

Abstract
CXCR2 is a G-protein-coupled receptor (GPCR) that binds the CXC chemokines, CXCL1–3 and CXCL5–8, and induces intra-cellular signals associated with chemotaxis. Many adaptor proteins are actively involved in the sequestration, internalization, and trafficking of CXCR2 and transduction of agonist-induced intracellular signaling. We have previously shown that adaptor protein β-arrestin-2 (βarr2) plays a crucial role in transducing signals mediated through CXCR2. To further investigate the role of βarr2 on CXCR2-mediated signaling during acute inflammation, zymosan-induced neutrophils were isolated from peritoneal cavities of βarr2-deficient (βarr2−/−) and their wild-type (βarr2+/+) littermate mice, and neutrophil CXCR2 signaling activities were determined by measurement of Ca2+ mobilization, receptor internalization, GTPase activity, and superoxide anion production. The results showed that the deletion of βarr2 resulted in increased Ca2+ mobilization, superoxide anion production, and GTPase activity in neutrophils, but decreased receptor internalization relative to wild-type mice. Two animal models, the dorsal air pouch model and the excisional wound healing model, were used to further study the in vivo effects of βarr2 on CXCR2-mediated neutrophil chemotaxis and on cutaneous wound healing. Surprisingly, the recruitment of neutrophils was increased in response to CXCL1 in the air pouch model and in the excisional wound beds of βarr2−/− mice. Wound re-epithelialization was also significantly faster in βarr2−/− mice than in βarr2+/+ mice. Taken together, the data indicate that βarr2 is a negative regulator for CXCR2 in vivo signaling.
Appropriate recruitment of neutrophils in tissues is essential for host defense in infection. This is exemplified by the fact that bacterial endotoxin-induced depression of neutrophil recruitment severely compromises host defense and exacerbates the clinical outcome of patients with septic shock (1– 4). Persistent accumulation of neutrophils in tissues is also an important provoking factor for many diseases including cardiac vascular disease, inflammatory bowel disease, chronic wounds, and lung injury (5–12). The trafficking of neutrophils after extravasation from the lumen of microvessels into peripheral tissues is based on the ability of cells to move toward the gradient of certain chemoattractants; this process is termed chemotaxis. A wealth of substances have been identified as chemotactic factors capable of initiating the chemotaxis of various cell types. Among these molecules, the bacterial-derived N-formylated peptides (fMLP), C5a, and the family of chemotactic cytokines or chemokines, which includes CXCL1–3 and CXCL5–8, have been identified as potent chemoattractants for neutrophils both in vitro and in vivo (13, 14).
Chemokines are small proteins that bind seven transmembrane G protein-coupled receptors (GPCRs),4 regulate leukocyte trafficking, and are highly important in the pathogenesis of inflammatory diseases. Based on the arrangement of the first two cysteine residues in the N terminus, chemokines can be classified into CC, CXC, CX3C, and C subfamilies (15). Chemokine receptors couple predominantly to pertussis toxin-sensitive Gαi proteins to activate phospholipase C, resulting in the generation of the intracellular messengers diacylglycerol and inositol 1,4,5-triphosphate. Whereas inositol 1,4,5-triphosphate mobilizes Ca2+, diacylglycerol in combination with elevated Ca2+ activates protein kinase C (16, 17). A series of responses in GPCR internalization and recycling are crucial for receptor signaling, and various groups of GPCRs are regulated differently during these processes.
Despite being extensively studied, the mechanisms underlying the signaling and trafficking of chemokine receptors are still not fully understood. The complexity of this issue is evident as more and more adaptor/scaffold proteins are being found to associate with GPCR endocytosis, recycling, signaling, and cell chemotaxis. Arrestin, a versatile family of adaptor proteins, forms complexes with most GPCRs after agonist binding and phosphorylation of receptors by G protein-coupled receptor kinases. These complexes are important for receptor desensitization as well as downstream signaling (18). Two members of the arrestin family, β-arrestin-1 (βarr1) and β-arrestin-2 (βarr2), which share a high degree of homology, bind to the CXC chemokine receptors CXCR1 and CXCR2 and regulate cellular responses to these receptors (19, 20). We have previously shown that βarr2 plays a crucial role in transducing signals of the CXCL8 receptors CXCR1 and CXCR2 (20). Clathrin-mediated CXCR2 endocytosis, followed by interaction with the Rab11-family interacting protein-2 and myosin Vb were also shown to regulate receptor recycling (21, 22). We have also shown that different domains of the C terminus of CXCR2 interact with protein phosphatase 2A, adaptin 2 (AP-2), and the heat shock cognate/heat shock protein 70-interacting protein to promote receptor sequestration, internalization, and trafficking (23–25). These processes likely play important roles in CXCR2-mediated downstream signaling.
This study was designed to further investigate the in vivo role of βarr2 in CXCR2 signaling and CXCR2-mediated neutrophil recruitment into inflamed sites. We compared neutrophil activation and chemotaxis in two mouse models of inflammation on βarr2+/+ and βarr2−/− mice: a dorsal air pouch and an excisional punch wound. The data show that βarr2 deficiency leads to enhanced CXCR2 signaling which is exemplified by a significant increase in neutrophil activation and superoxide production as well as neutrophil mobility within two differing in vivo models. The data also suggest that βarr2 deficiency promotes re-epithelization in the mouse excisional wound healing model.
