

Abstract
Cyclotides are a family of plant defense proteins that are highly resistant to adverse chemical, thermal, and enzymatic treatment. Here, we present the first crystal structure of a cyclotide, varv F, from the European field pansy, Viola arvensis, determined at a resolution of 1.8 Å. The solution state NMR structure was also determined and, combined with measurements of biophysical parameters for several cyclotides, provided an insight into the structural features that account for the remarkable stability of the cyclotide family. The x-ray data confirm the cystine knot topology and the circular backbone, and delineate a conserved network of hydrogen bonds that contribute to the stability of the cyclotide fold. The structural role of a highly conserved Glu residue that has been shown to regulate cyclotide function was also determined, verifying its involvement in a stabilizing hydrogen bond network. We also demonstrate that varv F binds to dodecylphosphocholine micelles, defining the binding orientation and showing that its structure remains unchanged upon binding, further demonstrating that the cyclotide fold is rigid. This study provides a biological insight into the mechanism by which cyclotides maintain their native activity in the unfavorable environment of predator insect guts. It also provides a structural basis for explaining how a cluster of residues important for bioactivity may be involved in self-association interactions in membranes. As well as being important for their bioactivity, the structural rigidity of cyclotides makes them very suitable as a stable template for peptide-based drug design.
Cyclotides are an intriguing family of plant-derived proteins (1, 2) that act in plant defense (3, 4) and display a range of interesting biological activities, including uterotonic (5), human immunodeficiency virus inhibitory (6), antimicrobial (7), cancer cell toxicity (8), and neurotensin antagonistic activities (9). Their natural function as plant defense agents was deduced from findings that cyclotides effectively inhibited the growth of two common cotton pests, Helicoverpa punctigera and Helicoverpa armigera, when the larvae of these pests were fed a cyclotide-containing diet (4). The ability of cyclotides to deliver their activity in the harsh environment of predator insect guts relies on them being exceptionally stable. Indeed, cyclotides have been shown to be resistant to harsh thermal, chemical, and enzymatic conditions in vitro (10, 11). Their remarkable stability also makes cyclotides potentially valuable scaffolds for pharmaceutical or agrochemical applications (12).
Cyclotides are the largest family of naturally occurring head-to-tail cyclized proteins, but other examples of circular proteins have been discovered in recent years in bacteria, plants, and animals (13, 14). The cyclotides are distinguished from other circular proteins in that their cysteine residues form three disulfide bonds that are arranged in a cystine knot motif (1). Positioned in the protein core, one of the disulfide bonds (CysIII–CysVI) threads through an embedded ring formed by the other two disulfide bonds (CysI–CysIV and CysII–CysV) and their connecting backbone sequences (15). Recent studies have shown that an intact cystine knot is required to maintain the remarkable stability of cyclotides (10, 11), highlighting the importance of a structural understanding of the cystine knot topology. The sequences between successive cysteine residues are referred to as loops and are considered to “display” the residues that define cyclotide bioactivities. Loops 1 and 4 are highly conserved in size and sequence across all cyclotides, reflecting their structural involvement in the cystine knot. By contrast, a variety of residues are displayed within the other loops, leading to the description of cyclotides as a natural combinatorial template (16), based around a cyclic cystine knot core. Current estimates suggest that there are at least 9000 cyclotides in the Violaceae (violet) plant family (17) and >50,000 in the Rubiaceae (coffee) family (18), making them a very large group of plant proteins.
The cyclotides have been divided into two main subfamilies, Möbius and bracelet (1), depending on the presence or absence, respectively, of a cis-Pro peptide bond in loop 5. Varv F, a previously uncharacterized cyclotide, belongs to the Möbius subfamily according to its sequence so it was of interest to see if the proposed cis-Pro peptide bond, which is the key structural feature used to classify the cyclotides, is present in its three-dimensional structure.
Until now, structural studies on cyclotides have been performed using nuclear magnetic resonance (NMR), a technique well suited to the study of small proteins (19). Although NMR has provided valuable insight into the nature of cyclotides, some structural features were initially difficult to unambiguously define. For example, there has been some debate over the disulfide bond connectivity. Apart from the knotted arrangement (CysI–CysIV, CysII–CysV, and CysIII–CysVI), which we initially suggested (20), a laddered arrangement was later proposed (CysI–CysVI, CysII–CysV, and CysIII–CysIV) based on an examination of local NOEs between Cys residues (21). Although the connectivity has been resolved on the basis of an analysis of high field NMR data relating to the side chain angles of the cysteine residues (22), and by a chemical analysis of partially reduced species (23, 24), a crystal structure would provide conclusive evidence of the disulfide bond topology. Another important structural feature of cyclotides is their hydrogen bond network, which is thought to stabilize the cyclic cystine knot framework. In particular, a highly conserved glutamic acid residue in loop 1, which has been shown to regulate cyclotide function in mutational studies (25, 26), is believed to be structurally important due to an interaction with the backbone amides of two residues in loop 3 (22). Again, a crystal structure would be useful for defining this interaction.
Generally, analysis of hydrogen bonding patterns in proteins using NMR relies on D2O exchange experiments or the temperature dependence of amide proton chemical shifts, quantified by so-called temperature coefficients (ΔδNH/ΔT). The sensitivity of backbone amide protons to temperature means that temperature coefficients correlate with the hydrogen bonding properties of the amides, with hydrogen-bonded amides generally showing smaller temperature coefficients than non-hydrogen bonded amides. In the current study we show that a comparison of temperature coefficients, slow exchange parameters, and direct determination of hydrogen bonds from a range of cyclotide structures (crystal and solution) can be used to derive general conclusions about the role of hydrogen bonds in stabilizing the generic cyclotide framework.
In this study, we structurally characterized varv F (27), a member of the Möbius subfamily of cyclotides that has been reported to have strong cytotoxic activity (28). The crystal structure that is presented here is the first crystal structure of a cyclotide. Comparison of the crystal structure with the NMR structure also determined in this study confirms the unique features of the cyclotide fold, including the cystine knot topology and β-sheet structure. We investigate the hydrogen bond network, believed to contribute to the stability of the cyclotides, based on an analysis of amide temperature coefficients, D2O exchange experiments, and the predicted hydrogen bonds from the crystal and NMR structures. We also determined the structural characteristics of micelle-bound varv F because cyclotides are believed to act through membrane interactions and so relating the solution and crystal structures to a structure representing the membrane-bound state provides an insight into the mode of action of cyclotides. Furthermore, a comprehensive understanding of the structural features that stabilize the cyclotide framework is important because the stability of cyclotides is key to their natural activity and also contributes to their potential as scaffolds of bioengineered drugs.
EXPERIMENTAL PROCEDURES
Varv F Isolation, Crystallization, and Data Collection—Varv F was isolated from Viola arvensis as described in Göransson et al. (27). Briefly, the dried aerial plant part was defatted with dichloromethane and then extracted with 50% aqueous ethanol. The aqueous extract was filtered through polyamide to remove tannins, followed by removal of low molecular weight substances by size exclusion chromatography on Sephadex G-10. Polysaccharides and buffer salts were then removed by reverse phase solid phase extraction. The resulting cyclotide-enriched fraction was separated with a Sephadex LH-20 column, and eluted with 30% methanol, 0.05% trifluoroacetic acid in water. Under these conditions this column separates cyclotides according to their content of aromatic amino acid residues. Varv F, which is distinguished from the majority of cyclotides by its content of two aromatic residues, could then be easily collected. Final purification was done using reverse phase (RP)2-high performance liquid chromatography (HPLC).
Crystal Screens 1 and 2 (Hampton Research, Aliso Viejo, CA) were used to screen for crystallization conditions using the hanging drop method. For the screen, varv F was dissolved in water at a concentration of 0.5 mm.1 μl of that solution was suspended in 1 μl of each reservoir solution, over reservoir/wells containing 500 μl of the 96 different buffers in the Crystal Screens. After 5 months at 20 °C, a single crystal was formed in buffer condition number 47 of Crystal Screen 2 containing 2.0 m magnesium chloride hexahydrate, 0.1 m Bicine, pH 9.0.
