

Abstract
Autosomal-recessive congenital sodium diarrhea (CSD) is characterized by perinatal onset of a persistent watery diarrhea with nonproportionally high fecal sodium excretion. Defective jejunal brush-border Na+/H+ exchange has been reported in three sporadic patients, but the molecular basis of the disease has not been elucidated. We reviewed data from a large cohort of CSD patients (n = 24) and distinguished CSD associated with choanal or anal atresia, hypertelorism, and corneal erosions—i.e., a syndromic form of CSD—occurring in ten families from an isolated form—i.e., classic CSD—presenting in seven families. Patients from both groups have a high risk of mortality due to immediate electrolyte imbalances and complications from long-term parenteral nutrition in the first years of life, but survivors can eventually adapt to partial or complete enteral nutrition. A genome-wide SNP scan was applied and identified a homozygous c.593−1G→A splicing mutation in SPINT2, encoding a Kunitz-type serine-protease inhibitor, in one extended kindred with syndromic CSD. The same mutation and four distinct, homozygous or compound heterozygous mutations (p.Y163C, c.1A→T, c.337+2T→C, c.553+2T→A) were identified in all syndromic patients. No SPINT2 mutations were found in classic-CSD patients. SPINT2 mutations were associated with loss of protein synthesis or failure to inhibit the serine protease trypsin in vitro. We delineate syndromic CSD as a distinct disease entity caused by SPINT2 loss-of-function mutations. SPINT2 mutations might lead to an excess of yet unknown serine protease activity in affected tissues.
Introduction
Congenital sodium diarrhea (CSD [MIM 270420]) is a rare, inherited diarrhea of infancy, first described in 1985.1,2 A diagnosis of CSD is made on the findings of a life-threatening secretory diarrhea, severe metabolic acidosis, and hyponatremia secondary to extraordinarily high fecal losses of sodium, with low or normal excretion of urinary sodium, in the absence of infectious, autoimmune, and endocrine causes. Intestinal biopsies from CSD patients generally reveal mild to moderate villus atrophy and do not show any specific histological changes, allowing differentiation from microvillus inclusion disease and congenital tufting enteropathy.1–3 Additional features, such as choanal atresia, anal atresia, corneal erosions, and hypertelorism, have been observed in association with CSD.3 Patients are initially dependent on parenteral nutrition in order to acquire adequate caloric and fluid intake and allow for normal growth and development. Prolonged parenteral nutrition is frequently complicated by bacteremia and liver disease and can confer a poor quality of life. Single-case reports have shown that CSD patients can be weaned completely from parenteral nutrition in childhood with oral sodium citrate supplementation while watery diarrhea persists.1,2
Defective jenunal brush-border Na+/H+ exchange was demonstrated in three unrelated patients with CSD.1,4,5 However, the pathogenesis of CSD remains poorly understood, and the molecular basis of the disease has not been identified. We previously considered and excluded an involvement of known jejunal genes coding for Na+/H+ exchangers (NHEs), in our study of a family with five affected children displaying autosomal-recessive inheritance of CSD,3 and Zachos et al. excluded such involvement using sequence analysis in one patient.6
We have now studied a large cohort of CSD patients to define the clinical phenotype and applied the positional-candidate approach to define the molecular basis of this disorder.
Material and Methods
Patients
Written informed consent from the parents or legal guardians of all patients was obtained, and the study was performed in accordance with national ethical standards on human experimentation. Patients fulfilling the following criteria were given a diagnosis of CSD:3 Newborns presenting with (a) intractable diarrhea that did not cease while infants were receiving nothing by mouth and (b) voluminous alkaline stools (fecal pH > 7.5, normal, 6.5–7.4) containing high concentrations of sodium (fecal Na+ > 70 mmol/l, normal, 20–35 mmol/l), leading to hyponatremia (plasma Na+ < 130 mmol/l, normal, 136–147 mmol/l) and profound metabolic acidosis (plasma pH < 7.30, normal, 7.35–7.45). Light- and/or electron-microscopic examinations revealed villus atrophy with no features of microvillus inclusion disease or tufting enteropathy. Anal atresia occurred in two patients. There was otherwise no radiological evidence of any anatomic anomaly in the gastrointestinal tract, and laboratory parameters indicating chronic infection or autoimmune disease were negative. Endocrine causes of secretory diarrhea were excluded by normal plasma concentrations of thyroid hormones, thyroid-stimulating hormone, vasoactive intestinal peptide, calcitonin, gastrin, and prostaglandin E2. Also, plasma concentrations of adrenocorticotropic hormone, cortisol, and 17-OH-progesterone were normal, excluding adrenogenital syndrome. Negative sweat tests excluded cystic fibrosis.