Materials and Methods
Animals
All experiments were approved by and confirmed to the guidelines of the Animal Care Committee of Meharry Medical College, Nashville, TN. Animals were housed five per cage in a room at 22 ± 5°C with an alternate 12-h light-dark cycle. βarr2−/− mice (C57BL/6 background) were kindly provided by Dr. Robert J. Lefkowitz (Howard Hughes Medical Institute, Duke University Medical Center, Durham, NC). Male and female mice were evaluated, and controls were age- and sex-matched littermates.
Genotyping
All mice were genotyped at the age of 3 wk; DNA samples were prepared from the tail tips with DNeasy tissue kit from Qiagen and subjected to triplex PCR as described (26 –28).
Murine skin air pouch model of inflammation
Mice (6 – 8 wk) were anesthetized with isofluorane, and dorsal air pouches were raised by injecting 3 ml of sterile air s.c. on days 0 and 3 as described previously (29 –31). On day 6 and while the mice were anesthetized with isofluorane, inflammation in the air pouch was induced by local injection of recombinant murine CXCL1 (100 pmol) dissolved in 0.5 ml of sterile PBS. Mice were sacrificed 4 h after CXCL1 injection by CO2 asphyxiation, and air pouches were lavaged three times with 3 ml of PBS. Cells were collected by centrifugation at 2000 rpm for 5 min at room temperature. The supernatants were removed, and the cells were resuspended in 10 ml of PBS and counted. Aliquots of the cell suspension were stained with Wright-Giemsa and enumerated by light microscopy (~90% of the exudates are neutrophils).
Peritoneal recruitment of leukocytes
Zymosan was prepared in PBS to a final concentration of 1 mg/ml, and 1.0 ml was injected into the peritoneum of control and βarr2−/− mice. Mice were euthanized by CO2 asphyxiation, and the peritoneal cavity was lavaged at 4 h postinjection with 8 ml of ice-cold RPMI containing 2% FBS and 2 mM EDTA. Cells were collected by centrifugation, counted, and stained with Diff-Quick to assess the percentage of neutrophils.
Intracellular Ca2+ mobilization, receptor internalization, and GTPase activity
Zymosan-elicited peritoneal neutrophils (3 × 106 cells) were washed with HEPES-buffered saline and loaded with 1 μM Indo-1-acetoxymethyl ester for 30 min at room temperature. The cells were washed and resuspended in 1.5 ml of HEPES-buffered saline, and intracellular Ca2+ mobilization was measured as described (32). For receptor internalization, 5 × 105 neutrophils were resuspended in 250 μl of HBSS containing 25 mM HEPES and 0.1% BSA. Cells were treated with recombinant murine CXCL1 (100 nM) at 37°C for different time periods. Reactions were stopped by adding 1 ml of ice-cold HBSS followed by centrifugation at 1000 rpm for 2 min. Then cells were washed three times and assayed for 125I-labeled CXCL1 binding as previously described (33). For GTPase activity, cells were isolated, pooled, and frozen at −80°C until use. Membranes were prepared and assayed for GTPase activity (10 μg of membrane preparations per assay) as described (32, 34) in the presence and absence of 1 μM recombinant murine CXCL1.
Superoxide anion production
The production of superoxide anion (O2−) was measured as described previously (35). Briefly, murine peritoneal neutrophils were resuspended in physiological buffer containing 130 mM NaCl, 4.6 mM KCl, 1.1 mM KH2PO4, 1 mM CaCl2, 5 mM glucose, and 20 mM HEPES (pH 7.4) at 1 × 107 cells/ml. Cells were seeded in 96-well plates, and then 25 μl of cytochrome c solution (type VI, 10 mg/ml) were added to the cells in the presence or absence of 20 μl of superoxide dismutase (1 mg/ml). After incubation at 37°C for 5 min, cells were stimulated with 100 nM CXCL1, RANTES, or PMA. After incubation for 60 min, the optical density of reduced cytochrome c in the supernatant was measured in a microplate reader (Bio-Rad) at 550 nm. Superoxide anion production was calculated as nanomoles of cytochrome c reduced per 106 cells with an extinction coefficient of E550 = 21 nM−1 cm−1.
Mouse skin excisional wounding procedures
Excisional punch wounds were made as described previously with slight modifications (36). Mice (6 – 8 wk of age and weighing 22–25 g) were anesthetized with an i.p. injection of ketamine (100 mg/kg body weight) and xylazine (10 mg/kg body weight) (Fort Dodge Animal Health). The dorsal surface of the mouse was cleaned, shaved, and sterilized with betadine solution (Purdue Frederick) and 70% ethanol. Two full-thickness excisional wounds were made in the dorsal paravertebral region with a 4-mm-diameter punch (Acu-Punch). Wounds were covered and kept under occlusion by Spray Bandage (Curad; Beiersdorf). Analgesics (Buprenex, Reckitt & Colman Pharmaceuticals) were administered i.p. with at a dosage of 4.5 μg/kg body weight. All wounds were visually monitored daily for signs of infection.