The crystal belongs to space group I4(1)32 with unit cell dimensions: a = b = c = 84.08 Å. There is one molecule in the asymmetric unit with a solvent content of 70% (29). Prior to data collection, crystals were removed from the crystallization drop and then flash-frozen in liquid nitrogen. Data to 1.8 Å resolution were collected at 100 K using monochromatic x-rays at a wavelength of 1.052 Å on beamline I711 at MAXlab (Lund, Sweden). The data were processed using programs DENZO and SCALEPACK (30, 31).
Crystal Structure Determination and Refinement—The structure was solved by molecular replacement using the program PHASER (32, 33). The search model was the high resolution NMR structure of kalata B1 (Protein Data Bandk code 1NB1) with three Cys residues (CysIV, CysV, and CysVI) replaced by alanine. The final structure was generated after several rounds of model building in O (34), and simulated annealing and positional and individual B-factor refinement in CNS (35). The 2Fo - Fc and Fo - Fc electron density maps clearly revealed three disulfide bonds formed by CysI and CysIV, CysII and CysV, and CysIII and CysVI. Water molecules were included where the difference electron density showed a peak above 3σ and the modeled water made stereochemically reasonable hydrogen bonds. The quality and geometry of the model were evaluated by PROCHECK. A Ramachandran plot showed that 85.7% of the residues were in the core allowed region and 14.3% were in the additionally allowed region. As will be discussed later, the x-ray structure of varv F was in excellent agreement with its NMR structure, suggesting that the determined conformation is not affected by solution conditions. The crystal structure has been assigned PDB code 3E4H.
Isolation of varv F for NMR Spectroscopy—Extraction of varv F from the aerial parts of V. arvensis was carried out using our previously established procedure (3). Fresh plant material (collected from the Northeastern region of Victoria, Australia) was ground in a kitchen blender and left overnight in 1:1 dichloromethane:methanol. The extract was filtered, and distilled water was added to promote the separation of the aqueous partition, which was collected and the residual methanol was evaporated in vacuo. Several steps of RP-HPLC were then employed to purify the protein. Preparative RP-HPLC was performed on a Waters 600 Controller system equipped with a Waters 484 Tuneable Absorbance Detector. The aqueous extract was loaded onto a Phenomenex Jupiter C18 column (250 × 22 mm, 5 μm, 300 Å) and eluted at a flow rate of 8 ml/min with a 1% buffer B (90% HPLC grade acetonitrile in H2O, 0.09% trifluoroacetic acid) per min of gradient. Semi-preparative RP-HPLC was performed on an Agilent 1100 series system with variable wavelength detector and a Phenomenex Jupiter C18 column (250 × 10 mm, 5 μm, 300 Å). Analytical RP-HPLC was performed using a Phenomenex Jupiter C18 column (250 × 4.6 mm, 5 μm, 300 Å). Masses were analyzed on a Micromass LCT mass spectrometer equipped with an electrospray ionization source.
NMR Spectroscopy and Structure Calculations—Varv F was dissolved in 90% H2O, 10% D2O at pH ∼6 giving a final concentration of 0.4 mm. A Bruker ARX 500, Bruker ARX 600, or Bruker DMX 750 was used to record spectra with sample temperatures within the range of 288–328 K. All spectra were recorded in phase-sensitive mode by using time-proportional phase incrementation (36). Two-dimensional experiments obtained included a TOCSY (37) with 80 ms mixing time, NOESY (38) with 200 ms mixing time, DQF-COSY (39), and E-COSY (40) in 100% D2O. Water suppression for TOCSY and NOESY experiments was achieved by using a modified WATERGATE sequence (41), whereas lower power irradiation during the relaxation delay was used in the DQF-COSY experiment. Spectra were acquired with 4096 data points in the F2 and 512 increments in the F1 dimension. The F1 and F2 dimensions were multiplied by a sine-squared function prior to Fourier transformation. Chemical shifts were internally referenced to sodium 2,2-dimethyl-2-silapentane-5-sulfonate.
Distance restraints were derived from cross-peaks in NOESY spectra recorded with a mixing time of 200 ms at 288 and 298 K. Spectra were analyzed with the program SPARKY (42). Backbone dihedral angle restraints were derived from 3JHNHα coupling constants measured from line shape analysis of antiphase cross-peak splitting in the DQF-COSY spectrum, whereas χ1 dihedral angles were derived from 3JHαHβ coupling constants from the E-COSY spectrum together with NOE intensities. After initial structure calculations were performed using CYANA (43), hydrogen bond restraints for slowly exchanging amides were added.
In total, 279 distance restraints, comprising 65 sequential, 63 non-sequential restraints, and 151 intra-residue restraints, were determined from NOESY spectra; 29 dihedral angle restraints (15 φ and 14 χ1) were derived based on coupling constants from DQF-COSY and E-COSY spectra, and slowly exchanging amides detected >16 h after dissolution of the sample in D2O were used to derive upper limit distance restraints for nine hydrogen bonds.
Final sets of 50 structures were calculated using a torsion angle-simulated annealing protocol within CNS (35) and were further refined in a water shell (44). The structures were analyzed with MOLMOL (45), PROMOTIF (46), and PROCHECK (47). For the 20 lowest energy structures, 81.8% of the residues were in the most favored and 18.2% in the additionally allowed regions of the Ramachandran plot. The NMR structure of varv F has been deposited in the PDB (code 2K7G).
Hydrogen Bond Analysis—To measure the temperature dependence of the amide chemical shifts (ΔδNH/ΔT), one-dimensional and TOCSY spectra were recorded at increasing temperatures in steps of 10 K from 280 to 330 K or from 278 to 328 K. The chemical shift movements were documented and fitted to a linear function. Temperature coefficients for the cyclotides were measured at two pH values, namely 3.3 and 6.4 for kalata B1, 3.2 and 6.0 for kalata B2, 2.8 and 6.0 for varv F, 2.9 and 5.6 for cycloviolacin O1, and 2.0 and 5.5 for kalata B5.
Amide protons detected in TOCSY spectra after a minimum of 16 h following dissolution in D2O were classified as slowly exchanging. TOCSY spectra of kalata B1, kalata B2, varv F, cycloviolacin O1 (all at pH ∼ 4), and kalata B5 (pH ∼ 5.5) were acquired after 16–72 h. Previously reported slowly exchanging amides were used for kalata B1 (20) and kalata B2 (4).
Hydrogen bond donor and acceptor pairs were determined using an in-house program. Each amide was classified as a hydrogen bond donor if for at least half of the corresponding ensemble of structures, they were able to satisfy empirical criteria (i.e. the distance between the hydrogen atom connected to the donor and the acceptor is less than 2.4 Å, and the angle subtended by the line from the hydrogen atom of the donor to the donor and the line from the hydrogen atom to the acceptor is greater than 90° (48)). For a few structures within the ensembles, certain amides, which normally belong to conserved hydrogen bonded positions, were not formally classified as hydrogen bonded but the failure to recognize these hydrogen bonds was found to be related to the stringency of the empirical criteria. For example, in kalata B1, the amide of Val-4 was not predicted to be a hydrogen bond donor using the criteria mentioned above, but after relaxing the distance restraint for a hydrogen bond to 2.75 Å, it was classified as being in a hydrogen bond. Similarly for Cys-19 of kalata B2 and Cys-20 of kalata B5, slight modification of the strict hydrogen bond criteria resulted in the respective amides being classified as hydrogen bond donors, consistent with the hydrogen bonding properties of the corresponding amides in the other cyclotides studied. These observations were taken into account when the temperature coefficients and slow exchanging nature of the amides were analyzed with respect to their hydrogen bonding classification. In kalata B5, the amide of Gly-16 did not have a predicted acceptor in more than half of the ensemble of structures, but had a number of possible bonding partners, including Glu-6, Cys-13, and Ile-14, and was classified as being involved in a hydrogen bond.