Linkage and Mutation Analysis
We carried out a genome-wide linkage scan using the Affymetrix GeneChip XbaI 10K 2.0 SNP arrays,7,8 using DNA samples from two affected and 11 unaffected individuals from one kindred (Figure 1A, Table 1 [family 1]).3 We obtained parametric multipoint LOD-score calculations and haplotypes with the Allegro program,9 using an autosomal-recessive, fully penetrant model. We selected ten genes from the candidate region (SCN1B [MIM 600235], KRTDAP, TMEM147, ATP4A [MIM 137216], RBM42, COX6B [MIM 124089], SPINT2 [MIM 605124], KCNK6 [MIM 603939], FBXO17 [MIM 609094], SNRPA [MIM 182285]) on the basis of putative function and expression patterns for mutation analysis. The coding regions and splice sites were PCR amplified and directly sequenced in the proband from the original family (primer sequences for SPINT2 are shown in Supplemental Data, available online, and data referring to the other genes are available from the authors). The sequencing reactions were analyzed on the ABI 3100 DNA sequencer, with BigDye terminator mix (Applied Biosystems, Vienna, Austria). The probands from 16 additional families were screened for SPINT2 mutations by direct sequencing. Relatives were tested for variants detected in a proband. A panel of DNA samples from anonymous healthy controls was analyzed for the presence of sequence variants. Highly degraded DNA samples extracted from formalin-fixed paraffin-embedded tissue from two patients of the original family were sequenced with PCR primers SPINT2 7F and SPINT2 7R3, generating an 89 bp PCR fragment encompassing the splice site found mutated in the proband. Multiplex ligation-dependent probe amplification (MLPA) was performed in samples without mutations, with the MRC-Holland SALSA MLPA P300-A1 control kit, and newly designed synthetic probes targeting three of the seven SPINT2 exons (Supplemental Data). Probes were designed according to the recommendations of MRC-Holland (Amsterdam).
Figure 1.
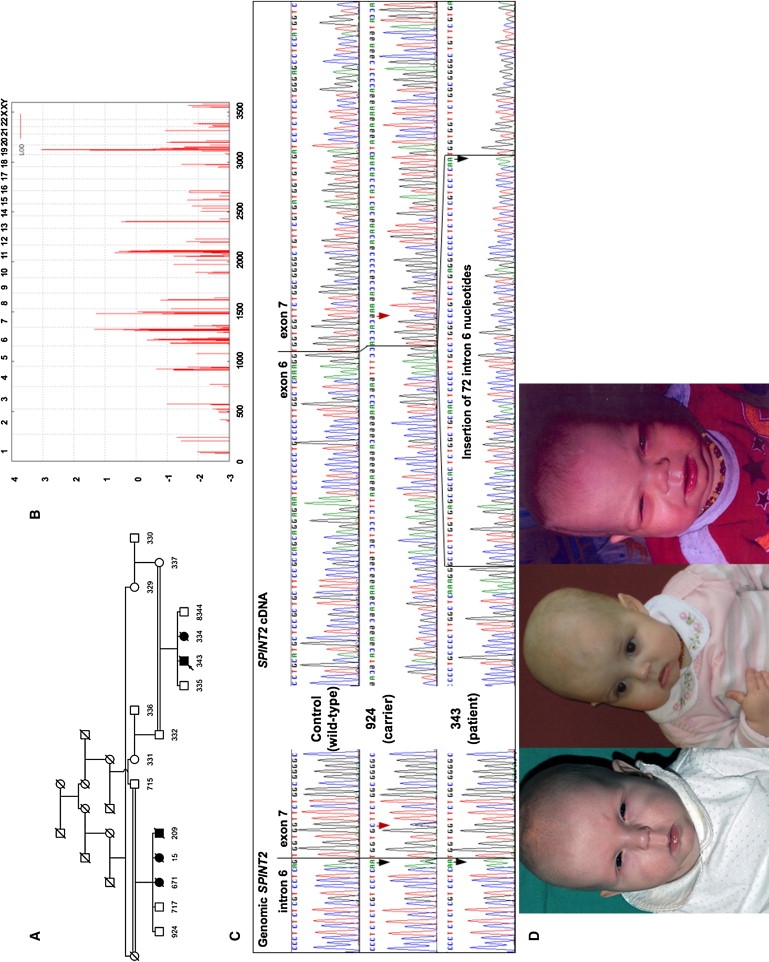
Genetic Mapping, an Analysis of the c.593-1G→A Splice-Site Mutation, and Clinical Findings in CSD
(A) Pedigree of CSD family no. 1. Numbers below symbols indicate individuals whose DNA was included in the genome-wide SNP-linkage scan, with the exception of patients 671 and 209, whose DNA samples were subsequently shown to harbor the SPINT2 mutation identified in the proband (arrow), and with the exception of individual 15.
(B) A LOD score above 3.0 indicates a single region on chromosome 19 that is significantly linked to the disease in this family. The graph represents a parametric LOD score on the y axis in relation to genetic position on the x axis. Human chromosomes are concatenated from p-ter (left) to q-ter (right) on the x axis, and the genetic distance is given in cM.
(C) Left: sequence chromatograms show the mutation c.593-1G→A (black arrows) in homozygous and heterozygous state in genomic DNA samples from individuals 343 and 924, respectively. Individual 924 also shows heterozygosity for c.598G→C (p.V200L, rs17850978, red arrow) on the genomic level. The c.598G→C variant and the c.593-1G→A mutation reside on different alleles, as revealed by haplotype analysis (not shown). Right: cDNA sequencing reveals apparent homozygosity for the c.598G→C variant in individual 924. Only an aberrant transcript containing 72 nucleotides of intron 6 is obtained from the patient with the homozygous c.593-1G→A mutation.
(D) Facial characteristics of patients CD1(1), A2HE(1), and 343. Note hypertelorism, long philtrum, sparse hair, and photophobia.
Table 1.