Immunohistochemical assessment of neutrophil infiltration in wound tissues
Mice were sacrificed, and the wound beds with adjacent margin skin tissues were harvested at postwounding days 1, 2, and 3. The tissues were fixed in 4% paraformaldehyde (Fisher) for 24 h and embedded in paraffin blocks. Tissue sections 6-μm thick were cut through the center of wounds, placed on charged slides, and baked overnight at 50°C. After a deparaffinization procedure, sections were rehydrated in TBS-Tween 20 for 5 min before immunostaining. Endogenous peroxidase was diminished by treatment with 0.03% hydrogen peroxide and samples were treated with diluted rabbit serum before the addition of primary Ab. Granulocytes expressing Ly-6G surface markers were immunostained with anti-mouse Ly-6G mAb (BD Pharmingen) diluted 1/50 for 60 min. Sections without primary Ab served as negative controls. The Vectastain ABC Elite (Vector Laboratories) system and NovaRed chromagen (Vector Laboratories) were used to produce localized, visible staining. Slides were lightly counterstained with Mayer’s hematoxylin, dehydrated, and coverslipped for observation under the microscope. For each type of either wild-type or βarr2−/− mouse, four wound samples from 2 mice were excised and analyzed for each time point after wounding. The extent of neutrophil infiltration into the wound tissues was evaluated as previously described (37, 38) on tissue slide images using Image Pro Plus software.
Myeloperoxidase (MPO) assay
MPO activity was assayed as described previously (36, 39). Briefly, wound tissues from postwounding days 1, 2, and 3 were harvested, snap frozen in liquid nitrogen, and homogenized in 1 ml of potassium phosphate buffer (50 mM, pH 6.0) containing 0.5% hexadecyltrimethylammonium bromide. Hexadecyltrimethylammonium bromide is a detergent that releases MPO from primary granules of neutrophils. After sonication on ice for 15 s (Sonic Model 300; Fisher Scientific), tissue homogenates were centrifuged at 20,000 × g for 20 min at 4°C. The supernatants were collected, and their protein concentrations were measured using the Bio-Rad protein determination assay. Aliquots of the supernatant containing equivalent amounts of 50 μg of protein were mixed with 500 μl of potassium phosphate buffer (50 mM, pH 6.0) containing 0.2% o-dianisidine hydrochloride (Sigma-Aldrich) and 0.0005% H2O2. The reaction was initiated by the addition of hydrogen peroxide. The change in OD490 during a 2-min reaction period was measured spectrophotometrically, using the Beckman-DU 7000 (Beckman Coulter). The optical densities were compared for each aliquot of wound tissue lysate from both wild-type and βarr2−/− mice. The MPO values of non-wounded skin tissues from each genotype served as controls. Wounds were collected on postwounding days 3, 5, 7, and 10. Each wound was centrally bisected, embedded in paraffin, and sectioned at 6 μm, followed by staining with Gomori’s trichrome. For either wild-type or βarr2−/− mice, eight wound tissues from four mice were excised and analyzed for each time point. The percentage of wound resurfacing was assessed by morphometric analysis (37, 38) on tissue slide images using Image Pro Plus software.
Statistical analyses
Results are expressed as mean ± SEM. Statistical differences between groups were determined by Student’s two-tailed paired t test, and the statistical difference was claimed for p < 0.05.
Results
Role of βarr2 in CXCR2-mediated signal transduction
Zymosan-elicited peritoneal neutrophils from βarr2−/− and βarr2+/+ animals were analyzed for CXCR2-mediated intracellular Ca2+ mobilization. As shown in Fig. 1A, CXCL1 induced Ca2+ mobilization in neutrophils from both βarr2−/− and βarr2+/+ mice. However, neutrophils from βarr2−/− animals showed a more sustained response to CXCL1 than cells from βarr2+/+ mice. CXCL1-induced G protein activation was also greater in membranes from βarr2−/− neutrophils than those from βarr2+/+ neutrophils as assessed by GTPase activity (Fig. 1B). βarr2 deletion also caused a marked decrease in CXCL1-mediated CXCR2 internalization in βarr2−/− mice relative to that of βarr2+/+ mice (Fig. 1C).
FIGURE 1.

Characterization of CXCR2 in βarr2−/− mice. A, For intracellular Ca2+ mobilization, zymosan-elicited peritoneal neutrophils (3 × 106 cells) were Indo-1-acetoxymethyl ester loaded and stimulated with 10 nM murine CXCL1 (MIP-2). Each tracing represents an analysis from a single mouse with the indicated βarr2 genotype, and the data shown are representative of at least five each of βarr2+/+ and βarr2−/− animals. B, For GTPase activity, membranes were prepared from neutrophils and assayed for time-dependent CXCL1-stimulated inorganic phosphate (Pi) released. Results shown are representative of one of two experiments performed in triplicate. C, For internalization, neutrophils (0.5 × 106 cells/assay) were treated with murine CXCL1 (100 nM) at different times, washed, and assayed for 125I-labeled CXCL1 binding. The values are presented as percentage of total which is defined as the total amount of 125I-labeled CXCL1 bound to control (untreated) cells. The experiment was repeated three times with similar results.