Cyclotide Binding to DPC Micelles—Titration of varv F (1.9 mm) in 10% D2O with dodecylphosphocholine (DPC) was performed at 30 °C, pH 2.9. The one-dimensional NMR spectrum was acquired and the diffusion rate (49) of the cyclotide-DPC complex was measured at each titration point, ranging from a DPC to cyclotide ratio of ∼2:1 to 60:1. Under the condition of fast exchange between the unbound and bound states, the chemical shift of an affected proton (δobs) depends on the chemical shift of the bound state (δbound), the chemical shift of the free state (δfree), and on the bound peptide to free peptide according to the following equation.
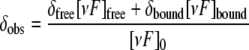 |
(Eq. 1) |
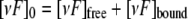 |
(Eq. 2) |
The Langmuir isotherm shown in the following equation was used for analysis (50) and provided values for Ka, the affinity constant, of the peptide for the DPC micelle, and N, the number of DPC molecules that form the site of the peptide binding.
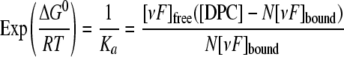 |
(Eq. 3) |
Modeling of Cyclotide-DPC Complex—Titration of varv F/DPC samples with 5- and 16-doxylstearate was performed at 50 °C. NOESY spectra (100 ms mixing time) were measured at 0 and 8 mm doxylstearate at pH 5. The paramagnetic attenuation induced by doxylstearate was qualitatively characterized by calculating the relative cross-peak intensity.
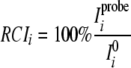 |
(Eq. 4) |
Here 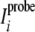 and
and
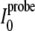 are the intraresidual
HN-Hα cross-peak intensities
(Hδ2–Hδ3 for prolines) in the spectra
of samples with and without a paramagnetic probe.
are the intraresidual
HN-Hα cross-peak intensities
(Hδ2–Hδ3 for prolines) in the spectra
of samples with and without a paramagnetic probe.
A geometric model of the varv F-micelle complex was built based on an approach described previously for kalata B1 (51), i.e. by constraining selected protons to be either inside or outside of a spherical micelle based on their relative broadening by the spin label. Using the 5-doxylstearate data, the HN and Hα atoms for which the relative intensity of their cross-peaks was <20% were restricted to be inside the micelle and other HN and Hα atoms were restricted to be outside the micelle.
RESULTS
The Crystal Structure of varv F—Like many small disulfide-rich proteins, cyclotides are difficult to crystallize. However, after many trials on a range of cyclotides from plants in Rubiaceae and Violaceae, we obtained a crystal of varv F, a member of the Möbius subfamily of cyclotides. The crystal diffracted to 1.8 Å resolution and belongs to space group I4(1)32 with unit cell dimensions: a = b = c = 84.08 Å. Crystallographic statistics for data collection and refinement are shown in Table 1. The data are 99.9% complete to 1.8 Å, and the overall merging Rsym is 6.1%. The crystal structure was determined by molecular replacement using the NMR structure of kalata B1 (PDB code 1NB1) as the search model, refined to a final Rfactor of 22.4% with an Rfree of 24.4% at 1.8-Å resolution. The mean coordinate error of the structure is estimated to be 0.22 Å, based on the Luzzati plot analysis (52).
TABLE 1.
Data collection and refinement statistics for the crystal structure of varv F
| Data collection | |
| Space group | I4(1)32 |
| Cell dimensions | |
| a, b, c (Å) | 84.08, 84.08, 84.08 |
| α, β, γ (°) | 90, 90, 90 |
| Resolution (Å) | 30-1.80 (1.86-1.80)a |
| No. observations | 178,276 |
| No. unique reflections | 4,978 |
| Rmergeb | 0.061 (0.252) |
| (I)/σ(I) | 15.1 (8.9) |
| Completeness (%) | 99.9 (99.6) |
| Redundancy | 35.8 (35.6) |
| Refinement | |
| Resolution (Å) | 30-1.80 (1.86-1.80) |
| No. reflections | 4,863 |
| Rfactorc | 0.224 (0.243) |
| Rfreed | 0.244 (0.261) |
| No. atoms | |
| Peptide | 202 |
| Water | 45 |
| B-factors | |
| Peptide | 21 |
| Water | 36 |
| Root mean square deviations | |
| Bond lengths (Å) | 0.004 |
| Bond angles (°) | 1.8 |
Highest resolution shell is shown in parentheses
Rmerge = Σhkl(Σi(|Ihkl,i – 〈Ihkl〉|))/Σhkl,i〈Ihkl〉, where Ihkl,i is the intensity of an individual measurement of the reflection with indices hkl, and 〈Ihkl〉 is the mean intensity of that reflection
Rfactor = Σ||Fo| – |Fc||/Σ|Fo|, where |Fo| and |Fc| are the observed and calculated structure factor amplitudes
Rfree is calculated as for Rfactor with 5% of the data excluded from refinement
Two key questions that we wanted to address by determining the crystal structure were the precise arrangement of the disulfide bonds (i.e. whether or not these formed a cystine knot) and the presence or absence of the cis-conformation of a conserved proline residue that NMR studies had identified as characteristic of the Möbius subfamily of cyclotides. The x-ray data clearly confirms the presence of both structural features. Fig. 1 shows an electron density map that highlights the topology of the cystine knot motif in which one disulfide bond threads through an embedded ring formed by two other disulfide bonds and their connecting backbone segments. Further analysis of the backbone ω dihedral angles shows that the bond between Trp-23 and Pro-24 has an ω angle of 0.4°, close to the ideal value of 0° for a cis-peptide bond (53, 54) and far from the value of 180° in a trans-peptide bond.
FIGURE 1.

Electron density maps of the cystine knot. Panel A shows the 2Fo-Fc electron density map, contoured at 1 σ, for the region of the structure around the cystine knot. The cystine residues are labeled. Panel B shows a Fo-Fc electron density map at 3σ where disulfide bonds are removed from the model. The position of backbone loops 1 and 4 are indicated.
As well as defining the knot and cis-Pro linkage, the x-ray data explains an unusual feature of the NMR spectra of cyclotides. Specifically, TOCSY peaks for Trp-23, which precedes the cis-Pro (residue 24), are typically very weak or undetectable in cyclotide NMR spectra (20). The crystal structure of varv F indicates that the φ angle for Trp-23 in loop 5 is -30°, giving rise to a dihedral angle of 90° between the NH and αH protons. This dihedral angle would lead to a small three-bond scalar coupling constant between the NH and αH protons and be expected to result in poor magnetization transfer during the mixing period of the TOCSY experiment. In summary, the crystal structure thus confirmed three key features that had been identified by NMR spectroscopy for other cyclotides.
Solution NMR Structure Determination of Varv F—For comparison with the crystal structure, the solution structure of varv F was determined using homonuclear two-dimensional NMR experiments. The peaks in the TOCSY spectrum were well dispersed in the amide region, with only a few clustered spin systems complicating initial resonance assignment (supplemental information). As discussed above, and consistent with NMR data for other cyclotides, the spin system for Trp-23 is notably absent in the TOCSY spectrum.
In combination with the TOCSY spectrum the NOESY spectrum was used to site-specifically assign spectral peaks to individual protons, and to then generate distance and angle restraints for structure calculations. Fig. 2 summarizes the NMR data used to define the secondary structure of varv F, from which it is apparent that it contains a β-sheet motif seen previously in other cyclotides and in the crystal structure reported here.
FIGURE 2.

Summary of structurally informative NMR data for varv F. Panel A shows the sequence (in a clockwise direction) and disulfide bonds as thick shaded lines between the cysteine residues (circled). Panels A and B, respectively, show short-range dαN NOEs and dNN NOEs. Black boxes represent sequential NOEs (the size of the box relates to the size of the NOE), whereas arcs represent NOEs between residues separated by 2 or 3 residues in the sequence. In panel B long-range NOE connectivities are shown as gray lines. Panel C focuses on the secondary structure. To illustrate the position of the residues in the cystine knot, three cysteines (CysIV, CysV, and CysVI) are highlighted with bold circles. The NOEs and predicted hydrogen bonds are included.
