

Abstract
Nonsyndromic autosomal-recessive optic neuropathies are rare conditions of unknown genetic and molecular origin. Using an approach of whole-genome homozygosity mapping and positional cloning, we have identified the first gene, to our knowledge, responsible for this condition, TMEM126A, in a large multiplex inbred Algerian family and subsequently in three other families originating from the Maghreb. TMEM126A is conserved in higher eukaryotes and encodes a transmembrane mitochondrial protein of unknown function, supporting the view that mitochondrial dysfunction may be a hallmark of inherited optic neuropathies including isolated autosomal-recessive forms.
Main Text
Hereditary optic atrophy is a group of neurodegenerative disorders characterized by a sudden or gradual loss of retinal ganglion cells function. The disease primarily involves the macular beam of the optic nerve and may progress to peripheral fibers. Leber hereditary optic neuropathy (LHON, [MIM 535000]) and autosomal-dominant optic atrophies (adOAs, [MIM 165500]) are by far more frequent than recessive forms.
Typically, patients with LHON and adOA have no relevant health problems apart from optic atrophy. However, the disease may be associated with a wide range of extraocular features of mitochondrial dysfunction, including brain, cardiac, muscle, or auditive signs. The clinical expression of extraocular manifestations varies from absence of expression to severe dysfunction, which defines the “plus” forms of LHON and OPA1.1–4
In contrast with LHON and adOA, autosomal-recessive optic atrophies (arOAs) usually involve the central nervous system and other organs. Nonsyndromic arOA are less common. Hitherto, one locus has been mapped but no disease gene has been identified (OPA6, ROA1 [MIM 258500]).5
Using a combination of Affymetrix GeneChip Human Mapping 10K 2.0 Arrays and microsatellite markers, we performed homozygosity mapping in a multiplex inbred nonsyndromic arOA family of Algerian ancestry. This approach identified a unique region of homozygosity on chromosome 11q14.1-q21 (ROA2, family 1, Figures S1 and S2 available online). The critical interval spanned 14.4 Mb and contained 40 known genes (Figure 1; maximum LOD score Zmax = 3.73 with no recombination event at the D11S4187 locus).
Figure 1.
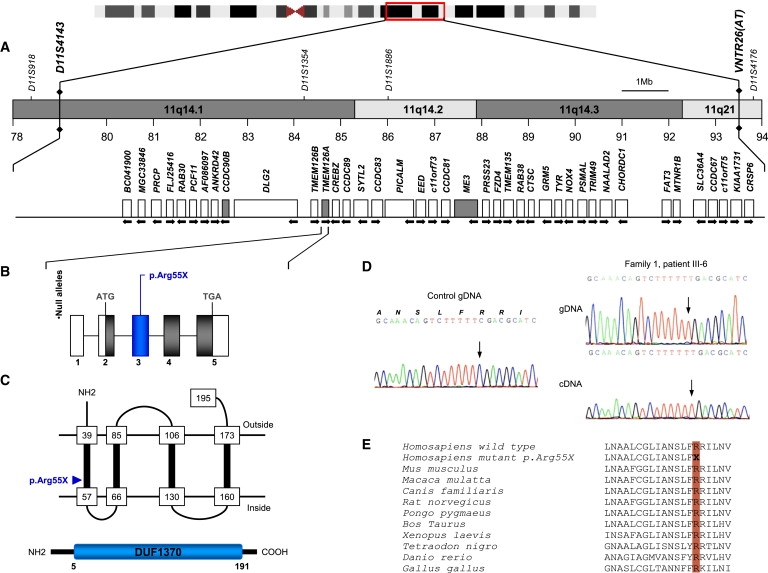
The Critical Region of the ROA2 Locus and the Structure of the TMEM126A Gene and Mutation Identified in Four arOA Families
(A) Physical map of human chromosome 11q14.1-q21 with selected genetic markers. The location and transcriptional direction of known genes are schematically represented, and distances are indicated relative to chromosome 11 according to the Ensembl and UCSC Genome Browser databases. Mitochondrial candidate genes tested in our study are in gray: coiled-coil domain containing (CCDC90B; Maestro score7 = +15), transmembrane protein 126A (TMEM126; Maestro score = +16), and malic enzyme 3 (ME3; Maestro score = +23).