To further assess the role of βarr2 in CXCR2 signaling, we determined the ability of CXCL1 to mediate superoxide production in neutrophils from βarr2−/− and βarr2+/+ mice by measuring the optical density of superoxide dismutase-inhibitable reduction of cytochrome c (35). As shown in Fig. 2, CXCL1 induced a 68.5-fold increase in H2O2 production in neutrophils from βarr2−/− mice compared with neutrophils from βarr2+/+ mice (1.78 ± 0.22 vs 0.026 ± 0.021-fold over basal for βarr2−/− and βarr2+/+, respectively). Superoxide production in response to RANTES (CCL5 chemokine which activates CCR5) and PMA (which is independent of receptor activation) were also included as controls. CCL5 induced a greater response in βarr2−/− mice (4.17 ± 0.2-fold over basal) relative to βarr2+/+ (0.9 ± 0.05-fold over basal). In contrast, PMA-mediated superoxide production was similar in both βarr2−/− (29.10 ± 1.42-fold over basal value) and βarr2+/+ (26.4 ± 0.57-fold over basal value) neutrophils.
FIGURE 2.

CXCL1-induced superoxide production in neutrophils from βarr2 deficient and wild-type mice. Zymosan-elicited peritoneal neutrophils (106 cells) from βarr2 deficient (βarr2−/−) and wild-type (βarr2+/+) mice were incubated in cytochrome c solution with or without superoxide dismutase and stimulated with 100 nM murine CXCL1, CCL5, or PMA for 60 min. Superoxide production was determined by monitoring the optical density of the reduced cytochrome c in the supernatant at 550 nm as described in Materials and Methods. Data are the averaged analysis from two animals (four wounds) with the indicated βarr2 genotype.
Role of βarr2 in CXCR2-mediated neutrophil migration in air pouch model
To determine the in vivo role of βarr2 in CXCR2-mediated neutrophil migration, air pouches were created in βarr2−/− mice and their littermates (βarr2+/+). PBS-induced neutrophil recruitment (~1.5 ×106 cells) was similar for mice of either genotype (Fig. 3). By contrast, injection of CXCL1 (100 pM) into the air pouch 6 days after it was raised induced greater neutrophil accumulation (8.26 ± 2.72 × 106 cells) in βarr2−/− mice than in control βarr2+/+ animals (4.04 ± 0.86 ± 106 cells; Fig. 3).
FIGURE 3.
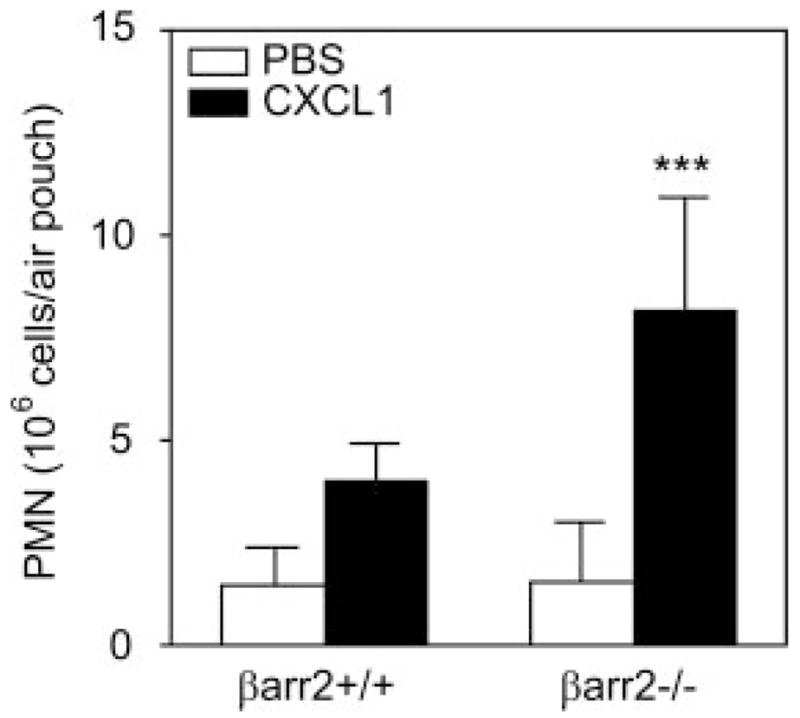
CXCL1-induced leukocyte migration into murine air pouch in βarr2−/− and wild-type control mice. Six-day air pouches were raised in the dorsum of 6- to 8-wk-old βarr2−/− mice and their littermates (βarr2+/+) as described in Materials and Methods. Mice were injected with 0.5 ml of PBS or PBS containing murine CXCL1 (100 pmol). Exudates were collected after 4 h, and the total number of leukocytes (~90% neutrophils (PMN)) was enumerated. Data represent means ± SD (n = 8). ***, p < 0.0001.
