35, 75). Phosphorylation of the N terminus of c-Jun results in conformational changes in this protein enabling the dephosphorylation of the C terminus of c-Jun by calcineurin to occur more easily, thus enhancing the activity of the AP-1 complex (37, 76). Increased calcineurin activity, which occurs under hypoxic conditions, in turn augments HIF-1α expression (77). Liu et al. (77) demonstrated that activated calcineurin promotes HIF-1α stabilization by dephosphorylating receptor for activated protein kinase C-1 (RACK-1). This event prevents RACK-1 from dimerizing to HIF-1α, resulting in inhibition of ubiquitination and subsequent degradation of HIF-1α. Thus, there may exist a positive feedback loop between calcineurin-dependent transcription factors (AP-1 and NFAT) and HIF-1 in regulating Cygb transcription. Further studies will be required to address this hypothesis.
Role of Other Transcription Factors in Regulating Cytoglobin Expression within the Myocyte—Multiple transcription factors and co-activators are responsible for the coordinated regulation of genes. In regards to Cygb, there may be other transcription factors that also play a role in regulating Cygb transcription. Guo et al. (78) recently demonstrated that the transcription factors c-Ets-1 and Sp1 may have a role in regulating Cygb transcription in the human CYGB gene. However, these elements are not conserved in other species. In the present study, we did discover evolutionarily conserved binding motifs for Sp1 in the 5′ upstream region of the Cygb gene, but the sites were not located within the 0.1-kb Cygb promoter fragment that contains the critical motif regulating Cygb transcription. However, it is also feasible that calcineurin not only increases the DNA binding affinity of AP-1 but also enhances the interaction of AP-1 with Sp1 to further stabilize the transcriptional machinery in activating Cygb transcription (37). Future studies will focus on the role of other transcription factors (e.g. Sp1) within the 5′ upstream region of the Cygb promoter and their potential interaction with calcineurin-dependent transcription factors (AP-1 and NFAT) in regulating Cygb transcription.
In summary, Cygb represents a stress-responsive hemoprotein that is abundantly expressed in the adult heart and brain. Its expression both at the transcript and protein levels is up-regulated in the hypoxia-induced hypertrophic heart. Although both hypoxia and activated calcineurin enhance the AP-1 and NFAT binding affinities to the Cygb promoter region, transcriptional activation of Cygb expression is regulated primarily by AP-1, and the precise role of NFAT in this regulatory process currently remains unknown. Finally, by virtue of the demonstrated regulatory role of calcineurin on Cygb transcription, our data establish that Cygb is a novel calcineurin-dependent tissue hemoprotein that may serve a functional role in cardiac growth (e.g. hypertrophy), cell survival, and/or apoptosis. Future studies using gene disruptive technology will be important to further explore the functional role of Cygb within the resting and stressed heart.
Acknowledgments
We thank Dr. Eric N. Olson for scientific advice and critical review of an earlier version of this manuscript.
This study was supported, in whole or in part, by National Institutes of Health Grants HL-63788 (to D. J. G.) and HL-076440 (to P. P. A. M.). This study was also supported by grants from the American Heart Association-Texas Affiliate to (P. P. A. M.), the Donald W. Reynolds Clinical Cardiovascular Research Center (to D. J. G. and P. P. A. M.), and the GlaxoSmithKline Research Foundation (to P. P. A. M.).
Footnotes
The abbreviations used are: Cygb, cytoglobin; Mb, myoglobin; NFAT, nuclear factor of activated T cell; AP-1, activator protein; HIF-1, hypoxia-inducible factor-1; RT, reverse transcription; qRT, quantitative reverse transcription; RIB, ribosome; ARE, AP-1-responsive element; NRE, NFAT-responsive element; CMV, cytomegalovirus; CsA, cyclosporin A; GFP, green fluorescent protein; EMSA, electrophoretic mobility shift assay; WT, wild type; ChIP, chromatin immunoprecipitation; dn, dominant negative.