(B) Structure of the TMEM126A gene and position of the disease-causing mutation identified in arOA families. The gene, located at 11q14.1, covers a genomic region of 8542 bp (GenBank NM_032273) and is composed of five exons. The transcript is 773 bp long, with coding sequence of 585 bp (exons 2–5, NM_032273.2). The coding region is indicated in gray and UTRs (5′ and 3′) are in white.
(C) The transmembrane 126A protein is predicted to contain four transmembrane domains (PredictProtein) and a domain of unknown function DUF1370 (Pfam).
(D) Chromatograms of TMEM126A genomic and cDNA sequences showing a homozygous nonsense c.163C→T; p.Arg55X identified in patient III6 - family 1.
(E) The arginine at residue 55 is highly conserved in different species (NCBI BLAST).
Considering that all known optic atrophy genes encode mitochondrial proteins,6 we selected in the interval three genes predicted to encode mitochondrial products7 (Figure 1). Systematic sequence analysis identified a homozygous nonsense mutation in the gene encoding TMEM126A, a transmembrane protein (c.163C→T; p.Arg55X; GenBank accession number NM_032273; Figure 1 and Figure S2). The mutation occurred in a CpG doublet, segregated with the disease in the family (Figure S2) and was not found in 700 control chromosomes (European, n = 600; Algerian, n = 100).
We screened 48 additional unrelated patients affected with nonsyndromic OA (arOA n = 14; adOA n = 10; sporadic n = 24) for TMEM126A mutations. All affected patients and their relatives, including all members of family 1, were recruited with their informed and written consent, as prescribed by the law on bioethics of the European Community and after approval by the local ethics committee (DC-2008-512, Paris-Necker). In all cases, OPA1 mutations and the most frequent LHON mutations (mtDNA G11778A, G3460A, T14484C, and G15257) were previously excluded by direct sequencing. None of the sporadic and adOA cases carried TMEM126A mutations. Conversely, the p.Arg55X mutation was identified in three additional arOA families of Maghrebian origin (Tunisa, family 2; Morocco, Families 3 and 4; Figure S2).
Segregation analysis of microsatellite markers flanking the mutation supported the hypothesis of a founder effect by showing the transmission of a small common haplotype with the disease in the four families (Figure S2). Haplotype studies as well as Bayesian calculations8 suggested that the TMEM126A c.163C→T mutation occurred 80 generations ago, i.e., about 2400 years ago (95% credible interval 35–150 generations).
The p.Arg55X mutation caused an early-onset severe bilateral deficiency in visual acuity (VA), optic disc pallor, and central scotoma (onset between 4 and 6 years; VA = counting fingers to 20/200; Figure S3). The peripheral visual field was strictly normal in all but the oldest patient, who lost it between the age of 30 and 37 (III6, family 1; Figure S3).
The phenotype differs from that of ROA1 patients (OPA6 [MIM 258500]) presenting with an early-onset but slowly progressive OA affecting the macular beam of optic nerve with preservation of the peripheral bundles in advanced stage.5
Polarographic tests and spectrophotometric assays on cultured skin fibroblasts9 showed normal respiratory chain function in patient III6 - family 1 but partial deficiency of Complex I in patient V1 - family 2 (17 nmol/min/mg of protein, mean = 37 ± mean standard deviation 5 nmol/min/mg of protein). Patient V1 - family 2 presented with normal brain MRI but moderate hypertrophic cardiomyopathy. These features, along with the minor brain MRI alterations (Figure S4) and mild hearing loss in patient III6 - family 1, are suggestive of a mitochondrial dysfunction as previously noted in patients with LHON or OPA1 mutations.1–4,10
The TMEM126A gene spans 8.5 kb on chromosome 11 and encodes a single ubiquitous transcript (770 bp) made of one noncoding and four coding exons (Figure 1).