Determination of neutrophil infiltration of wound tissues
In addition to the demonstration of differential leukocyte recruitment into chemokine concentrated sites in the air pouch model, a second in vivo mouse model was included. Neutrophil chemotaxis was evaluated in the mouse skin excisional wound healing model. In both wild-type and βarr2−/− mice, the infiltration of Ly-6G-positive neutrophils was prominent within the wound bed as early as postwounding day 1 and during the early phase after wounding (Fig. 4, A and B), then decreased steadily through day 10 (Fig. 4, C and D). With higher magnification, a dense accumulation of infiltrating neutrophils in βarr2−/− wound tissue was evident at the interface between the newly formed wound scab and its underlying s.c. tissue, whereas Ly-6G-positive cells scattered in wound granulation tissue (Fig. 4, E and F). By postwounding days 2 and 3, increased numbers of neutrophils had accumulated at the scab-granulation tissue interface (Fig. 4E, inset b), and Ly-6G-positive cells showed a sporadic distribution in the areas immediately adjacent to the wound bed (Fig. 4E, inset a). The leading epidermal tips advanced from wound edges toward the center of the wound through the Ly-6G-positive neutrophil-dominant zone at the scab-granulation tissue interface (Fig. 4E, inset b). This immunohistological observation indicated that the neutrophil chemotaxis in βarr2−/− mice followed the identical spatial and temporal pattern observed in the excisional wound bed of the wild-type mice (Fig. 4F, insets a and b).
FIGURE 4.

Histological observation of excisional wounds in βarr2−/− (A, C, and E) and βarr2+/+ mice (B, D, and F). A and B, Day 3 excisional injuries show a wound bed filled with a provisional matrix and epithelial tips that are advancing down the sides of the crater while massive inflammatory infiltrates containing polymorphonuclear neutrophils have streamed into the defect (trichrome staining; size bars, 1100 μm). C and D, Day 10 wounds are 100% resurfaced, but the βarr2−/− mouse reveals an epidermis that is greatly thickened by comparison. βarr2−/− neodermis is also thickened and shows diminished cellularity that typically accompanies the resolution of the granulation tissue (trichrome staining; size bars, 50 μm). E and F, Day 2 wound edges are immunostained for neutrophils using Ly-6G antisera. Epithelial cells have begun to migrate down the side of the deficit. (size bars, 250 μm). Inset Box a shows the density of Ly-6G immunopositive cells at the dermal-s.c. interface within the adjacent tissue (size bars, 80 μm). Inset Box b shows the extreme density of Ly-6G-immunoreactive cells that have amassed at the interface between the scab and the provisional matrix of the granulation tissue nearer the central region of the excisional wound (size bar, 15 μm).*, Wound edges; arrowheads, advancing epithelial tips.
Neutrophil infiltration was further quantitatively determined by MPO assays performed on excisional wound tissue extracts. The data showed that the MPO activity of wild-type mouse wounds increased sharply by 24 h postwounding and was sustained over postwounding day 3 (Fig. 5). The MPO activity at 24 h after wounding (postwounding day 1) in βarr2−/− mice was equivalent to that of the wild-type mice. However, by postwounding day 2, the MPO activity in wounds from the βarr2−/− mice was ~1-fold higher than that of wounds from wild-type mice (p < 0.01). This was followed by a marked decrease in MPO activity to the level exhibited in wounds from wild-type mice at postwounding day 3.
FIGURE 5.
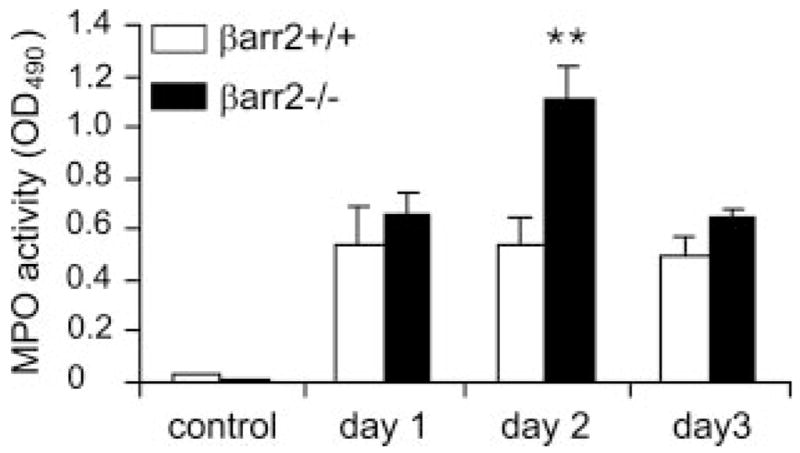
MPO activity assay from wound extracts. MPO activity within each wound bed was determined spectrophotometrically as described in Materials and Methods. Mean ± SEM of four wounds for each time point of each mouse genotype is shown. Statistical difference is evaluated by Student’s t test. **, p < 0.01.
Increased wound re-epithelialization in βarr2−/− mice
The ability of wild-type and βarr2−/− mice to close the wound was assessed histologically from trichrome-stained sections by comparing the percentage of wound resurfacing at different time points (Fig. 4, A and B). As shown in Fig. 6, the percentages of epidermal resurfacing for wild-type mouse wounds at days 3, 5, and 7 were 32, 38, and 72%, respectively, whereas wound resurfacing percentages from βarr2−/− mice were significantly increased to 40, 61, and 89% at days 3, 5, and 7, respectively (p < 0.05 for comparison between βarr2−/− and βarr2+/+ mouse wounds of days 3 and 7, and p < 0.01 for comparison of wounds of day 5). Wounds from both genotypes were each completely re-epithelialized by postwounding day 10 (Fig. 4, C and D). Wound contraction rates (defined histologically as the distance between wound edges at different time points) showed no significant difference between the two mouse groups (data not shown).
FIGURE 6.