Clinical and Molecular Findings of CSD Families
| Family No., Ancestry | Patient Identifier | Sex, Parental Consan-guinity | Polyhy-dramnios | Age, Outcome, Current Treatment | Malformations and Dysmorphological Features | SPINT2 Nucleotide Alteration | Coding Sequence, Protein Alteration | Exon |
|---|---|---|---|---|---|---|---|---|
| 1, Austria3 | 343 | M, yes | unknown | died at 13 mo | bilateral choanal atresia, hypertelorism, corneal erosions | c.593-1G→A, homozygous | decreased and aberrant expression | 7a |
| 334 | F, yes | unknown | died at 7 mo | hypertelorism, corneal erosions | c.593-1G→A, homozygous | decreased and aberrant expression | 7a | |
| 671 | M, yes | unknown | died at 6 mo | not recorded | c.593-1G→A, homozygous | decreased and aberrant expression | 7a | |
| 15 | F, yes | unknown | died at 10 mo | double kidney | no DNA sample | - | - | |
| 209 | F, yes | unknown | died at 4 mo | bilateral choanal atresia, cleft palate, aortic-valve hamartoma | c.593-1G→A, homozygous | decreased and aberrant expression | 7a | |
| 2, Austria | A2HE(1) | F, yes | no | 3 yrs, alive, EN | hypertelorism, corneal erosions | c.593-1G→A, homozygous | decreased and aberrant expression | 7a |
| 3, Sweden | S1H(1) | F, no | unknown | died at 6 mo | unilateral choanal atresia, broad thumbs, corneal erosions | c.488A→G, homozygous | p.Y163C | 5 |
| S1H(2) | F, no | no | died at 14 mo | bilateral choanal atresia, extra digit on right hand, low-sitting thumbs, corneal erosions | c.488A→G, homozygous | p.Y163C | 5 | |
| 4, Sweden | S2L(1) | M, no | unknown | 13 yrs, alive, PPN | corneal erosions | c.488A→G, homozygous | p.Y163C | 5 |
| 5, Sweden | S3SH(1) | F, yes | no | 12 yrs, alive, PPN | not recorded | c.488A→G | p.Y163C | 5 |
| 6, Sweden | S4EB(1) | F, no | no | 12 yrs, alive, PPN | bilateral choanal atresia, corneal erosions | c.337+2T→C, c.488A→G; compound heterozygous | p.Y163C, abnormal splicing | 3b, 5 |
| 7, Sweden | S5AA(1) | M, no | yes | died at 31 mo | bilateral choanal atresia | c.488A→G, homozygous | p.Y163C | 5 |
| S5AA(2) | F, no | no | died at 11 mo | bilateral choanal atresia, anal atresia | no DNA | - | - | |
| 8, Sweden | S6TJ(1) | F, no | no | died at 22 mo | bilateral choanal atresia, corneal erosions | c.488A→G, c.553+2T→A; compound heterozygous | p.Y163C, abnormal splicing | 5, 5b |
| 9, The Netherlands | N1P(1) | M, no | no | died at 9 yrs | bilateral choanal atresia, long philtrum, anteverted nares, short and brittle hair, mild psychomotor delay | c.1A→T, c.488A→G; compound heterozygous | p.Y163C, start-codon removal | 1, 5 |
| 10, Canada—Amish | CD1(1) | F, yes | yes | 5 mo, alive, TPN | bilateral choanal atresia, imperforate anus, rectovaginal fistula, corneal erosions, short and brittle hair | c.488A→G, homozygous | p.Y163C | 5 |
| 11, UK1 | UK1P | F, no | yes | 29 yrs, alive, EN | none | none detected | none | - |
| 12, UK5 | UK2B | M, no | yes | 20 yrs, alive, EN | none | none detected | none | - |
| 13, Algeria | F1B | F, yes | no | 21 mo, EN | none | none detected | none | - |
| 14, Italy | D1 | M, no | no | 5 yrs, alive, PPN | none | none detected | none | - |
| 15, Finland2 | SF | F, no | yes | 32 yrs, alive, EN | none | none detected | none | - |
| 16, Roma | SLK1SZ | F, yes | no | died at age 23 days | none | none detected | none | - |
| 17, Roma | SLK2K(1) | F, yes | unknown | died at age 20 days | none | none detected | none | - |
| SLK2K(2) | F, yes | unknown | died at age 9 days | none | no DNA | - | - |
Abbreviations are as follows: TPN, total parenteral nutrition; PPN, partial parenteral nutrition; EN, enteral nutrition; F, female; M, male.
Mutation deletes the consensus splice acceptor.
Mutation deletes the consensus splice donor.
SPINT2 mRNA Analysis and Transient Transfection
mRNA was extracted from blood samples with the PAXgene Blood RNA Kit (PreAnalytiX, QIAGEN, Vienna, Austria) and reversely transcribed into cDNA with the SuperScript III First-Strand Synthesis System (Invitrogen, Lofer, Austria). SPINT2 primers c4f and 7R were used for amplification and direct sequencing of an aberrantly spliced fragment in the patient with the c.593−1G→A mutation. SPINT2 cDNA from patients harboring the p.Y163C or c.593−1G→A mutation and from a control was amplified with the use of primers TopoF and V5R. For cloning of c.1A→T, the forward primer used was TopoM1F (Supplemental Data). The PCR products were cloned into the pcDNA3.2/V5/GW/D-Topo vector (Invitrogen, Leogang, Austria) so that C-terminally fused V5 epitopes could be obtained. The constructs were verified by DNA sequencing, and COS-7 cells grown to ∼70% confluency were transfected via electroporation. Cells were lysed 48 hr after transfection by the addition of 0.5 ml radioimmunoassay precipitation buffer (RIPA) without protease inhibitors. After 20 min incubation on ice, cells were scratched off with a rubber policeman, and the postnuclear supernatant was prepared by centrifugation at 12.000 × g for 30 min. Supernatants were concentrated on Amicon 10.000 Da molecular-weight-cutoff membrane ultrafiltration tubes (Millipore, Vienna, Austria).