The TMEM126 protein includes the Pfam domain of unknown function defined by the homology between the TMEM126A p.Lys5-Gly191 and TMEM126B p.Asn45-229Glu amino acid sequences, respectively. Four transmembrane domains with the N-terminal and C-terminal sequences on the outside of the membrane were predicted in the TMEM126A DUF1370 domain (PredictProtein, Figure 1). Domain of unknown function (DUF) proteins are hypothetical eukaryotic proteins of around 200 residues in length. Members of this family are regarded as specific to mammals, but phylogenetic analyses contradict this notion. Indeed, orthologs of several DUF proteins, including TMEM126A, have been found in many vertebrate and invertebrate species as well as in plant organisms.11,12 No ortholog could be identified in yeast and fungi.
To determine its subcellular localization, we overexpressed a TMEM126A-myc fusion protein into COS-7 cells. Epitope-tagged wild-type TMEM126A colocalized with mitochondrial Complex II subunit 70 kDa Fp (SDHA [MIM 600857]), Complex IV subunit 1 (MTCO1 [MIM 516030]), ATP synthase subunit beta (ATP5B [MIM 102910]), and ATP synthase subunit alpha (ATP5A [MIM 164360]), supporting the mitochondrial localization of the protein (Figure 2). The fusion protein did not significantly colocalize with the markers of the Golgi apparatus, lysosomes, early endosomes, cytoskeleton, microtubule, and centrosome (Figure S5).
Figure 2.
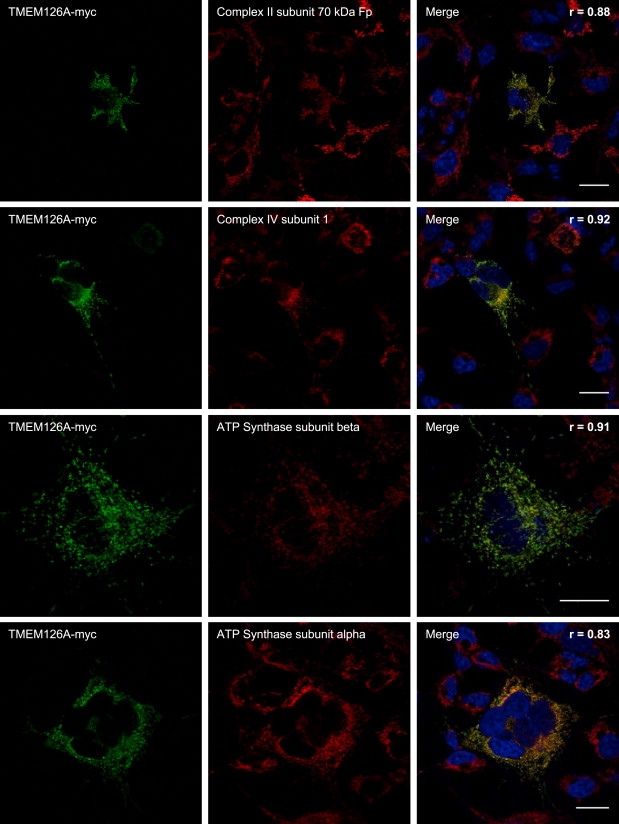
Localization of Transmembrane 126A Protein to Mitochondria of Cultured Cells
Expression in COS-7 cells of a TMEM126-myc fusion protein labeled 48 hr after transfection with an antibody against the myc tag (green) compared to mitochondrial markers (red) via the following primary antibodies: mouse monoclonal anti-Complex II subunit 70 kDa Fp (1/2000, MitoSciences) for succinate dehydrogenase complex, subunit A, flavoprotein (Fp); mouse monoclonal anti-Complex IV subunit 1 (1/200, MitoSciences) for mitochondrially encoded cytochrome c oxidase I; mouse monoclonal anti-ATP synthase subunit beta (1/400, MitoSciences) for ATP synthase, H+ transporting, mitochondrial F1 complex, beta polypeptide; and mouse monoclonal anti-ATP synthase subunit alpha (1/500, MitoSciences) for ATP synthase, H+ transporting, mitochondrial F1 complex, alpha subunit 1. The Pearson's coefficient (r) was calculated with the ImageJ 1.42d software to estimate the degree of colocalization between TMEM126-myc and organelle markers. Its value can range from 1 to −1, with 1 standing for complete positive correlation and −1 for a negative correlation, and with zero standing for no correlation. Pearson's coefficients between TMEM126-myc and mitochondria markers are > 0.83, showing a strong colocalization. These results were confirmed by JACoP Van Steenel's CCF coefficients21 and Pearson's coefficients calculated with Imaris 6.1.5 software (data not shown). Images were acquired with a Leica SP5 confocal microscope (objective 63×; scale bar represents 20 μm) and Leica Application Suite Advanced Fluorescence Lite software.