Comparison of wound re-epithelialization rates. The percentages of wound resurfacing at postwound days 3, 5, and 7 were 32, 38, and 72% in βarr2+/+ mice, whereas that in βarr2−/− mice were 40, 61, and 89%, respectively. The value of each time point was obtained from eight wounds and is expressed as mean ± SEM. *, p < 0.05; **, p < 0.01 by Student’s t test.
Discussion
β-Arrestins play a pivotal role in the termination of GPCR-mediated signaling by binding to the phosphorylated form of the receptor, thereby uncoupling the receptor from its trimeric G protein. β-Arrestins also act as scaffold proteins by forming complexes with other proteins, which upon receptor activation and/or internalization activate downstream effectors (40 – 42). Previous studies from our laboratories and others using transfected cell lines expressing wild-type and mutant receptors have shown that CXCR2 undergoes internalization via two mechanisms: one is receptor phosphorylation/arrestin-dependent; and the other is receptor phosphorylation/arrestin independent. The later mechanism seems to be mediated by adaptor proteins such as AP-2 and heat shock cognate/heat shock protein 70 interacting protein and requires the C-terminal leucine-rich motifs of the receptors (LLKIL and KILAIH GLI) (23, 24). In this study, we sought to determine the precise role of βarr2 in CXCR2-mediated leukocyte functions using a βarr2−/− mouse model (28). The data herein demonstrated that βarr2 negatively modulates CXCR2-mediated neutrophil activation and motility. First, upon activation by CXCL1, zymosan-elicited peritoneal neutrophils from βarr2−/− displayed greater G protein activation, sustained intracellular Ca2+ mobilization, and robust superoxide production compared with βarr2+/+ animals (Figs. 1 and 2). Second, βarr2−/− mice showed a 2-fold increase in CXCL1-mediated neutrophil accumulation in the dorsal air pouch relative to wild-type animals (Fig. 3). Third, βarr2−/− mice showed greater MPO activity at the site of the wounds (Fig. 5).
An important question addressed in this study deals with the in vivo role of receptor internalization in CXCR2-mediated leukocyte motility. Previous studies from our laboratory have shown that agonist-induced receptor internalization is necessary for CXCR2-mediated cell migration. First, in HEK293 cells expressing CXCR2, overexpression of a dominant negative mutant of dynamin blocked receptor internalization and inhibited CXCL1- and CXCL8-mediated chemotaxis (21). Second, a carboxyl tail-deleted mutant of CXCR2, 331T, resistant to receptor phosphorylation, displayed decreased chemotaxis in RBL-2H3 cell lines, relative to wild-type CXCR2 (20). In RBL-2H3 cells, the decreased chemotaxis was correlated with an increase in phosphoinositide hydrolysis and intracellular Ca2+ mobilization, suggesting that second messenger production rather than internalization plays a feedback regulatory role in chemotaxis (20, 43). The in vivo data, however, indicate that βarr2 is likely involved in CXCR2-mediated neutrophil chemotaxis in a pathway that is independent of its role in receptor internalization. βarr2−/− neutrophils showed a ~50% decrease in CXCL1-mediated CXCR2 internalization but a significant increase in CXCL1-induced neutrophil accumulation in the air pouch relative to wild-type animals (Figs. 1 and 3). Furthermore, in cutaneous excisional wounds, the arrestin status had an impact on two disparate aspects of wound repair. MPO activity in the wound bed as well as re-epithelialization were significantly increased in βarr2−/− mice as compared with βarr2+/+ mice (Figs. 5 and 6).
Lymphocytes from βarr2−/− mice showed an increase in CXCR4-mediated G protein activation but a decreased chemotaxis in response to CXCL12, relative to βarr2+/+ (28). These data likely indicate that βarr2 uses different cellular pathways to mediate migration of lymphocytes, and these may be different in 1) CXCR4 vs CXCR2-mediated responses, 2) cell types, and 3) various assays for chemotaxis. Supporting this contention is the observation that inhibition of βarr2 expression in HEK293 decreased CXCR4 mediated chemotaxis and p38MAPK activation, but not CXCR2-mediated chemotaxis (42, 44). Furthermore, in mouse embryonic fibroblast (MEF) deficient in both βarr1 and βarr2 (βarr1/2−/−), CXCR4-mediated p38MAPK, JNK, and ERK1/2 phosphorylation were significantly attenuated, whereas CXCR2 activation produced a robust increase in MAPK activation. Interestingly, CXCR4 internalization was totally inhibited in MEF βarr1/2−/− cells, whereas CXCR2 internalization was simply delayed relative to control MEF (βarr1/2+/+). These data further suggest that CXCR2 internalization can occur, at least in part, independent of βarr1/2.
An interesting observation is that the in vivo migration of βarr2−/− neutrophils is increased relative to that of neutrophils from wild-type animals. These results contrasted with our previous in vitro studies using stably transfected RBL-2H3 cells where cells expressing CXCR2 mutant receptors deficient in internalization showed decreased chemotaxis in response to CXCL8 (20). These data likely indicate that the in vivo environment in the air pouch and the cutaneous wound healing models are different from those of the in vitro Transwell assay. Supporting that contention is the observation that zymosan-elicited peritoneal neutrophils from βarr2−/− mice displayed reduced chemotaxis in vitro relative to βarr2+/+ animals (data not shown).