Using the NOE, dihedral, and hydrogen bond restraints, a set of 50 structures was calculated using torsion angle-simulated annealing. A backbone superimposition of the 20 lowest energy structures after energy minimization is presented in Fig. 3. Most regions of the structure are well defined, with an average root mean square deviation of 0.37 Å for the backbone atoms and 0.91 Å for the heavy atoms. The structural statistics, as summarized in Table 2, show that the experimental restraints were fitted with minimal violations.
FIGURE 3.

Structure of varv F. A stereoview of the crystal structure and the 20 lowest-energy NMR structures is presented in panels A and B, respectively. Every tenth residue is labeled. Panel C shows a ribbon diagram of the solution NMR varv F structure, highlighting the β-sheet hairpin loop and cystine knot. A superimposition of the crystal structure and solution structures is shown in panel D, where the backbone of the crystal structure is colored in red.
TABLE 2.
Geometric and energetic statistics for the 20 lowest energy structures of varv F
| NMR distance and dihedral constraints | |
|---|---|
| Distance constraints | |
| Total NOE | 279 |
| Intra-residue | 151 |
| Inter-residue | 128 |
| Sequential (|i-j| = 1) | 65 |
| Medium range (|i-j| ≤ 4) | 34 |
| Long-range (|i-j| ≥ 5) | 29 |
| Intermolecular | 0 |
| Hydrogen bonds | 9 |
| Total dihedral angles | 29 |
| φ | 15 |
| χ1 | 14 |
| Structure statistics | |
| Violations (mean ± S.D.) | |
| Distance constraints (Å) | –0.12 ± 0.06 |
| Dihedral angle constraints (°) | –2.34 ± 0.32 |
| Max. dihedral angle violation (°) | –3.39 |
| Max. distance constraint violation (Å) | –0.30 |
| Deviations from idealized geometry | |
| Bond lengths (Å) | 0.004 ± 0.0002 |
| Bond angles (°) | 0.53 ± 0.05 |
| Impropers (°) | 0.46 ± 0.05 |
| Average pairwise root mean square deviationa (Å) | |
| Backbone | 0.37 ± 0.14 |
| Heavy | 0.91 ± 0.23 |
Pairwise room mean square deviation was calculated among 20 refined structures
Structural analysis using the programs PROMOTIF (46) and MOLMOL (45) confirmed that the major secondary structure feature is a β-hairpin over residues 19–28. As expected, the crystal and NMR structures are in excellent agreement, and can be superimposed with backbone root mean square deviations ranging from 0.61 to 1.08 Å for individual structures within the NMR ensemble.
Geometry and Dynamics of Cyclotide Hydrogen Bonds—One of the advantages of obtaining both x-ray and NMR data for a single protein is that high resolution geometrical data on hydrogen bonds from the former can be combined with kinetic information from the latter. To characterize the location of the hydrogen bonds, believed to be important for stabilizing the cyclotide fold, a program to predict hydrogen bond donor-acceptor pairs was written (refer to “Experimental Procedures”) and used to analyze the structures of a representative group of five cyclotides, including three from the Möbius subfamily (kalata B1, kalata B2, and varv F) and two from the bracelet subfamily (cycloviolacin O1 and kalata B5). Their sequences are summarized in Fig. 4A. The crystal structure of varv F shows a network of 13 intramolecular hydrogen bonds, which are also present in the NMR structure of varv F, and nine additional hydrogen bonds involving external water molecules. Across the five cyclotides studied, the pattern of hydrogen bonds was conserved. Four of the hydrogen-bonded amides are involved in defining the β-hairpin secondary structure that is conserved among the cyclotide structures examined in this work. The conserved hydrogen bond network is schematically illustrated in Fig. 4B.
FIGURE 4.

Hydrogen bond properties for a selection of cyclotides, including three from the Möbius subfamily: kalata B1 (1NB1), kalata B2 (1PT4), varv F (crystal structure presented in this work), and two from the bracelet subfamily, cycloviolacin O1 (1NBJ) and kalata B5. In Panel A, an alignment of the sequences is shown at the top of the diagram, where the conserved cysteine residues are boxed and labeled, and the location of the loops is also shown in the Panel B. Residues with amides that are confidently predicted to be hydrogen bond donors (according to the criteria described by the inset in panel B and present in at least half of the ensemble of structures) are bolded. Amides that are slowly exchanging are shaded with a gray background. Conserved hydrogen bond donors are denoted with asterisks underneath the alignment. Panel B shows a schematic representation of hydrogen bond donor-acceptor pairs that are conserved throughout the cyclotide family; dotted arrows are directed toward the acceptor from the donor. Additional Möbius/bracelet subfamily-specific hydrogen bonds not illustrated here are also present. These are particularly located in loop 3, which in the bracelet subfamily, fold into a short helix. Panel C shows amide temperature coefficients of the selected cyclotides. The temperature coefficient data are separated into three categories depending on whether the corresponding amides are slow exchanging (SE) and/or are hydrogen bonded (H-bond) according to the structures. Data points that move from one side to the other of the nominal -4.6 ppb/K cut-off when the pH is lowered are colored gray. Residues discussed in the text are labeled.
To analyze dynamic aspects of the hydrogen bond network, D2O exchange parameters and amide temperature coefficients (ΔδNH/ΔT) were measured for the five selected cyclotides. Amides involved in intramolecular hydrogen bonds are typically less accessible than nonhydrogen-bonded amides and exchange slowly with solvent protons. The D2O exchange data summarized in Fig. 4, A and B, show that all of the observed slowly exchanging amides can be classified as hydrogen bond donors according to the structures. Fig. 4C shows that there is a wide distribution of ΔδNH/ΔT for any individual cyclotide, with a maximum value of 2.1 ppb/K and a minimum of -12.2 ppb/K, but they cluster nicely into groups characteristic of the hydrogen-bonded state of the amide. For proteins in general, low numerical values for amide temperature coefficients (i.e. ΔδNH/ΔT ≥ -4.6 ppb/K) are suggestive of the presence of a hydrogen bond, whereas high coefficients (i.e. ΔδNH/ΔT <-4.6 ppb/K) suggest that the measured amide is accessible to solvent, i.e. not intramolecularly hydrogen bonded (55). This trend is observed in the current study, confirming the value of temperature coefficients as markers of hydrogen bonding in cyclotides. Notably, the ΔδNH/ΔT values for amides that are both slowly exchanging and hydrogen bonded cluster mainly on one side of the nominal -4.6 ppb/K cut off (Fig. 4C, panel c) and those that are neither slowly exchanging nor hydrogen bonded cluster mainly on the other side (Fig. 4C, panel a). Those amides that are formally hydrogen bonded in the calculated structures but are not slowly exchanging typically have ΔδNH/ΔT values on the high side of the -4.6 ppb/K cut-off (Fig. 4C, panel b). This observation shows that temperature coefficients are a sensitive indicator of even relatively weak hydrogen bonds in cyclotides.
Exceptions to the trends noted above occur for the amides of Glu-7, Asn/Tyr-15, and Thr-16 (boxed in Fig. 4C, panels d and e), belonging to Möbius subfamily cyclotides. Residue 7 (a conserved Glu) has a low value of ΔδNH/ΔT, whereas residues 15 and 16 have high values, both trends apparently inconsistent with the hydrogen-bonded state of the corresponding amides. However, these exceptions are consistent with the observation in other peptides that temperature coefficients also depend on the absolute chemical shift of the amide proton: extremely downfield shifted protons tend to have high temperature coefficients and upfield shifted protons tend to have low coefficients (56). For the Möbius cyclotides, the amide chemical shift of residue 7 is extremely upfield shifted at ∼7 ppm, whereas the amides of residues 15 and 16 are typically downfield at ∼11 ppm.