Purification of Recombinant Spint2 Proteins
Fusion proteins were purified from cell lysates by immunoaffinity chromatography with the use of a monoclonal mouse anti-V5 antibody (ABP Affinity BioReagents, Golden, CO, USA) immobilized to NHS-activated sepharose (1 m HiTrap column, GE Healthcare, Vienna, Austria), according to the manufacturer's directions. Protein extracts were loaded in lysis buffer onto the immunoaffinity column and eluted in 0.05 M glycine pH 2.7, containing 0.15 M NaCl and 0.1% Triton X, after extended washing. Elution fractions were neutralized by addition of saturated Tris HCl pH 8.9 to a final concentration of 2%. Anti-V5 immunoreactivity in elution fractions was assayed by western blotting: equal amounts of protein (50 μg) were separated by 12.5% SDS-PAGE10 and blotted onto PVDF membranes (GE Healthcare, Vienna, Austria), and SPINT2-V5 fusion protein was detected with mouse anti-V5 antibody at 1:500 dilution in 0.3% skimmed milk in PBST (Invitrogen, Leogang, Austria) and with a secondary horseradish-peroxidase-labeled goat anti-mouse antibody (DAKO, Vienna, Austria) at 1:1000 dilution. Immunological complexes were visualized by enhanced chemiluminescence with the use of an ECL-kit (GE Healthcare, Vienna, Austria) and a CCD camera (Chemidoc XRS, BioRad, Munich, Germany).
Trypsin-Activity Assays
The trypsin inhibitory activity of SPINT2 WT and SPINT2 Y163C proteins was tested by trypsin-mediated para-nitroaniline (pNA) release from the artificial substrate Tos-Gly-Pro-Lys-para-nitroaniline. After preincubation of 4μg/ml trypsin with equal amounts of immunoaffinity-purified SPINT2 WT or SPINT2 Y163C in 5 mM morpholinoethanesulfonic acid (MES), pH 6.5, the reactions were mixed with artificial substrate at a final concentration of 100 μM, and the absorbance at 405 nm was monitored over 10 min in a Beckmann DU64 spectrophotometer equipped with a cuvette heated at 37°C. Trypsin activity was calculated from the slope of the linear increase in absorbance over time. The experiments were performed in quadruplicate, and results were analyzed with the Mann-Whitney U Test.
Results
Homozygosity mapping in a large kindred (Figure 1A) identified a single genomic region of extended homozygosity (Figure 1B) and haplotypes consistent with the inheritance of the mutation from a common ancestor in the proband and affected sib. The critical interval spanned 6.754 Mb on chromosome 19q13, with boundaries set between SNPs rs1363364 and rs2317314 by two recombinants (data not shown). Among the 220 genes contained in the interval (National Center for Biotechnology Information map build 36.2), ten genes were considered as plausible candidates (see Material and Methods).
Sequencing revealed no mutations in nine candidate genes, whereas analysis of the seven SPINT2 exons and splice sites identified a homozygous c.593-1G→A mutation affecting a canonical splice site in the index case (Figure 1A). A faint amount of an aberrant fragment of SPINT2 RNA from leukocytes of this patient, compared with controls, was seen by RT-PCR. Sequencing this cDNA species revealed the activation of a cryptic splice site upstream in intron 6 and the abnormal inclusion of 72 nucleotides in the message (Figure 1C). This fragment was not amplified in cDNA from mutation carriers (Figure 1C), suggesting preferential transcription from the normal allele or, rather, nonsense-mediated mRNA decay of additional abnormal RNA species resulting from this splice-site mutation, thus leading to loss of normal gene expression.
A SPINT2-mutation analysis was subsequently performed in all available DNA samples from our series of CSD patients. The clinical baseline characteristics and results of mutation analysis of patients are summarized in Table 1. The same homozygous c.593-1G→A mutation was identified in an apparently unrelated CSD patient originating from the same rural area in Austria as the original family. Four distinct germline SPINT2 mutations were detected in eight additional families (Table 1 and Supplemental Data). Each of these mutations segregated with the disease status in families, and none was present among 188 Austrian and 100 Swedish controls. Six apparently unrelated families of Swedish origin and an Amish patient (originating from Alsace-Lorraine) shared a c.488A→G (p.Y163C) mutation. Genotyping eight SNPs within and flanking SPINT2 in these families provided evidence for a common haplotype, suggesting a founder effect (data not shown). The mutated tyrosine 163 is highly conserved within the conserved catalytic Kunitz domain (Figure 2).
Figure 2.
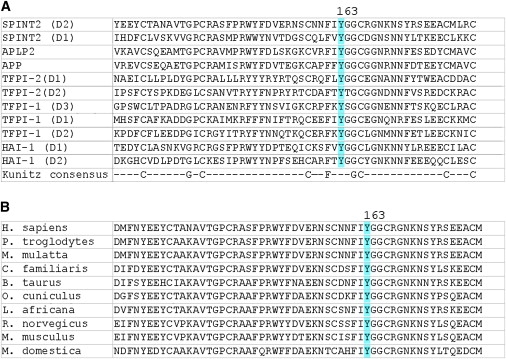
Conservation of the SPINT2 p.Y163C Mutation
(A) Alignment of multiple human Kunitz domains from different proteins. The mutated tyrosine is invariantly conserved within the highly conserved catalytic Kunitz domain.