The negative effect of arrestin in CXCR2 chemotaxis could be explained in several ways. First, the two mechanisms of internalization are additive, causing the rapid internalization of the receptor upon agonist activation (~95% in the first 2–5 min) relative to CXCR4 or CXCR1 (~50% after 30 – 60 min) (20). This rapid internalization diminishes signal length and, as a consequence, MAP kinase activation and chemotaxis. Inhibition of either pathway delays receptor internalization, resulting in increased intracellular signal activation in response to ligand binding and thus cell migration. Second, it is possible that only the AP-2-dependent mechanism for receptor internalization and signal amplification is required for chemotaxis. Inhibition of the arrestin-dependent pathway may promote AP-2-mediated CXCR2 internalization and signal amplification and thus chemotaxis. Third, leukocyte migration in vivo is a complex mechanism, which requires the interaction of the cells with adaptor molecules such as E-selectin for complete arrest, preceding transendothelial migration (45). CXCR2 was recently shown to mediate leukocyte arrest under conditions of vascular flow independent of E-selectin (46). Thus, the absence of β-arrestins, which increases receptor activity, may increase the ability of CXCR2 to mediate neutrophil arrest and chemotaxis in vivo. A fourth explanation could be that the increase in receptor activity results in greater secretion of proteases and tissue permeability thereby facilitating cell migration.
Excisional wounding breaches the skin and represents a formidable challenge to the animal. The result is typically an immediate and intense inflammatory response. We have previously shown that deletion of the gene encoding CXCR2 or inhibition of CXCR2 function significantly delays the recruitment of neutrophils, decreases proliferation of keratinocytes, and delays wound closure in the mouse excisional wound healing model (36, 39). In the present study, deficiency in βarr2 results in enhanced activation of in vivo murine CXCR2 signaling, as illustrated by higher neutrophil infiltration into wounds. This observation is consistent with the increased CXCL1-induced neutrophil chemotaxis in the air pouch model. Altogether the enhanced wound repair in the βarr2−/− mice may result from the combined effects of increased CXCR2-mediated keratinocyte motility, enhanced neutrophil infiltration, as well as effects of other cell types in the wound bed. With regard to the time course of MPO data, MPO activity usually peaks around 24 h after wounding and declines thereafter. We observed that the MPO value was not significantly higher in wounds from βarr2−/− mice as compared with wounds from wild-type mice at day 1. However, in wounds from βarr2−/− mice, MPO activity peaked at a significantly higher level on post wound day 2 than for those from wild-type mice on either day 1 or day 2. We cannot be certain why the MPO activity remains high in βarr2−/− mice on post-wounding day 2. There are several possible explanations: 1) neutrophils that migrate into the wound bed in βarr2−/− mice remain viable for a longer period of time, resulting in higher levels of MPO activity in the wound bed on postwound day 2; 2) neutrophils of βarr2−/− mice are slower to degranulate in vivo and therefore retain higher levels of measurable MPO activity in the wound bed on postwound day 2; 3) neutrophils from βarr2−/− mice exhibit higher levels of MPO activity than those from wild-type mice; or 4) neutrophils from βarr2−/− mice continue to respond to the chemokine gradient for a longer period of time, peaking at day 2. We favor possibility 4, based on data from the air pouch experiments described here. Future studies will be directed toward clarifying the mechanism for enhanced/prolonged MPO activity in wounded βarr2−/− mice.
An unanswered question in this study is, “What is the role played by βarr1 in CXCR2-mediated chemotaxis in vivo and in wound healing.” In vitro studies using mouse embryonic fibroblasts deficient in βarr1, βarr2, or βarr1/2 have shown that CXCR2 couples to either βarr1 or βarr2 to inhibit CXCL8-mediated respiratory burst (44). Double knockout of βarr1 and βarr2 results in embryonic lethality and thus cannot be tested in in vivo wound healing models (47). Although βarr1−/− mice have been generated, their genotype can be determined only by Southern blotting (51), making it difficult to conduct studies that require a large number of animals. Nonetheless, several studies have shown that there are differences between βarr1 and βarr2 with regard to their affinities for different GPCRs, subcellular distribution, function in GPCR recycling through the endosome, and the activation of receptor mediated downstream signaling (47). CXCR2 undergoes endocytosis in a clathrin/AP2-dependent fashion (22, 28). βarr2 has 6-fold greater affinity for clathrin than βarr1, and AP-2 preferentially binds to βarr2 (48, 49). Therefore, it is likely that CXCR2 interacts preferentially with βarr2 to mediate cellular functions. Supporting this contention is the observation that CXCR2 undergoes rapid ligand-induced endocytosis (within several minutes) with rapid recruitment of βarr2 to cytoplasmic membrane (20). These features are consistent with the conclusion that for class A GPCRs, βarr2 is more efficient than βarr1 in binding and regulating endocytosis (50).
In summary, the data herein indicate that β-arrestin-2 negatively regulates CXCR2-mediated neutrophil functions. Several questions, however, remain to be addressed. Arrestin was shown to bind to both AP-2 and the phosphorylated form of the receptor. It will be important to determine whether the receptor/AP-2/arrestin or receptor/arrestin/AP-2 complex is more important for rapid receptor internalization, polarization of intracellular signals, and optimal chemotaxis. Moreover, further studies are needed to define the functional interaction between βarr2, CXCR2, and other interacting proteins to more fully target CXCR2-mediated chemotaxis in chronic inflammatory disease and in wound healing.