The temperature coefficient data are also valuable for discerning trends in hydrogen bonding that may vary with pH. For example, Fig. 4C shows that several residues in or adjacent to loop 3 have temperature coefficients that change from one side to the other of the cut-off value of -4.6 ppb/K and appear to become hydrogen bonded at high pH, consistent with the fact that the protonation of Glu-7 affects its ability to stabilize loop 3. These include: Thr-16 in varv F, Gly-12 and Asn-29 in kalata B1, Ser-22 in cycloviolacin O1, and Val-17, Ile-18, Gly-19, and Cys-22 in kalata B5.
A significant benefit of the crystal structure of varv F over previous NMR structures of cyclotides is that it provides additional information about the interaction of cyclotides with their surrounding aqueous environment. Based on distances between observed water molecules and potential hydrogen bond partners in varv F, 60 possible hydrogen bond interactions were identified (supplemental information Table S1). The temperature factors of the water molecules vary significantly, with the most stable water molecule (WAT30) forming putative hydrogen bonds with the amides of both CysIII and CysV. A relatively stable water molecule (WAT49), which sits in a cavity near the amide of Glu-7, provides an explanation for the low temperature coefficient of Glu-7 mentioned above. This cavity appears to be a conserved feature of Möbius cyclotides; for example, it is present in the structure of kalata B1, a Möbius cyclotide, but absent from the structure of the bracelet cyclotides cycloviolacin O1.
Binding to DPC Micelles—The mechanism of action of cyclotides is believed to involve interactions with biological membranes (57, 58). To determine whether the cyclotide fold is affected by membrane binding, we studied the interaction of varv F with DPC micelles. Titration of DPC into a sample of varv F caused the chemical shift of several varv F protons to change systematically, consistent with a binding interaction. Fig. 5A shows the changes in chemical shift of the Hε1 proton of Trp-23, and indicates that when the DPC to peptide mole ratio (D:P) reaches 30, almost all the varv F is in a micelle-bound state. Fitting the binding curve to a Langmuir isotherm yields a binding constant (Ka) of 2.20 × 103 m-1, slightly weaker than was recently reported for kalata B1 binding to DPC micelles (50).
FIGURE 5.

Binding of varv F to DPC micelles. Panel A shows the titration of 1.9 mm varv F with DPC in H2O (10% D2O), pH 2.9, at 30 °C. The chemical shift, δ, of the Hε1 proton of Trp-23 (filled diamonds) and the diffusion coefficient (DT, open circles) are shown as a function of the DPC/varv F mole ratio. The binding curves were analyzed using the Langmuir isotherm. The dotted line shows the extrapolation of the DT to zero DPC concentration. The inset shows one-dimensional NMR spectra monitoring the change in the Trp-23 Hε1 signal as DPC was titrated into a sample of varv F. Panel B compares the Hα chemical shifts of varv F in the free and micelle-bound states. Panel C shows the relative cross-peak intensity after 5-doxylstearate (black squares) and 16-doxylstearate (gray circles) were added to separate samples of varv F (1.9 mm, pH 5, 50 °C) and DPC at a DPC to peptide ratio of 60:1. The relative cross-peak intensities (HN-Hα for nonproline residues and Hδ2-Hδ3 for prolines) in 100 ms NOESY spectra are shown.
The translational diffusion coefficient (DT) of the varv F-DPC complex was measured at each step of the titration (Fig. 5A). Extrapolation of the DT values obtained after saturation of binding back to infinite dilution to correct for viscosity effects allowed the DT of the varv F-DPC complex to be determined as 1.15 × 10-10 m2 s-1. Substitution of this value into the Stokes-Einstein relation suggested that the varv F-DPC complex had a hydrodynamic radius, RH of 24 Å, which is slightly larger than the RH of pure DPC micelle (DT = 1.2 × 10-10 m2 s-1;RH = 23.1 Å) (59), consistent with complex formation. Thus, both chemical shift and diffusion measurements confirmed a binding interaction of varv F with DPC micelles.
To determine whether the structure of varv F is affected by membrane binding, the Hα chemical shifts for each residue in the free and bound states were compared, as shown in Fig. 5B. Hα chemical shifts are very sensitive to the backbone topology of a protein and so the observed similarity in shifts strongly suggests that the overall structure of varv F does not change upon binding to the micelle surface, confirming that the cyclotide fold is rigid and not influenced by membrane interactions.
To provide an insight into how cyclotides bind to the micelle surface, we embedded the relaxation probes, 5- and 16-doxylstearate, into separate varv F/DPC samples to assess how deeply varv F penetrated the micelle. By comparing NOESY spectra of samples with and without relaxation probes, the broadening of specific proton signals of varv F was quantified. Fig. 5C shows that broadening occurs principally in loops 1, 2, and 5, consistent with only minor penetration of varv F into the micelle. The orientation of binding was calculated based on the observed broadening and is shown in Fig. 6, along with the positions of key bound water molecules, the hydrogen bonding network, and a patch of surface-exposed hydrophobic residues.
FIGURE 6.

Structural features of cyclotides implicated in their membrane binding and bioactivity. Panel A shows the location of water molecules (black spheres) that lie in a cavity of varv F near the highly conserved Glu-7, which is involved via its side chain carboxyl in a hydrogen bond network involving the amides of Tyr-15 and Thr-16 as shown in panel B. One of the water molecules forms a hydrogen bond to the Glu-7 amide. Other water molecules are also present in the crystal structure but are not illustrated here (see supplementary material). Panel A also highlights the surface of varv F and its hydrophobic patch (shaded in black), which significantly overlaps with its membrane binding face. For clarity this view is rotated 90° from the view in panel B, which shows the membrane binding orientation based on 5-doxylstearate attenuation data in dodecylphosphocholine micelles. This orientation leaves a cluster of residues surrounding Glu-7, and known to be important for bioactivity, exposed and available for self-association interactions that are proposed to facilitate further membrane insertion.
DISCUSSION
In this paper we report the first crystal structure of a cyclotide, varv F, and compare it with the solution NMR structure of the same cyclotide. Landmark structural features observed in NMR-derived solution structures of other cyclotides, such as the cystine knot topology and associated β-sheet core, were confirmed in the crystal structure. Additionally, the hydrogen bond network observed in the crystal structure was used to delineate a conserved set of hydrogen bonds, which were consistent with NMR amide temperature coefficients and slow exchange parameters in a representative group of cyclotides. Overall, the results show that the cyclotide fold is bound by a tight disulfide knot at the protein core, which is flanked by an array of hydrogen bonds that restrict the movement of the exposed loops. This detailed analysis of the stabilizing structural features explains the structural mechanism by which cylotides maintain their natural activity in harsh conditions and provides a basis for mutagenesis studies investigating the mode of action of cyclotides. Furthermore, this analysis may potentially assist in molecular design applications for the engineering of novel functions into the rigid cyclotide framework.
The Cyclotide Structural Framework—To date, 11 cyclotide structures have been published, all determined by NMR, including kalata B1 (20, 22), kalata B2 (4, 24), kalata B8 (60), circulin A (61), circulin B (62), cycloviolacin O1 (1, 22), cycloviolacin O14 (63), palicourein (64), tricyclon A (65), vhr1 (66), and vhl-1 (67), along with several computation models built on these solution structures (e.g. vodo M and vodo N (68)). In all of the structures the circular backbone is folded back onto itself and stabilized by a cystine knot, composed of three disulfide bonds (CysI–CysIV, CysII–CysV, and CysIII–CysVI) and the backbone sequences of loops 1 and 4. This topology is also present in the crystal structure of varv F reported here, thereby confirming the key features of the cyclotide fold by an independent structural method. Furthermore, the crystal structure confirms the main element of the secondary structure observed in all previous NMR structures of cyclotides, namely two β-strands that form a β-hairpin centered in loop 5. The crystal structure confirms that many of the residues project away from the cystine core and participate in interactions that are important for cyclotide bioactivity (50).