(B) Alignment of known and predicted SPINT2 orthologs shows high evolutionary conservation of this mutated tyrosine residue.
SPINT2 mutations were detected in ten of 17 families (group 1). Neither mutations nor copy-number changes (Supplemental Data) were present in the remaining seven families (group 2), leading us to establish a genotype-phenotype correlation: We noted that malformations, dysmorphic features, or corneal erosions were recorded in 14 of 16 patients from families with SPINT2 mutations (Figure 1D); i.e., a syndromic form of CSD was present. In contrast, malformations or corneal disease were not recorded in any of eight CSD patients from group 2, i.e., indicating classic CSD. Nine of 16 patients from group 1 have had cyclically recurrent, painful punctate epithelial corneal lesions. Local diclofenac treatment once a day prolonged relapse intervals. Peripheral neovascularization of the corneas, as well as hyperopia and astigmatism, were found in two patients. One of these patients has a reduced visual acuity of 0.5 in both eyes at the age of 12 years.
A history of polyhydramnios is a frequent finding in both CSD groups. Patients from both groups were dependent on total parenteral nutrition in the first years of life and frequently died from related complications. However, survivors who could be partially or completely weaned from parenteral nutrition were observed in both groups (Table 1).
To investigate the functional consequences of the SPINT2 missense alterations, as well as the c.593-1G→A mutation, we expressed recombinant WT and mutant proteins in COS-7 cells through transient transfection. Immunoblotting of cell extracts showed two bands matching the predicted molecular weight of SPINT2, with and without cleaved signal peptide for WT and p.Y163C mutation. No immunoreactivity was visible in SPINT2-overexpressing cells encoding the start-codon mutation (c.1A→T) or the intronic variant c.593-1G→A (Figures 3A and 3B). No immunoreactivity for the SPINT2 fusion protein could be detected in concentrated media from SPINT2-overexpressing cells, suggesting that SPINT2 is retained intracellularly in vitro (data not shown). Compared with WT, Cys-163-mutated SPINT2 caused a significant decrease (p < 0.05) in the ability to inhibit trypsin, a prototype serine protease (Figure 3C).
Figure 3.
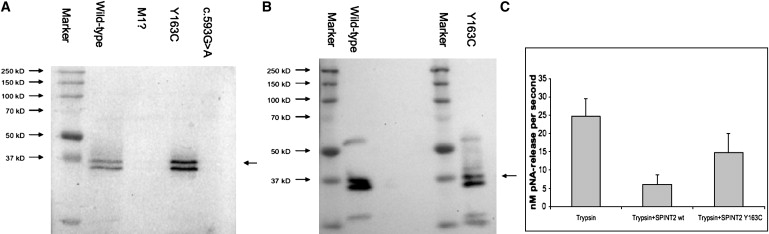
Expression and Trypsin-Inhibition Analysis of WT and Mutated SPINT2 Proteins
(A) Expression of SPINT2 WT and c.1A→T-, p.Y163C-, and c.593-1G→A-mutated proteins in COS-7 cells.
(B) Purification of SPINT2 WT and Cys-163-mutated protein showed no quantitative or qualitative differences.
(C) Trypsin activity was significantly less inhibited in COS-7 cells transfected with the Y163C SPINT2 mutant as compared with WT protein.
Discussion
Here, we present clinical and genetic evidence to delineate a syndromic form of CSD distinct from the originally described, classic-CSD type.1,2 Choanal atresia, hypertelorism, and corneal erosions are particularly frequent in syndromic CSD, and anal atresia occurred in two of 16 patients. Although both types of the disease can be fatal in the neonatal period and in the first years of life as a result of dehydration, severe electrolyte imbalances, and long-term complications of parenteral nutrition, we present mounting evidence that patients from both groups can eventually adapt to partial or complete enteral nutrition during childhood. A successful outcome appears to be independent of the disease-causing SPINT2 genotype, as reflected by our identification of six syndromic patients with a SPINT2 c.593-1G→A mutation. Of these patients, five died before their 13th month of life, from complications of parenteral nutrition, and a sixth patient could be weaned from parenteral nutrition during her 3rd year of life. This was the only syndromic-CSD patient in whom at the age of 18 months oral sodium (2.5 mmol/kg/d) and potassium citrate (1.0 mmol/kg/d) supplementation was introduced. A similar oral therapy regimen was considered beneficial in patients with classic CSD.1,2 Early diagnosis and adequate treatment might therefore confer a favorable long-term prognosis in syndromic and classic CSD. Alternatively, either adverse or beneficial effects of genetic modifiers might ultimately determine the outcome of these diseases.
The positional candidate approach led to the identification of a total of five distinct splicing and missense mutations in homozygous or compound-heterozygous state in SPINT2, encoding a Kunitz-type serine-protease inhibitor, in syndromic-CSD families, but not in classic-CSD families. SPINT2, also known as placental bikunin and hepatocyte growth factor activator inhibitor type 2 (HAI-2), has been shown to be a potent inhibitor of a number of serine proteases, such as pancreatic trypsin, plasmin, kallikrein,11 and hepatocyte growth factor activator,12 in vitro. Serine proteases carry out several physiological and cellular functions, ranging from degradation and digestive processes to protein processing and tissue remodeling.13 Loss of SPINT2 protein expression or its function would therefore be expected to increase the activity of target serine proteases. Indeed, we showed that three SPINT2 mutations cause either a decrease in the ability to inhibit trypsin or undetectable amounts of SPINT2 protein in vitro. We also found that SPINT2 proteins were not secreted in vitro, consistent with the prediction of a transmembrane domain (residues 198–221). This suggests either insertion of SPINT2 into the plasma membrane, with the Kunitz domains localizing extracellularly, or an intracellular role of the protein. Together, our data show that syndromic CSD is caused by SPINT2 loss of function. The physiological target of SPINT2 and the molecular pathology caused by SPINT2 deficiency are currently unknown.