RT-PCR on total RNA from various adult and fetal human tissues showed that TMEM126A is strongly expressed in the brain (whole), cerebellum, fetal brain, skeletal muscle, testis, fetal retinal pigmentary epithelium (RPE), and fetal retina (Figure S6). In situ hybridization on adult mouse retina (8 months) detected significant levels of specific mRNA in the ganglion cell layer (GCL), the optic nerve head (ON), the outer plexiform layer (OPL), and in the outer ellipsoide (oe) length of photoreceptor inner segments (IS). Faint to no labeling was noted in the outer nuclear layer (ONL) and photoreceptor outer segments. Immunolocalization of the mitochondria-specific alpha subunit of the ATP synthase (ATP5A) on retinal sections of the same animal showed the same pattern of expression, supporting the view that TMEM126A transcripts colocalized with mitochondria (Figure 3). This result suggests that similarly to the dynamin-related GTPase protein Mgm1, of which OPA1 is the human ortholog, TMEM126A is a mitochondria-localized mRNA (MLR) proteins and may be essential in the early nucleation process of large mitochondrial complexes.13–16
Figure 3.
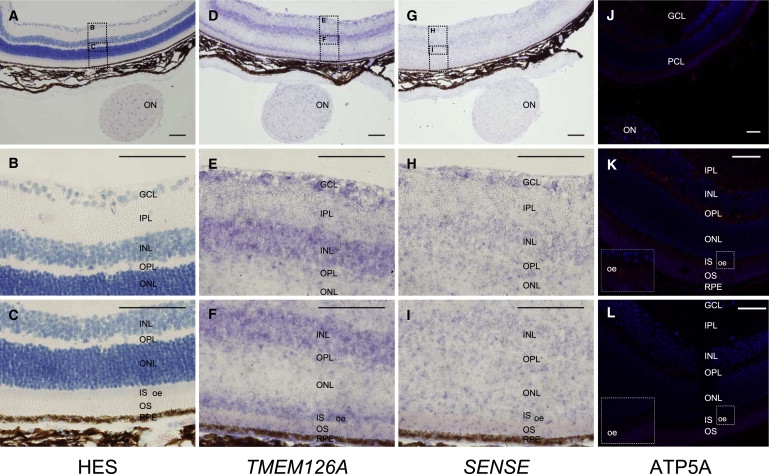
Analysis of TMEM126 mRNA Expression by In Situ Hybridization Compared to Localization of Mitochondria in Adult Mouse Retina
Murine retina cDNA was amplified with primers 5′-T7-ACAGAGTCACGTGTGAGCCAA-3′ and 5′-CCATTGACGGGTATAGCCAA-3′ (sense probe) and 5′-T7-CCATTGACGGGTATAGCCAA-3′ and 5′-ACAGAGTCACGTGTGAGCCAA-3′ (antisense probe) to produce a 524 bp product of the mouse Tmem126a coding region (NM_025460, primer T7: 5′-TAATACGACTCACTATAGGGAGA-3′). The PCR products were purified (QIAquick PCR Purification Kit), verified by sequencing, and subsequently amplified by PCR with T7-polymerase-specific primers (Invitrogen) and Digoxigenin (DIG) RNA labeling mix (Roche). The DIG-labeled cRNA probes were hybridized to the mouse retina sections and detected with an anti-digoxygenin alkaline phosphatase-linked antibody (Roche) and visualized with NBT/BCIP (Roche).