Previously, the connectivity of the disulfide bonds in cyclotides, which is key to their stability, has been the topic of some debate. De novo determination of the disulfide connectivity by NMR is difficult because cyclotides are disulfide-rich, with all of the cysteine side chains packed tightly together in the molecular core. The proximity of bonded and non-bonded cysteine residues means that NOE distance restraints between the cysteine residues could potentially agree with many alternative disulfide bond arrangements. The difficulty in determining the correct disulfide connectivity was emphasized by the fact that two disulfide topologies, a knotted arrangement and a laddered one, were proposed in two independent studies (20, 21). Although subsequent studies involving the analysis of side chain angle restraints (22) and the chemical analysis of partially reduced species (23, 24) provided strong support for the knotted arrangement, the crystal structure of varv F now provides conclusive evidence that the knotted arrangement we proposed originally is indeed correct. A clear definition of the cystine knot is important because it appears that this motif is primarily responsible for the thermal, enzymatic, and chemical stability of the cyclotides (11).
In general, peptide bonds can adopt either the cis (ω = 0°) or trans (ω = 180°) conformation, although the cis conformation is higher in energy and not often observed except in X-Propeptide bonds (about 9% of Pro are cis-conformers (69)). Varv F was predicted to belong to the Möbius cyclotide subfamily because its sequence includes a Pro residue that, in other members of the family, forms a cis-peptide bond in loop 5 creating a conceptual twist in the circular backbone. Members of the bracelet cyclotide subfamily lack this Pro residue and have an all-trans circular backbone ribbon. Using NMR, the conformation of Pro-peptide bonds can be determined by the intensity of X-Pro NOE cross-peaks, where a strong sequential Hα-Hδ cross-peak is indicative of a trans-peptide bond, whereas a strong sequential Hα-Hα cross-peak confirms a cis-peptide bond. The latter cross-peak was observed in the NOESY spectrum of varv F, and the crystal structure confirmed the presence of a cis-peptide bond preceding Pro-24. Together, the crystallographic and NMR data verify the classification of varv F as a member of the Möbius cyclotide subfamily. Furthermore, the residue immediately before the conserved Pro in loop 5 is always either a Tyr or a Trp in the Möbius subfamily (with the exception of kalata B11, which has a Asp-Pro combination), consistent with findings that aromatic residues are favored as the preceding residue of a cis-Pro (69).
It has been proposed (22) that the Gly residue in the last position of loop 3 of cyclotides is highly conserved because it has a positive φ angle (unfavorable in non-Gly residues) that helps it link loop 3 to the cystine knot. The crystal and solution structures both confirm that the equivalent Gly residue in varv F (residue 18) has a positive φ angle. It is also interesting to note that the three other Gly residues in the varv F structures also adopt positive φ angles. These data agree with the conformations observed for most Gly residues in other cyclotide NMR structures, suggesting that the less-conserved Gly residues, where present, are important for minimizing strain in the cyclotide scaffold, allowing the disulfides in the core to be tightly packed. This creates a compact peptide structure that is less susceptible to conformational changes or degradation than a loosely packed peptide.
In addition to the cystine knot motif and Gly residues at strategic points in the framework, a network of hydrogen bonds is also thought to be critical for stabilizing the compact cyclotide fold (70). A conserved pattern of hydrogen bonds was observed in the crystal structure of varv F and in the solution structures of the five cyclotides examined by NMR. The hydrogen bonds are distributed throughout the cyclotide fold and help stabilize every loop in the structure. Of particular interest is a hydrogen bond network between the side chain oxygen atoms of the conserved Glu residue (e.g. Glu-7 for varv F) in loop 1 and the backbone amides of two residues in loop 3 (e.g. Tyr-15 and Thr-16 for varv F) (22). This H-bond network reported previously in NMR structures of cyclotides is also present in the crystal structure of varv F, supporting the proposal that this Glu in loop 1 is highly conserved because its network of hydrogen bonds stabilizes the cyclotide fold (70). In a recent study, replacement of Glu-7 with an Ala in kalata B1 was shown to result in a peptide that was more susceptible to thermal unfolding and was more flexible (26).
In general, both D2O exchange data and ΔδNH/ΔT data have been used in NMR studies of proteins to identify amides that can potentially act as hydrogen bond donors. For the five cyclotides examined in this study, both approaches were able to predict the presence of hydrogen bonds. All of the experimentally detected slowly exchanging amides are involved in a hydrogen bond network, confirming that slow exchange data are reliable for determining hydrogen bonded amides for cyclotides. There is also a clear trend observed for the ΔδNH/ΔT data, hydrogen-bonded amides have low temperature coefficients, whereas uncommitted amides have high temperature coefficients. Although subject to some limitations due to exceptions based on chemical shifts, the advantage of temperature coefficients over slow exchange data is that a larger number of hydrogen-bonded amides can potentially be identified, including those with weaker hydrogen bonds. The ΔδNH/ΔT analysis has another advantage over exchange experiments: generally, temperature coefficients are not pH-dependent unless a conformational change in the structure is induced by the change in pH (55). Thus by recording and comparing ΔδNH/ΔT at different pH values it is possible to probe structural changes in the hydrogen bond network as a function of pH. This possibility is particularly important for the cyclotides given the involvement of Glu-7 in the hydrogen bond network.
Insights into the Biological Function of Cyclotides—The excellent agreement between the crystal and solution structures of varv F and also to the high-resolution NMR structure of kalata B1 emphasizes the rigidity and stability of the cyclotide framework. This rigidity is crucial because cyclotides must be able to maintain their structure and function in the harsh environment of insect guts, where they deliver their plant-defense activity through membrane interactions (58). In this study we have shown that the structure of varv F is unaffected by binding to micelles, which were used to mimic a membrane environment. The ability of cyclotide structures to remain rigid regardless of the surrounding environment is further supported by two recent NMR studies, which also used micelles to mimic a membrane environment (50, 71). In all binding studies of cyclotides to DPC micelles so far, the membrane-binding site of cyclotides overlaps significantly with their surface-exposed hydrophobic patch, as illustrated in Fig. 6. The crystal structure of varv F provides a high-resolution structural picture of this crucial membrane-binding region.
Membrane binding interactions are, however, only part of the mechanism explaining the mode of action of cyclotides. A recent alanine mutagenesis study of the prototypical cyclotide, kalata B1, identified a cluster of residues, which are not part of the membrane binding surface, as crucial for cyclotide insecticidal activity (26). These residues are believed to be involved in a self-association interface. Cyclotides have been shown to have a weak tendency to form tetramers (and octamers) in solution (72) and it is tempting to speculate that cyclotides exert their bioactivity by accumulating at the membrane surface and subsequently self-associating to form pores that disrupt the membrane, or conversely first self-associating and then binding to membranes. The x-ray structural data presented here supports the former suggestion, because proteins with a tendency to multimerize frequently crystallize in their complexed state, but no evidence for a potential tetramer/octamer structure could be discerned from the crystal symmetry packing of varv F. This finding supports the view that a membrane environment may facilitate self-association of cyclotides. The model presented in Fig. 6, which represents the orientation of the first encounter of a monomeric cyclotide with a membrane surface, is consistent with this proposal, as it shows that the “bioactive face,” which surrounds the conserved Glu residue in loop 1, is free to become involved in self-association interactions to produce multimeric structures, which apparently then form membrane pores.
This conserved Glu residue is the most conserved feature of the cyclotide family (apart from the disulfide knot) and is functionally important for both insecticidal and cytotoxic activity (25, 26). However, the reason for its high conservation and importance for function is still a key question in structure-function studies of cyclotides. One suggestion is that it is involved in a metal binding site, as NMR experiments using Mn2+ ions show significant quenching of signal intensities in the region around Glu-7 (21, 50, 71). However, despite varv F being crystallized with high concentrations of Mg2+, a metal bound form of varv F was not detected, suggesting that metal ion interactions with cyclotides may not play a significant role in their mode of action. Instead it may be the important structural role of Glu-7 in the distinctive hydrogen bond network that we have identified here that is needed for retaining the rigid fold, which in turn is important for the self-association and mode of action.