References
- 1.Levy, D., Garrison, R. J., Savage, D. D., Kannel, W. B., and Castelli, W. P. (1990) N. Engl. J. Med. 322 1561–1566 [DOI] [PubMed] [Google Scholar]
- 2.Heineke, J., and Molkentin, J. D. (2006) Nat. Rev. 7 589–600 [DOI] [PubMed] [Google Scholar]
- 3.Seddon, M., Looi, Y. H., and Shah, A. M. (2007) Heart 93 903–907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Burmester, T., Ebner, B., Weich, B., and Hankeln, T. (2002) Mol. Biol. Evol. 19 416–421 [DOI] [PubMed] [Google Scholar]
- 5.Trent, J. T., III, and Hargrove, M. S. (2002) J. Biol. Chem. 277 19538–19545 [DOI] [PubMed] [Google Scholar]
- 6.Kawada, N., Kristensen, D. B., Asahina, K., Nakatani, K., Minamiyama, Y., Seki, S., and Yoshizato, K. (2001) J. Biol. Chem. 276 25318–25323 [DOI] [PubMed] [Google Scholar]
- 7.Mammen, P. P., Shelton, J. M., Ye, Q., Kanatous, S. B., McGrath, A. J., Richardson, J. A., and Garry, D. J. (2006) J. Histochem. Cytochem. 54 1349–1361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Garry, D. J., Kanatous, S. B., and Mammen, P. P. (2003) Trends Cardiovasc Med. 13 111–116 [DOI] [PubMed] [Google Scholar]
- 9.Schmidt, M., Gerlach, F., Avivi, A., Laufs, T., Wystub, S., Simpson, J. C., Nevo, E., Saaler-Reinhardt, S., Reuss, S., Hankeln, T., and Burmester, T. (2004) J. Biol. Chem. 279 8063–8069 [DOI] [PubMed] [Google Scholar]
- 10.Weiland, T. R., Kundu, S., Trent, J. T., III, Hoy, J. A., and Hargrove, M. S. (2004) J. Am. Chem. Soc. 126 11930–11935 [DOI] [PubMed] [Google Scholar]
- 11.Fordel, E., Thijs, L., Martinet, W., Lenjou, M., Laufs, T., Van Bockstaele, D., Moens, L., and Dewilde, S. (2006) Neurosci. Lett. 410 146–151 [DOI] [PubMed] [Google Scholar]
- 12.Xu, R., Harrison, P. M., Chen, M., Li, L., Tsui, T. Y., Fung, P. C., Cheung, P. T., Wang, G., Li, H., Diao, Y., Krissansen, G. W., Xu, S., and Farzaneh, F. (2006) Mol. Ther. 13 1093–1100 [DOI] [PubMed] [Google Scholar]
- 13.Parsons, W. J., Richardson, J. A., Graves, K. H., Williams, R. S., and Moreadith, R. W. (1993) Proc. Natl. Acad. Sci. U. S. A. 90 1726–1730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bassel-Duby, R., Grohe, C. M., Jessen, M. E., Parsons, W. J., Richardson, J. A., Chao, R., Grayson, J., Ring, W. S., and Williams, R. S. (1993) Circ. Res. 73 360–366 [DOI] [PubMed] [Google Scholar]
- 15.Chin, E. R., Olson, E. N., Richardson, J. A., Yang, Q., Humphries, C., Shelton, J. M., Wu, H., Zhu, W., Bassel-Duby, R., and Williams, R. S. (1998) Genes Dev. 12 2499–2509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grayson, J., Williams, R. S., Yu, Y. T., and Bassel-Duby, R. (1995) Mol. Cell. Biol. 15 1870–1878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yan, Z., Serrano, A. L., Schiaffino, S., Bassel-Duby, R., and Williams, R. S. (2001) J. Biol. Chem. 276 17361–17366 [DOI] [PubMed] [Google Scholar]