(A–L) Magnification of the mouse retinal cell layers.
(D–L) In adult mouse retina, TMEM126 gene is expressed in the ganglion cell layer (GCL), the inner nuclear layer (INL), the outer plexiform layer (OPL), and the outer ellipsoide (oe) length of photoreceptors inner segments (IS). The staining is slightly less pronounced in the optic nerve (ON) and in the inner plexiform layer. TMEM126A mRNA was not detected in the outer nuclear layer (ONL), outer plexiform layer, or the inner plexiform layer.
(G–I) A sense DIG-cRNA probe revealed no staining in all cellular layers indicating the specificity of the assay. (J and K) Immunolocalization of mitochondria in mouse retinal cell layers with an anti-ATP5A. Mitochondria are sequestered in the ganglion cell layer, the outer plexiform layer, the outer ellipsoid length of the photoreceptors' inner segments, the retinal pigment epithelium cells, and the optic nerve.
(L) Negative control. Note that outer segments display an autofluorescence. PCL denotes photoreceptor cell layer, OS denotes outer segments, and RPE denotes retinal pigment epithelium. Images were recorded on an Olympus IX81 microscope (objective 10× and 40×; scale bar represents 100 μm) with an Olympus DP70 camera, processed with Soft Imaging System Cell̂P and Leica SP5 confocal microscope (objective 40× oil-immersion objective; scale bar represents 50 μm) and Leica Application Suite Advanced Fluorescence Lite software for immunohistochemistry.
In contrast to OPA1 mutations, we found no fragmentation of the mitochondrial network and/or in depletions of the mitochondrial DNA (mtDNA)17,18 in the fibroblasts of patients III6 - family 1 and V1 - family 2 (data not shown), suggesting that TMEM126A and OPA1 may not be functionally related.
Recently, mutations in another TMEM protein have been reported to cause neonatal mitochondrial encephalocardiomyopathy.12 Interestingly, the absence of TMEM70 in patients resulted in the loss of ATP synthase,12 whose subunits have also been shown to be MLR proteins.16
As a probable MLR protein, TMEM126A may be important to the assembly of a protein complex required for the function of retinal ganglion cells in higher eukaryotes. In other tissues, it is possible that another protein of the TMEM family could adequately substitute for the defective function of TMEM126A. It is also possible that TMEM126A may be required to accelerate the assembly of a protein complex. In the absence of TMEM126A, the complex would constitute slowly, and only high-energy-demanding retinal ganglion cells would be affected.
We have identified TMEM126A as a gene that encodes a mitochondrial protein of higher eukaryotes and whose mutations are to our knowledge the first known genetic cause of nonsyndromic autosomal-recessive optic atrophy. This finding gives further support to the view that mitochondrial dysfunctions are a hallmark of optic neuropathies, suggesting that common therapeutic approaches may be developed to improve the long-term prognosis of patients whatever the gene involved.19,20
Acknowledgments
We are grateful to the families who participated. We thank E. Martin, V. Dufresne, K. Bigot, and N. Goudin at the IFR94 Imaging Platform for technical assistance and P. Nitschké and J.M. Plaza at the University Paris-Descartes Bioinformatics Platform. This work has been supported by the Retina France and Fédération des Aveugles de France Associations, the Foundation Fighting Blindness (Grant FFB-BR-GE-0406-0335-INSERM), and the European Community (EC-IP-EVI-Genoret LSHG-CT-2005-51236).
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Ensembl, http://www.ensembl.org
NCBI BLAST, http://www.ncbi.nlm.nih.gov/BLAST
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
Pfam Sanger Institute, http://pfam.sanger.ac.uk
PredictProtein, http://www.predictprotein.org
UCSC Genome Browser, http://genome.ucsc.edu
Accession Numbers
The GenBank accession number reported for Homo sapiens transmembrane protein 126A (TMEM126A) mRNA in this paper is NM_032273.