This study has identified hydrogen bonding interactions of cyclotides with surrounding water molecules. Water molecules associated with protein surfaces or buried in crevices are integral parts of protein structures and interact primarily through hydrogen bonds with polar groups. Although cyclotides have a surface-exposed hydrophobic patch (Fig. 6A) they still contain large numbers of hydroxyl-bearing residues. In fact, after Cys and Gly, Thr and Ser are the most abundant residues in cyclotides, and it is around these residues that electron densities from water molecules can be seen in the x-ray data. The precise definition of solvent molecules will help define starting models for computer simulations of cyclotides in the presence of lipids, which will be an important next step to gain insight into the structure of the proposed cyclotide pore, which has been proven difficult to characterize by NMR or crystallography.
Conclusion—In summary, this study has shown that the crystal, solution, and membrane-bound structures of varv F, a prototypic Möbius cyclotide, are almost identical, confirming that cyclotides are rigid molecules that are unaffected by external conditions such as binding to membrane surfaces. This structural integrity allows the cyclotides to maintain their activity in the harsh environment of predator insect guts. In addition, this rigidity confirms the potential value of cyclotides as promising templates for the development of engineered protein drugs. They are small, structurally well defined proteins that are remarkably stable. The engineering of novel bioactivities onto exposed loops in cyclotides should be greatly facilitated by the understanding of their structural features elucidated here. In particular, it is clear that the cystine knot and associated β-sheet structure is stabilized by a conserved network of hydrogen bonds and the strategic placement of glycine residues.
Supplementary Material
The atomic coordinates and structure factors (code 3E4H) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
This work was supported by a grant from the Australian Research Council (to D. J. C.).
The on-line version of this article (available at http://www.jbc.org) contains supplemental Table S1 and Figs. S1 and S2.
Footnotes
The abbreviations used are: RP, reverse phase; HPLC, high performance liquid chromatography; PDB, Protein data bank; DPC, dodecylphosphocholine; Bicine, N,N-bis(2-hydroxyethyl)glycine.
References
- 1.Craik, D. J., Daly, N. L., Bond, T., and Waine, C. (1999) J. Mol. Biol. 294 1327-1336 [DOI] [PubMed] [Google Scholar]
- 2.Craik, D. J., Daly, N. L., Mulvenna, J., Plan, M. R., and Trabi, M. (2004) Curr. Protein Pept. Sci. 5 297-315 [DOI] [PubMed] [Google Scholar]
- 3.Jennings, C., West, J., Waine, C., Craik, D., and Anderson, M. (2001) Proc. Natl. Acad. Sci. U. S. A. 98 10614-10619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jennings, C. V., Rosengren, K. J., Daly, N. L., Plan, M., Stevens, J., Scanlon, M. J., Waine, C., Norman, D. G., Anderson, M. A., and Craik, D. J. (2005) Biochemistry 44 851-860 [DOI] [PubMed] [Google Scholar]
- 5.Gran, L. (1973) Lloydia 36 174-178 [PubMed] [Google Scholar]
- 6.Gustafson, K. R., McKee, T. C., and Bokesch, H. R. (2004) Curr. Protein Pept. Sci. 5 331-340 [DOI] [PubMed] [Google Scholar]
- 7.Tam, J., Lu, Y., Yang, J., and Chiu, K. (1999) Proc. Natl. Acad. Sci. U. S. A. 96 8913-8918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Svangård, E., Göransson, U., Hocaoglu, Z., Gullbo, J., Larsson, R., Claeson, P., and Bohlin, L. (2004) J. Nat. Prod. 67 144-147 [DOI] [PubMed] [Google Scholar]
- 9.Witherup, K., Bogusky, M., Anderson, P., Ramjit, H., Ransom, R., Wood, T., and Sardana, M. (1994) J. Nat. Prod. 57 1619-1625 [DOI] [PubMed] [Google Scholar]
- 10.Daly, N. L., Gustafson, K. R., and Craik, D. J. (2004) FEBS Lett. 574 69-72 [DOI] [PubMed] [Google Scholar]
- 11.Colgrave, M. L., and Craik, D. J. (2004) Biochemistry 43 5965-5975 [DOI] [PubMed] [Google Scholar]
- 12.Craik, D. J., Simonsen, S., and Daly, N. L. (2002) Curr. Opin. Drug Discovery Dev. 5 251-260 [PubMed] [Google Scholar]
- 13.Trabi, M., and Craik, D. J. (2002) Trends Biochem. Sci. 27 132-138 [DOI] [PubMed] [Google Scholar]
- 14.Craik, D. J. (2006) Science 311 1563-1564 [DOI] [PubMed] [Google Scholar]
- 15.Craik, D. J., Daly, N. L., and Waine, C. (2001) Toxicon 39 43-60 [DOI] [PubMed] [Google Scholar]
- 16.Craik, D. J., Cemazar, M., and Daly, N. L. (2007) Curr. Opin. Drug Discovery Dev. 10 176-184 [PubMed] [Google Scholar]
- 17.Simonsen, S. M., Sando, L., Ireland, D. C., Colgrave, M. L., Bharathi, R., Goransson, U., and Craik, D. J. (2005) Plant Cell 17 3176-3189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gruber, C. W., Elliott, A. G., Ireland, D. C., Delprete, P. G., Dessein, S., Goransson, U., Trabi, M., Wang, C. K., Kinghorn, A. B., Robbrecht, E., and Craik, D. J. (2008) Plant Cell 20 2471-2483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Craik, D. J., and Daly, N. L. (2007) Mol. Biosyst. 3 257-265 [DOI] [PubMed] [Google Scholar]
- 20.Saether, O., Craik, D. J., Campbell, I. D., Sletten, K., Juul, J., and Norman, D. G. (1995) Biochemistry 34 4147-4158 [DOI] [PubMed] [Google Scholar]
- 21.Skjeldal, L., Gran, L., Sletten, K., and Volkman, B. (2002) Arch. Biochem. Biophys. 399 142-148 [DOI] [PubMed] [Google Scholar]
- 22.Rosengren, K. J., Daly, N. L., Plan, M. R., Waine, C., and Craik, D. J. (2003) J. Biol. Chem. 278 8606-8616 [DOI] [PubMed] [Google Scholar]
- 23.Göransson, U., and Craik, D. J. (2003) J. Biol. Chem. 278 48188-48196 [DOI] [PubMed] [Google Scholar]
- 24.Nair, S. S., Romanuka, J., Billeter, M., Skjeldal, L., Emmett, M. R., Nilsson, C. L., and Marshall, A. G. (2006) Biochim. Biophys. Acta 1764 1568-1576 [DOI] [PubMed] [Google Scholar]
- 25.Herrmann, A., Svangård, E., Claeson, P., Gullbo, J., Bohlin, L., and Göransson, U. (2006) Cell Mol. Life Sci. 63 235-245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Simonsen, S. M., Sando, L., Rosengren, K. J., Wang, C. K., Colgrave, M. L., Daly, N. L., and Craik, D. J. (2008) J. Biol. Chem. 283 9805-9813 [DOI] [PubMed] [Google Scholar]