CSD is a secretory diarrhea characterized by hyponatremia due to enormous fecal losses of large amounts of sodium, metabolic acidosis, and no evidence of primary structural abnormalities in the intestinal epithelium. Intestinal absorption of sodium occurs along the entire gut and is provided by a variety of transport mechanisms: co-transporters, i.e., proteins that manage the simultaneous absorption of Na+ with nutrients such as glucose14 or amino acids;15 coupled exchangers, such as the Na+/H+- and Cl−/HCO3-transport proteins, allowing electroneutral absorption of NaCl;16 and the amiloride-sensitive epithelial sodium channel (ENaC [MIM 600760]).17,18
In syndromic CSD, SPINT2 mutations can affect proteolytic activity in the functional regulation of intestinal epithelial absorption or secretion of sodium. Possible targets include the membrane-bound extracellular serine proteases, the channel-activating proteases (CAP)/prostasin [MIM 600823]19 and TMPRSS2 [MIM 602060],20 and the intracellular protease furin21 all of which have been shown to regulate ENaC activity through limited proteolysis. In other studies, however, the serine-protease inhibitor aprotinin19 and a recombinant, secreted form of SPINT222 blocked ENaC-mediated Na+ transport in vitro. A direct role of SPINT2 as an inhibitor of serine proteases involved in the activation of ENaC thus appears unlikely.
Alternatively, the serine protease dipeptidyl peptidase IV (DPPIV [MIM 126141]) has been suggested to affect the activity of NHE3 (sodium/hydrogen exchanger 3, SLC9A3 [MIM 182307]), which localizes to the apical epithelial plasma membrane and contributes largely to NaCl absorption.6,23 Lack of inhibition of DPPIV would be expected to lead to decreased NaCl absorption. Notably, NHE3-knockout mice recapitulate the intestinal human-CSD phenotype,24 and a defect of sodium/proton exchange as a cause of CSD was suggested in three patients with the classic type.1,4 We did not identify a SPINT2 mutation in one patient with defective NHE,1 compatible with the possibility that CSD can result from different mechanisms and consistent with genetic-locus heterogeneity.
SPINT2 might be the first serine-protease inhibitor to be involved in an intestinal disease. SPINT2 is apparently not being secreted, and its deficiency is not associated with overt histological changes of the intestine, contrasting with, e.g., deficiency of serine-protease inhibitors Kazal type 5 (SPINK5 [MIM 605010]) and type 1 (SPINK1 [MIM 167790]). SPINK5 deficiency causes severe abnormalities of cell-layer desquamation and inflammation in congenital, autosomal-recessive dermatosis associated with Netherton syndrome,25 and heterozygous and homozygous SPINK1 mutations are associated with chronic pancreatitis.26
The fact that complete loss of SPINT2 protein causes severe clefting of the embryonic ectoderm and early intrauterine death in mice27 supports the idea that either some residual SPINT2 activity is retained in patients or other proteins can compensate for loss of SPINT2 in humans. Notably, the presence of choanal and intestinal atresia implicates SPINT2 activity in human prenatal development. However, choanal and intestinal atresia might result from a secondary phenomenon after normal embryonal development, as a result of a disturbance of the optimal surface-liquid volume, which is maintained by the regulated transport of sodium and chloride across the airway epithelium.28 This situation might be similar to that of disturbed CFTR activity causing primary or secondary aplasia of the vas deferens prenatally.29
In conclusion, we have characterized syndromic CSD as a new entity, which differs from classic CSD by loss-of-function mutations in SPINT2 and the presence of congenital malformations and dysmorphological features or corneal erosions. The determination of the molecular basis of syndromic CSD and the recent identification of the genetic defects of the differential diagnoses, microvillus inclusion disease [MIM 251850]30 and tufting enteropathy,31 will facilitate rapid diagnosis of patients and contribute to the counseling of affected families. The consequences of alterations in SPINT2 function on the intestinal absorption of sodium and human development in syndromic CSD remain to be elucidated.
Supplemental Data
Supplemental Data include two figures and four tables and can be found with this paper online at http://www.ajhg.org/.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
Entrez Gene, http://www.ncbi.nlm.nih.gov/sites/entrez
MRC-Holland, http://www.mrc-holland.com/pages/support_desing_synthetic_probespag.html
Accession Numbers
The GenBank accession number for the SPINT2 mRNA sequence reported in this paper is NM_021102.2.
The GenBank accession number for the genomic SPINT2 sequence reported in this paper is NC_000019.8.
Acknowledgments
We thank Gabriele Rammesmayer and Silvia Lechner for technical assistance. This work was supported by grant no. 99 from Tiroler Medizinischer Forschungsfond to A.R.J.; grants P18470 to A.R.J. and P19579 to H.Z., from the Fonds zur Förderung der Wissenschaftlichen Forschung in Österreich (FWF); and a grant from Tiroler Wasserkraft AG (TIWAG) to T.M.