References
- 1.Lev D., Yanoov-Sharav M., Watemberg N., Leshinsky-Silver E., Lerman-Sagie T. White matter abnormalities in Leber's hereditary optic neuropathy due to the 3460 mitochondrial DNA mutation. Eur. J. Paediatr. Neurol. 2002;6:121–123. doi: 10.1053/ejpn.2001.0558. [DOI] [PubMed] [Google Scholar]
- 2.Vinkler C., Lev D., Kalish H., Watemberg N., Yanoov-Sharav M., Leshinsky-Silver E., Lerman-Sagie T. Familial optic atrophy with white matter changes. Am. J. Med. Genet. A. 2003;121A:263–265. doi: 10.1002/ajmg.a.20238. [DOI] [PubMed] [Google Scholar]
- 3.Yu-Wai-Man P., Griffiths P.G., Hudson G., Chinnery P.F. Inherited Mitochondrial Optic Neuropathies. J. Med. Genet. 2009;46:145–158. doi: 10.1136/jmg.2007.054270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Amati-Bonneau P., Valentino M.L., Reynier P., Gallardo M.E., Bornstein B., Boissière A., Campos Y., Rivera H., de la Aleja J.G., Carroccia R. OPA1 mutations induce mitochondrial DNA instability and optic atrophy ‘plus’ phenotypes. Brain. 2008;131:338–351. doi: 10.1093/brain/awm298. [DOI] [PubMed] [Google Scholar]
- 5.Barbet F., Gerber S., Hakiki S., Perrault I., Hanein S., Ducroq D., Tanguy G., Dufier J.L., Munnich A., Rozet J.M. A first locus for isolated autosomal recessive optic atrophy (ROA1) maps to chromosome 8q. Eur. J. Hum. Genet. 2003;11:966–971. doi: 10.1038/sj.ejhg.5201070. [DOI] [PubMed] [Google Scholar]
- 6.Carelli V., La Morgia C., Iommarini L., Carroccia R., Mattiazzi M., Sangiorgi S., Farne S., Maresca A., Foscarini B., Lanzi L. Mitochondrial optic neuropathies: How two genomes may kill the same cell type? Biosci. Rep. 2007;27:173–184. doi: 10.1007/s10540-007-9045-0. [DOI] [PubMed] [Google Scholar]
- 7.Calvo S., Jain M., Xie X., Sheth S.A., Chang B., Goldberger O.A., Spinazzola A., Zeviani M., Carr S.A., Mootha V.K. Systematic identification of human mitochondrial disease genes through integrative genomics. Nat. Genet. 2006;38:576–582. doi: 10.1038/ng1776. [DOI] [PubMed] [Google Scholar]
- 8.Hanein S., Perrault I., Gerber S., Delphin N., Benezra D., Shalev S., Carmi R., Feingold J., Dufier J.L., Munnich A. Population history and infrequent mutations: How old is a rare mutation? GUCY2D as a worked example. Eur. J. Hum. Genet. 2008;16:115–123. doi: 10.1038/sj.ejhg.5201905. [DOI] [PubMed] [Google Scholar]
- 9.Rustin P., Chretien D., Bourgeron T., Gérard B., Rötig A., Saudubray J.M., Munnich A. Biochemical and molecular investigations in respiratory chain deficiencies. Clin. Chim. Acta. 1994;228:35–51. doi: 10.1016/0009-8981(94)90055-8. [DOI] [PubMed] [Google Scholar]
- 10.Zanna C., Ghelli A., Porcelli A.M., Karbowski M., Youle R.J., Schimpf S., Wissinger B., Pinti M., Cossarizza A., Vidoni S. OPA1 mutations associated with dominant optic atrophy impair oxidative phosphorylation and mitochondrial fusion. Brain. 2008;131:352–367. doi: 10.1093/brain/awm335. [DOI] [PubMed] [Google Scholar]
- 11.Smith U.M., Consugar M., Tee L.J., McKee B.M., Maina E.N., Whelan S., Morgan N.V., Goranson E., Gissen P., Lilliquist S. The transmembrane protein meckelin (MKS3) is mutated in Meckel-Gruber syndrome and the wpk rat. Nat. Genet. 2006;38:191–196. doi: 10.1038/ng1713. [DOI] [PubMed] [Google Scholar]