- 18.Molkentin, J. D., Lu, J. R., Antos, C. L., Markham, B., Richardson, J., Robbins, J., Grant, S. R., and Olson, E. N. (1998) Cell 93 215–228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rothermel, B. A., McKinsey, T. A., Vega, R. B., Nicol, R. L., Mammen, P., Yang, J., Antos, C. L., Shelton, J. M., Bassel-Duby, R., Olson, E. N., and Williams, R. S. (2001) Proc. Natl. Acad. Sci. U. S. A. 98 3328–3333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mammen, P. P., Kanatous, S. B., Yuhanna, I. S., Shaul, P. W., Garry, M. G., Balaban, R. S., and Garry, D. J. (2003) Am. J. Physiol. 285 H2132—H2141 [DOI] [PubMed] [Google Scholar]
- 21.Mammen, P. P., Shelton, J. M., Goetsch, S. C., Williams, S. C., Richardson, J. A., Garry, M. G., and Garry, D. J. (2002) J. Histochem. Cytochem. 50 1591–1598 [DOI] [PubMed] [Google Scholar]
- 22.Chaturvedi, M. M., Mukhopadhyay, A., and Aggarwal, B. B. (2000) Methods Enzymol. 319 585–602 [DOI] [PubMed] [Google Scholar]
- 23.Bruick, R. K., and McKnight, S. L. (2001) Science 294 1337–1340 [DOI] [PubMed] [Google Scholar]
- 24.Brown, P. H., Chen, T. K., and Birrer, M. J. (1994) Oncogene 9 791–799 [PubMed] [Google Scholar]
- 25.Oka, T., Dai, Y. S., and Molkentin, J. D. (2005) Mol. Cell. Biol. 25 6649–6659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Livak, K. J., and Schmittgen, T. D. (2001) Methods (Amst.) 25 402–408 [DOI] [PubMed] [Google Scholar]
- 27.van Rooij, E., Doevendans, P. A., de Theije, C. C., Babiker, F. A., Molkentin, J. D., and de Windt, L. J. (2002) J. Biol. Chem. 277 48617–48626 [DOI] [PubMed] [Google Scholar]
- 28.Maemura, K., Hsieh, C. M., Jain, M. K., Fukumoto, S., Layne, M. D., Liu, Y., Kourembanas, S., Yet, S. F., Perrella, M. A., and Lee, M. E. (1999) J. Biol. Chem. 274 31565–31570 [DOI] [PubMed] [Google Scholar]
- 29.Fan, M., Goodwin, M. E., Birrer, M. J., and Chambers, T. C. (2001) Cancer Res. 61 4450–4458 [PubMed] [Google Scholar]
- 30.Paasinen-Sohns, A., Kielosto, M., Kaariainen, E., Eloranta, T., Laine, A., Janne, O. A., Birrer, M. J., and Holtta, E. (2000) J. Cell Biol. 151 801–810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang, N., Verna, L., Hardy, S., Zhu, Y., Ma, K. S., Birrer, M. J., and Stemerman, M. B. (1999) Circ. Res. 85 387–393 [DOI] [PubMed] [Google Scholar]
- 32.Loots, G. G., Ovcharenko, I., Pachter, L., Dubchak, I., and Rubin, E. M. (2002) Genome Res. 12 832–839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Crabtree, G. R., and Olson, E. N. (2002) Cell 109 (suppl.) S67–S79 [DOI] [PubMed] [Google Scholar]
- 34.Bruick, R. K. (2003) Genes Dev. 17 2614–2623 [DOI] [PubMed] [Google Scholar]
- 35.Laderoute, K. R. (2005) Semin. Cell Dev. Biol. 16 502–513 [DOI] [PubMed] [Google Scholar]
- 36.Liu, H., Colavitti, R., Rovira, I. I., and Finkel, T. (2005) Circ. Res. 97 967–974 [DOI] [PubMed] [Google Scholar]