- 27.Göransson, U., Luijendijk, T., Johansson, S., Bohlin, L., and Claeson, P. (1999) J. Nat. Prod. 62 283-286 [DOI] [PubMed] [Google Scholar]
- 28.Lindholm, P., Göransson, U., Johansson, S., Claeson, P., Gullbo, J., Larsson, R., Bohlin, L., and Backlund, A. (2002) Mol. Cancer Ther. 1 365-369 [PubMed] [Google Scholar]
- 29.Matthews, B. W. (1968) J. Mol. Biol. 33 491-497 [DOI] [PubMed] [Google Scholar]
- 30.Minor, W. (1993) XDISPLAYF, Purdue University, West Lafayette, IN
- 31.Otwinowski, Z. (1993) in Proceedings of the CCP4 Study Weekend: Data Collections and Processing (Sawyer, L., Isaacs, N., and Bailey, S., eds) SERC Daresbury Laboratory, Warrington, UK
- 32.Read, R. J. (2001) Acta Crystallogr. Sect. D Biol. Crystallogr. 57 1373-1382 [DOI] [PubMed] [Google Scholar]
- 33.Storoni, L. C., McCoy, A. J., and Read, R. J. (2004) Acta Crystallogr. Sect. D Biol. Crystallogr. 60 432-438 [DOI] [PubMed] [Google Scholar]
- 34.Jones, T. A., Zou, J. Y., Cowan, S. W., and Kjeldgaard, M. (1991) Acta Crystallogr. Sect. A Found. Crystallogr. 47 110-119 [DOI] [PubMed] [Google Scholar]
- 35.Brunger, A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J. S., Kuszewski, J., Nilges, M., Pannu, N. S., Read, R. J., Rice, L. M., Simonson, T., and Warren, G. L. (1998) Acta Crystallogr. Sect. D Biol. Crystallogr. 54 905-921 [DOI] [PubMed] [Google Scholar]
- 36.Marion, D., and Wüthrich, K. (1983) Biochem. Biophys. Res. Commun. 113 967-974 [DOI] [PubMed] [Google Scholar]
- 37.Braunschweiler, L., and Ernst, R. R. (1983) J. Magn. Reson. 53 521-528 [Google Scholar]
- 38.Jeener, J., Meier, B. H., Bachmann, P., and Ernst, R. R. (1979) J. Chem. Phys. 71 4546-4553 [Google Scholar]
- 39.Rance, M., Sorensen, O. W., Bodenhausen, G., Wagner, G., Ernst, R. R., and Wüthrich, K. (1983) Biochem. Biophys. Res. Commun. 27 157-162 [Google Scholar]
- 40.Griesinger, C., Sorensen, O. W., and Ernst, R. R. (1987) J. Magn. Reson. 75 474-492 [Google Scholar]
- 41.Piotto, M., Saudek, V., and Sklenar, V. (1992) J. Biomol. NMR 2 661-665 [DOI] [PubMed] [Google Scholar]
- 42.Goddard, T. D., and Kneller, D. G. (2005) SPARKY 3, University of California, San Francisco, CA
- 43.Güntert, P., Mumenthaler, C., and Wüthrich, K. (1997) J. Mol. Biol. 273 283-298 [DOI] [PubMed] [Google Scholar]
- 44.Linge, J. P., and Nilges, M. (1999) J. Biomol. NMR 13 51-59 [DOI] [PubMed] [Google Scholar]
- 45.Koradi, R., Billeter, M., and Wüthrich, K. (1996) J. Mol. Graph. 14 51-55 [DOI] [PubMed] [Google Scholar]
- 46.Hutchinson, E. G., and Thornton, J. M. (1996) Protein Sci. 5 212-220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Laskowski, R. A., MacArthur, M. W., Moss, D. S., and Thornton, J. M. (1993) J. Appl. Crystallogr. 26 283-291 [Google Scholar]
- 48.McDonald, I. K., and Thornton, J. M. (1994) J. Mol. Biol. 238 777-793 [DOI] [PubMed] [Google Scholar]
- 49.Altieri, A. S., Hinton, D. P., and Byrd, R. A. (1995) J. Am. Chem. Soc. 117 7566-7567 [Google Scholar]
- 50.Shenkarev, Z. O., Nadezhdin, K. D., Sobol, V. A., Sobol, A. G., Skjeldal, L., and Arseniev, A. S. (2006) FEBS J. 273 2658-2672 [DOI] [PubMed] [Google Scholar]
- 51.Dubovskii, P. V., Dementieva, D. V., Bocharov, E. V., Utkin, Y. N., and Arseniev, A. S. (2001) J. Mol. Biol. 305 137-149 [DOI] [PubMed] [Google Scholar]
- 52.Luzzati, D. (1953) Ann. Inst. Pasteur (Paris) 85 277-281 [PubMed] [Google Scholar]
- 53.Pauling, L. (1960) The Nature of the Chemical Bond, 3 Ed., Cornell University Press, Ithaca, NY
- 54.Ramachandran, G. N., and Sasisekharan, V. (1968) Adv. Protein Chem. 23 283-437 [DOI] [PubMed] [Google Scholar]
- 55.Cierpicki, T., and Otlewski, J. (2001) J. Biomol. NMR 21 249-261 [DOI] [PubMed] [Google Scholar]
- 56.Andersen, N. H., Neidigh, J. W., Harris, S. M., Lee, G. M., Liu, Z., and Tong, H. (1997) J. Am. Chem. Soc. 119 8547-8561 [Google Scholar]
- 57.Kamimori, H., Hall, K., Craik, D. J., and Aguilar, M. I. (2005) Anal. Biochem. 337 149-153 [DOI] [PubMed] [Google Scholar]
- 58.Barbeta, B. L., Marshall, A. T., Gillon, A. D., Craik, D. J., and Anderson, M. A. (2008) Proc. Natl. Acad. Sci. U. S. A. 105 1221-1225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shenkarev, Z. O., Balashova, T. A., Efremov, R. G., Yakimenko, Z. A., Ovchinnikova, T. V., Raap, J., and Arseniev, A. S. (2002) Biophys. J. 82 762-771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Daly, N. L., Clark, R. J., Plan, M. R., and Craik, D. J. (2006) Biochem. J. 393 619-626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Daly, N. L., Koltay, A., Gustafson, K. R., Boyd, M. R., Casas-Finet, J. R., and Craik, D. J. (1999) J. Mol. Biol. 285 333-345 [DOI] [PubMed] [Google Scholar]
- 62.Koltay, A., Daly, N. L., Gustafson, K. R., and Craik, D. J. (2005) Int. J. Pept. Res. Therapeutics 11 99-106 [Google Scholar]
- 63.Ireland, D. C., Colgrave, M. L., and Craik, D. J. (2006) Biochem. J. 400 1-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Barry, D. G., Daly, N. L., Bokesch, H. R., Gustafson, K. R., and Craik, D. J. (2004) Structure 12 85-94 [DOI] [PubMed] [Google Scholar]
- 65.Mulvenna, J. R., Sando, L., and Craik, D. J. (2005) Structure 13 691-701 [DOI] [PubMed] [Google Scholar]
- 66.Trabi, M., and Craik, D. J. (2004) Plant Cell 16 2204-2216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chen, B., Colgrave, M. L., Daly, N. L., Rosengren, K. J., Gustafson, K. R., and Craik, D. J. (2005) J. Biol. Chem. 280 22395-22405 [DOI] [PubMed] [Google Scholar]
- 68.Svangård, E., Göransson, U., Smith, D., Verma, C., Backlund, A., Bohlin, L., and Claeson, P. (2003) Phytochemistry 64 135-142 [DOI] [PubMed] [Google Scholar]
- 69.Pahlke, D., Freund, C., Leitner, D., and Labudde, D. (2005) BMC Struct. Biol. 5 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Craik, D. J., Cemazar, M., Wang, C. K., and Daly, N. L. (2006) Biopolymers 84 250-266 [DOI] [PubMed] [Google Scholar]
- 71.Shenkarev, Z. O., Nadezhdin, K. D., Lyukmanova, E. N., Sobol, V. A., Skjeldal, L., and Arseniev, A. S. (2008) J. Inorg. Biochem. 102 1246-1256 [DOI] [PubMed] [Google Scholar]
- 72.Nourse, A., Trabi, M., Daly, N. L., and Craik, D. J. (2004) J. Biol. Chem. 279 562-570 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.