- 12.Cizkova A., Stránecký V., Mayr J.A., Tesarová M., Havlícková V., Paul J., Ivánek R., Kuss A.W., Hansíková H., Kaplanová V. TMEM70 mutations cause isolated ATP synthase deficiency and neonatal mitochondrial encephalocardiomyopathy. Nat. Genet. 2008;40:1288–1290. doi: 10.1038/ng.246. [DOI] [PubMed] [Google Scholar]
- 13.Heinrich P.C., Schmelzer E., Northemann W., Kaiser C., Witt I. Rat liver cytochrome c oxidase subunits IV and V: Cell-free synthesis as larger molecular weight precursors, mRNA sizes and sites of synthesis. Prog. Clin. Biol. Res. 1982;102:149–159. [PubMed] [Google Scholar]
- 14.Rings E.H., Büller H.A., de Boer P.A., Grand R.J., Montgomery R.K., Lamers W.H., Charles R., Moorman A.F. Messenger RNA sorting in enterocytes. Co-localization with encoded proteins. FEBS Lett. 1992;300:183–187. doi: 10.1016/0014-5793(92)80192-j. [DOI] [PubMed] [Google Scholar]
- 15.Sylvestre J., Vialette S., Corral Debrinski M., Jacq C. Long mRNAs coding for yeast mitochondrial proteins of prokaryotic origin preferentially localize to the vicinity of mitochondria. Genome Biol. 2003;4:R44. doi: 10.1186/gb-2003-4-7-r44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Garcia M., Darzacq X., Delaveau T., Jourdren L., Singer R.H., Jacq C. Mitochondria-associated yeast mRNAs and the biogenesis of molecular complexes. Mol. Biol. Cell. 2007;18:362–368. doi: 10.1091/mbc.E06-09-0827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Frezza C., Cipolat S., Martins de Brito O., Micaroni M., Beznoussenko G.V., Rudka T., Bartoli D., Polishuck R.S., Danial N.N., De Strooper B., Scorrano L. OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell. 2006;126:177–189. doi: 10.1016/j.cell.2006.06.025. [DOI] [PubMed] [Google Scholar]
- 18.Olichon A., Landes T., Arnauné-Pelloquin L., Emorine L.J., Mils V., Guichet A., Delettre C., Hamel C., Amati-Bonneau P., Bonneau D. Effects of OPA1 mutations on mitochondrial morphology and apoptosis: Relevance to ADOA pathogenesis. J. Cell. Physiol. 2007;211:423–430. doi: 10.1002/jcp.20950. [DOI] [PubMed] [Google Scholar]
- 19.Delettre C., Lenaers G., Griffoin J.M., Gigarel N., Lorenzo C., Belenguer P., Pelloquin L., Grosgeorge J., Turc-Carel C., Perret E. Nuclear gene OPA1, encoding a mitochondrial dynamin-related protein, is mutated in dominant optic atrophy. Nat. Genet. 2000;26:207–210. doi: 10.1038/79936. [DOI] [PubMed] [Google Scholar]
- 20.Chevrollier A., Guillet V., Loiseau D., Gueguen N., de Crescenzo M.A., Verny C., Ferre M., Dollfus H., Odent S., Milea D. Hereditary optic neuropathies share a common mitochondrial coupling defect. Ann. Neurol. 2008;63:794–798. doi: 10.1002/ana.21385. [DOI] [PubMed] [Google Scholar]
- 21.Bolte S., Cordelières F.P. A guided tour into subcellular colocalization analysis in light microscopy. J. Microsc. 2006;224:213–232. doi: 10.1111/j.1365-2818.2006.01706.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.