- 37.Chen, B. K., Huang, C. C., Chang, W. C., Chen, Y. J., Kikkawa, U., Nakahama, K., Morita, I., and Chang, W. C. (2007) Mol. Biol. Cell 18 1118–1127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rao, A., Luo, C., and Hogan, P. G. (1997) Annu. Rev. Immunol. 15 707–747 [DOI] [PubMed] [Google Scholar]
- 39.Vega, R. B., Bassel-Duby, R., and Olson, E. N. (2003) J. Biol. Chem. 278 36981–36984 [DOI] [PubMed] [Google Scholar]
- 40.Rothermel, B., Vega, R. B., Yang, J., Wu, H., Bassel-Duby, R., and Williams, R. S. (2000) J. Biol. Chem. 275 8719–8725 [DOI] [PubMed] [Google Scholar]
- 41.Olson, E. N., and Williams, R. S. (2000) Cell 101 689–692 [DOI] [PubMed] [Google Scholar]
- 42.Olson, E. N., and Williams, R. S. (2000) BioEssays 22 510–51910842305 [Google Scholar]
- 43.Leonard, D. A., Rajaram, N., and Kerppola, T. K. (1997) Proc. Natl. Acad. Sci. U. S. A 94 4913–4918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Taimor, G., Rakow, A., and Piper, H. M. (2001) FASEB J. 15 2518–2520 [DOI] [PubMed] [Google Scholar]
- 45.Vega, R. B., Yang, J., Rothermel, B. A., Bassel-Duby, R., and Williams, R. S. (2002) J. Biol. Chem. 277 30401–30407 [DOI] [PubMed] [Google Scholar]
- 46.Ito, H., Adachi, S., Tamamori, M., Fujisaki, H., Tanaka, M., Lin, M., Akimoto, H., Marumo, F., and Hiroe, M. (1996) J. Mol. Cell. Cardiol. 28 1271–1277 [DOI] [PubMed] [Google Scholar]
- 47.Yousef, Z. R., Redwood, S. R., and Marber, M. S. (2000) Cardiovasc. Drugs Ther. 14 243–252 [DOI] [PubMed] [Google Scholar]
- 48.Koulmann, N., Novel-Chate, V., Peinnequin, A., Chapot, R., Serrurier, B., Simler, N., Richard, H., Ventura-Clapier, R., and Bigard, X. (2006) Am. J. Respir. Crit. Care Med. 174 699–705 [DOI] [PubMed] [Google Scholar]
- 49.Jacob, M. H., Pontes, M. R., Araujo, A. S., Barp, J., Irigoyen, M. C., Llesuy, S. F., Ribeiro, M. F., and Bello-Klein, A. (2006) Life Sci. 79 2187–2193 [DOI] [PubMed] [Google Scholar]
- 50.Frey, N., Katus, H. A., Olson, E. N., and Hill, J. A. (2004) Circulation 109 1580–1589 [DOI] [PubMed] [Google Scholar]
- 51.Wilkins, B. J., and Molkentin, J. D. (2004) Biochem. Biophys. Res. Commun. 322 1178–1191 [DOI] [PubMed] [Google Scholar]
- 52.Schaeffer, P. J., Wende, A. R., Magee, C. J., Neilson, J. R., Leone, T. C., Chen, F., and Kelly, D. P. (2004) J. Biol. Chem. 279 39593–39603 [DOI] [PubMed] [Google Scholar]
- 53.Baines, C. P., and Molkentin, J. D. (2005) J. Mol. Cell. Cardiol. 38 47–62 [DOI] [PubMed] [Google Scholar]
- 54.Erin, N., Bronson, S. K., and Billingsley, M. L. (2003) Neuroscience 117 541–555 [DOI] [PubMed] [Google Scholar]
- 55.Erin, N., Lehman, R. A., Boyer, P. J., and Billingsley, M. L. (2003) Neuroscience 117 557–565 [DOI] [PubMed] [Google Scholar]
- 56.De Windt, L. J., Lim, H. W., Taigen, T., Wencker, D., Condorelli, G., Dorn, G. W., II, Kitsis, R. N., and Molkentin, J. D. (2000) Circ. Res. 86 255–263 [DOI] [PubMed] [Google Scholar]