References
- 1.Booth I.W., Stange G., Murer H., Fenton T.R., Milla P.J. Defective jejunal brush-border Na+/H+ exchange: a cause of congenital secretory diarrhoea. Lancet. 1985;1:1066–1069. doi: 10.1016/s0140-6736(85)92369-4. [DOI] [PubMed] [Google Scholar]
- 2.Holmberg C., Perheentupa J. Congenital Na+ diarrhea: a new type of secretory diarrhea. J. Pediatr. 1985;106:56–61. doi: 10.1016/s0022-3476(85)80465-0. [DOI] [PubMed] [Google Scholar]
- 3.Muller T., Wijmenga C., Phillips A.D., Janecke A., Houwen R.H., Fischer H., Ellemunter H., Fruhwirth M., Offner F., Hofer S. Congenital sodium diarrhea is an autosomal recessive disorder of sodium/proton exchange but unrelated to known candidate genes. Gastroenterology. 2000;119:1506–1513. doi: 10.1053/gast.2000.20514. [DOI] [PubMed] [Google Scholar]
- 4.Keller K.M., Wirth S., Baumann W., Sule D., Booth I.W. Defective jejunal brush border membrane sodium/proton exchange in association with lethal familial protracted diarrhoea. Gut. 1990;31:1156–1158. doi: 10.1136/gut.31.10.1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fell J.M., Miller M.P., Finkel Y., Booth I.W. Congenital sodium diarrhea with a partial defect in jejunal brush border membrane sodium transport, normal rectal transport, and resolving diarrhea. J. Pediatr. Gastroenterol. Nutr. 1992;15:112–116. doi: 10.1097/00005176-199208000-00002. [DOI] [PubMed] [Google Scholar]
- 6.Zachos N.C., Tse M., Donowitz M. Molecular physiology of intestinal Na+/H+ exchange. Annu. Rev. Physiol. 2005;67:411–443. doi: 10.1146/annurev.physiol.67.031103.153004. [DOI] [PubMed] [Google Scholar]
- 7.Kennedy G.C., Matsuzaki H., Dong S., Liu W.M., Huang J., Liu G., Su X., Cao M., Chen W., Zhang J. Large-scale genotyping of complex DNA. Nat. Biotechnol. 2003;21:1233–1237. doi: 10.1038/nbt869. [DOI] [PubMed] [Google Scholar]
- 8.Janecke A.R., Thompson D.A., Utermann G., Becker C., Hubner C.A., Schmid E., McHenry C.L., Nair A.R., Ruschendorf F., Heckenlively J. Mutations in RDH12 encoding a photoreceptor cell retinol dehydrogenase cause childhood-onset severe retinal dystrophy. Nat. Genet. 2004;36:850–854. doi: 10.1038/ng1394. [DOI] [PubMed] [Google Scholar]
- 9.Gudbjartsson D.F., Thorvaldsson T., Kong A., Gunnarsson G., Ingolfsdottir A. Allegro version 2. Nat. Genet. 2005;37:1015–1016. doi: 10.1038/ng1005-1015. [DOI] [PubMed] [Google Scholar]
- 10.Laemmli U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 11.Marlor C.W., Delaria K.A., Davis G., Muller D.K., Greve J.M., Tamburini P.P. Identification and cloning of human placental bikunin, a novel serine protease inhibitor containing two Kunitz domains. J. Biol. Chem. 1997;272:12202–12208. doi: 10.1074/jbc.272.18.12202. [DOI] [PubMed] [Google Scholar]
- 12.Kawaguchi T., Qin L., Shimomura T., Kondo J., Matsumoto K., Denda K., Kitamura N. Purification and cloning of hepatocyte growth factor activator inhibitor type 2, a Kunitz-type serine protease inhibitor. J. Biol. Chem. 1997;272:27558–27564. doi: 10.1074/jbc.272.44.27558. [DOI] [PubMed] [Google Scholar]
- 13.Turk B. Targeting proteases: successes, failures and future prospects. Nat. Rev. Drug Discov. 2006;5:785–799. doi: 10.1038/nrd2092. [DOI] [PubMed] [Google Scholar]
- 14.Wright E.M., Loo D.D.F., Hirayama B.A., Turk E. Sugar absorption. In: Johnson L.R., Barrett K.E., Ghishan F.K., Merchant J.L., Said H.M., Wood J.D., editors. Physiology of the Gastrointestinal Tract. Elsevier Academic Press; Amsterdam, The Netherlands: 2006. pp. 1653–1665. [Google Scholar]
- 15.Ganapathy V., Gupta N., Martindale R.G. Protein digestion and absorption. In: Johnson L.R., Barrett K.E., Ghishan F.K., Merchant J.L., Said H.M., Wood J.D., editors. Physiology of the Gastrointestinal Tract. Elsevier Academic Press; Amsterdam, The Netherlands: 2006. pp. 1667–1692. [Google Scholar]
- 16.Kunzelmann K., Mall M. Electrolyte transport in the mammalian colon: mechanisms and implications for disease. Physiol. Rev. 2002;82:245–289. doi: 10.1152/physrev.00026.2001. [DOI] [PubMed] [Google Scholar]
- 17.Greig E.R., Mathialahan T., Boot-Handford R.P., Sandle G.I. Molecular and functional studies of electrogenic Na(+) transport in the distal colon and rectum of young and elderly subjects. Gut. 2003;52:1607–1615. doi: 10.1136/gut.52.11.1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rossier B.C. The epithelial sodium channel: activation by membrane-bound serine proteases. Proc. Am. Thorac. Soc. 2004;1:4–9. doi: 10.1513/pats.2306007. [DOI] [PubMed] [Google Scholar]
- 19.Planes C., Leyvraz C., Uchida T., Angelova M.A., Vuagniaux G., Hummler E., Matthay M., Clerici C., Rossier B. In vitro and in vivo regulation of transepithelial lung alveolar sodium transport by serine proteases. Am. J. Physiol. Lung Cell. Mol. Physiol. 2005;288:L1099–L1109. doi: 10.1152/ajplung.00332.2004. [DOI] [PubMed] [Google Scholar]
- 20.Donaldson S.H., Hirsh A., Li D.C., Holloway G., Chao J., Boucher R.C., Gabriel S.E. Regulation of the epithelial sodium channel by serine proteases in human airways. J. Biol. Chem. 2002;277:8338–8345. doi: 10.1074/jbc.M105044200. [DOI] [PubMed] [Google Scholar]
- 21.Sheng S., Carattino M.D., Bruns J.B., Hughey R.P., Kleyman T.R. Furin cleavage activates the epithelial Na+ channel by relieving Na+ self-inhibition. Am. J. Physiol. Renal Physiol. 2006;290:F1488–F1496. doi: 10.1152/ajprenal.00439.2005. [DOI] [PubMed] [Google Scholar]
- 22.Bridges R.J., Newton B.B., Pilewski J.M., Devor D.C., Poll C.T., Hall R.L. Na+ transport in normal and CF human bronchial epithelial cells is inhibited by BAY 39–9437. Am. J. Physiol. Lung Cell. Mol. Physiol. 2001;281:L16–L23. doi: 10.1152/ajplung.2001.281.1.L16. [DOI] [PubMed] [Google Scholar]
- 23.Girardi A.C., Degray B.C., Nagy T., Biemesderfer D., Aronson P.S. Association of Na(+)-H(+) exchanger isoform NHE3 and dipeptidyl peptidase IV in the renal proximal tubule. J. Biol. Chem. 2001;276:46671–46677. doi: 10.1074/jbc.M106897200. [DOI] [PubMed] [Google Scholar]
- 24.Schultheis P.J., Clarke L.L., Meneton P., Miller M.L., Soleimani M., Gawenis L.R., Riddle T.M., Duffy J.J., Doetschman T., Wang T. Renal and intestinal absorptive defects in mice lacking the NHE3 Na+/H+ exchanger. Nat. Genet. 1998;19:282–285. doi: 10.1038/969. [DOI] [PubMed] [Google Scholar]
- 25.Chavanas S., Bodemer C., Rochat A., Hamel-Teillac D., Ali M., Irvine A.D., Bonafe J.L., Wilkinson J., Taieb A., Barrandon Y. Mutations in SPINK5, encoding a serine protease inhibitor, cause Netherton syndrome. Nat. Genet. 2000;25:141–142. doi: 10.1038/75977. [DOI] [PubMed] [Google Scholar]
- 26.Witt H., Luck W., Hennies H.C., Classen M., Kage A., Lass U., Landt O., Becker M. Mutations in the gene encoding the serine protease inhibitor, Kazal type 1 are associated with chronic pancreatitis. Nat. Genet. 2000;25:213–216. doi: 10.1038/76088. [DOI] [PubMed] [Google Scholar]
- 27.Mitchell K.J., Pinson K.I., Kelly O.G., Brennan J., Zupicich J., Scherz P., Leighton P.A., Goodrich L.V., Lu X., Avery B.J. Functional analysis of secreted and transmembrane proteins critical to mouse development. Nat. Genet. 2001;28:241–249. doi: 10.1038/90074. [DOI] [PubMed] [Google Scholar]
- 28.Myerburg M.M., Butterworth M.B., McKenna E.E., Peters K.W., Frizzell R.A., Kleyman T.R., Pilewski J.M. Airway surface liquid volume regulates ENaC by altering the serine protease-protease inhibitor balance: a mechanism for sodium hyperabsorption in cystic fibrosis. J. Biol. Chem. 2006;281:27942–27949. doi: 10.1074/jbc.M606449200. [DOI] [PubMed] [Google Scholar]
- 29.Shin D., Gilbert F., Goldstein M., Schlegel P.N. Congenital absence of the vas deferens: incomplete penetrance of cystic fibrosis gene mutations. J. Urol. 1997;158:1794–1798. doi: 10.1016/s0022-5347(01)64131-4. [DOI] [PubMed] [Google Scholar]
- 30.Muller T., Hess M.W., Schiefermeier N., Pfaller K., Ebner H.L., Heinz-Erian P., Ponstingl H., Partsch J., Rollinghoff B., Kohler H. MYO5B mutations cause microvillus inclusion disease and disrupt epithelial cell polarity. Nat. Genet. 2008;40:1163–1165. doi: 10.1038/ng.225. [DOI] [PubMed] [Google Scholar]
- 31.Sivagnanam M., Mueller J.L., Lee H., Chen Z., Nelson S.F., Turner D., Zlotkin S.H., Pencharz P.B., Ngan B.Y., Libiger O. Identification of EpCAM as the gene for congenital tufting enteropathy. Gastroenterology. 2008;135:429–437. doi: 10.1053/j.gastro.2008.05.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.