- 57.Molkentin, J. D. (2001) Circ. Res. 88 1220–1222 [DOI] [PubMed] [Google Scholar]
- 58.Shaulian, E., and Karin, M. (2001) Oncogene 20 2390–2400 [DOI] [PubMed] [Google Scholar]
- 59.Shaulian, E., and Karin, M. (2002) Nat. Cell Biol. 4 E131–136 [DOI] [PubMed] [Google Scholar]
- 60.Taimor, G., Schluter, K., and Piper, H. M. (2001) J. Mol. Cell. Cardiol. 33 503–511 [DOI] [PubMed] [Google Scholar]
- 61.Wenzel, S., Taimor, G., Piper, H. M., and Schluter, K. D. (2001) FASEB J. 15 2291–2293 [DOI] [PubMed] [Google Scholar]
- 62.Cummins, E. P., and Taylor, C. T. (2005) Pfluegers Arch. Eur. J. Physiol. 450 363–371 [DOI] [PubMed] [Google Scholar]
- 63.Coronella-Wood, J., Terrand, J., Sun, H., and Chen, Q. M. (2004) J. Biol. Chem. 279 33567–33574 [DOI] [PubMed] [Google Scholar]
- 64.Kudoh, S., Komuro, I., Mizuno, T., Yamazaki, T., Zou, Y., Shiojima, I., Takekoshi, N., and Yazaki, Y. (1997) Circ. Res. 80 139–146 [DOI] [PubMed] [Google Scholar]
- 65.Wu, S., Gao, J., Ohlemeyer, C., Roos, D., Niessen, H., Kottgen, E., and Gessner, R. (2005) Free Radic. Biol. Med. 39 1601–1610 [DOI] [PubMed] [Google Scholar]
- 66.Chinenov, Y., and Kerppola, T. K. (2001) Oncogene 20 2438–2452 [DOI] [PubMed] [Google Scholar]
- 67.Crabtree, G. R. (2001) J. Biol. Chem. 276 2313–2316 [DOI] [PubMed] [Google Scholar]
- 68.Im, S. H., and Rao, A. (2004) Mol. Cells 18 1–9 [PubMed] [Google Scholar]
- 69.Macian, F., Lopez-Rodriguez, C., and Rao, A. (2001) Oncogene 20 2476–2489 [DOI] [PubMed] [Google Scholar]
- 70.Graef, I. A., Mermelstein, P. G., Stankunas, K., Neilson, J. R., Deisseroth, K., Tsien, R. W., and Crabtree, G. R. (1999) Nature 401 703–708 [DOI] [PubMed] [Google Scholar]
- 71.Semenza, G. L. (1999) Cell 98 281–284 [DOI] [PubMed] [Google Scholar]
- 72.Semenza, G. L. (2000) J. Appl. Physiol. 88 1474–1480 [DOI] [PubMed] [Google Scholar]
- 73.Wystub, S., Ebner, B., Fuchs, C., Weich, B., Burmester, T., and Hankeln, T. (2004) Cytogenet. Genome Res. 105 65–78 [DOI] [PubMed] [Google Scholar]
- 74.Guo, X., Philipsen, S., and Tan-Un, K. C. (2007) Biochem. Biophys. Res. Commun. 364 145–150 [DOI] [PubMed] [Google Scholar]
- 75.Laderoute, K. R., Calaoagan, J. M., Gustafson-Brown, C., Knapp, A. M., Li, G. C., Mendonca, H. L., Ryan, H. E., Wang, Z., and Johnson, R. S. (2002) Mol. Cell. Biol. 22 2515–2523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Derijard, B., Hibi, M., Wu, I. H., Barrett, T., Su, B., Deng, T., Karin, M., and Davis, R. J. (1994) Cell 76 1025–1037 [DOI] [PubMed] [Google Scholar]
- 77.Liu, Y. V., Hubbi, M. E., Pan, F., McDonald, K. R., Mansharamani, M., Cole, R. N., Liu, J. O., and Semenza, G. L. (2007) J. Biol. Chem. 282 37064–37073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Guo, X., Philipsen, S., and Tan-Un, K. C. (2006) Biochim. Biophys. Acta 1759 208–215 [DOI] [PubMed] [Google Scholar]