Acknowledgments
We thank Kelly S. Parman of Mouse Pathology and IHC Core Laboratory, Vanderbilt University Medical Center, for technical support with mouse tissue processing and immunostaining. We also thank Dr. Robert J. Lefkowitz, Howard Hughes Medical Institute, Duke University Medical Center, Durham, NC, for providing β-arrestin-2−/− mice and for critically reading the manuscript.
Footnotes
This work was supported by a Historically Black Colleges and Universities Grant (to A.R. and R.M.R.); by a Senior Career Scientist Award from the Department of Veterans Affairs (to A.R.); and by the National Institutes of Health through Skin Disease Research Center Grant 2P30 AR41943 (to L.B.N.).
Abbreviations used in this paper: GPCR, G protein-coupled receptor; βarr1, β-arrestin-1; βarr2, β-arrestin-2; AP-2, adaptin 2; MPO, myeloperoxidase; MEF, mouse embryonic fibroblast.
Disclosures
The authors have no financial conflict of interest.
References
- 1.Bignold LP, Rogers SD, Siaw TM, Bahnisch J. Inhibition of chemotaxis of neutrophil leukocytes to interleukin-8 by endotoxins of various bacteria. Infect Immun. 1991;59:4255–4258. doi: 10.1128/iai.59.11.4255-4258.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Goya T, Morisaki T, Torisu M. Immunologic assessment of host defense impairment in patients with septic multiple organ failure: relationship between complement activation and changes in neutrophil function. Surgery. 1994;115:145–155. [PubMed] [Google Scholar]
- 3.Wagner JG, Roth RA. Neutrophil migration during endotoxemia. J Leukocyte Biol. 1999;66:10–24. doi: 10.1002/jlb.66.1.10. [DOI] [PubMed] [Google Scholar]
- 4.Chishti AD, Shenton BK, Kirby JA, Baudouin SV. Neutrophil chemotaxis and receptor expression in clinical septic shock. Intensive Care Med. 2004;30:605–611. doi: 10.1007/s00134-004-2175-y. [DOI] [PubMed] [Google Scholar]
- 5.Miller AP, Feng W, Xing D, Weathington NM, Blalock JE, Chen YF, Oparil S. Estrogen modulates inflammatory mediator expression and neutrophil chemotaxis in injured arteries. Circulation. 2004;110:1664–1669. doi: 10.1161/01.CIR.0000142050.19488.C7. [DOI] [PubMed] [Google Scholar]
- 6.Ross R. Atherosclerosis: an inflammatory disease. N Engl J Med. 1999;340:115–126. doi: 10.1056/NEJM199901143400207. [DOI] [PubMed] [Google Scholar]
- 7.Libby P, Ridker PM, Maseri A. Inflammation and atherosclerosis. Circulation. 2002;105:1135–1143. doi: 10.1161/hc0902.104353. [DOI] [PubMed] [Google Scholar]
- 8.Wetzler C, Kampfer H, Stallmeyer B, Pfeilschifter J, Frank S. Large and sustained induction of chemokines during impaired wound healing in the genetically diabetic mouse: prolonged persistence of neutrophils and macrophages during the late phase of repair. J Invest Dermatol. 2000;115:245–253. doi: 10.1046/j.1523-1747.2000.00029.x. [DOI] [PubMed] [Google Scholar]
- 9.Ajuebor MN, Zagorski J, Kunkel SL, Strieter RM, Hogaboam CM. Contrasting roles for CXCR2 during experimental colitis. Exp Mol Pathol. 2004;76:1–8. doi: 10.1016/j.yexmp.2003.08.004. [DOI] [PubMed] [Google Scholar]
- 10.Keshavarzian A, Fusunyan RD, Jacyno M, Winship D, MacDermott RP, Sanderson IR. Increased interleukin-8 (IL-8) in rectal dialysate from patients with ulcerative colitis: evidence for a biological role for IL-8 in inflammation of the colon. Am J Gastroenterol. 1999;94:704–712. doi: 10.1111/j.1572-0241.1999.00940.x. [DOI] [PubMed] [Google Scholar]
- 11.Sue RD, Belperio JA, Burdick MD, Murray LA, Xue YY, Dy MC, Kwon JJ, Keane MP, Strieter RM. CXCR2 is critical to hyperoxia-induced lung injury. J Immunol. 2004;172:3860–3868. doi: 10.4049/jimmunol.172.6.3860. [DOI] [PubMed] [Google Scholar]
- 12.Belperio JA, Keane MP, Burdick MD, Londhe V, Xue YY, Li K, Phillips RJ, Strieter RM. Critical role for CXCR2 and CXCR2 ligands during the pathogenesis of ventilator-induced lung injury. J Clin Invest. 2002;110:1703–1716. doi: 10.1172/JCI15849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Luster AD. Chemokines: chemotactic cytokines that mediate inflammation. N Engl J Med. 1998;338:436–445. doi: 10.1056/NEJM199802123380706. [DOI] [PubMed] [Google Scholar]
- 14.Baggiolini M. Chemokines and leukocyte traffic. Nature. 1998;392:565–568. doi: 10.1038/33340. [DOI] [PubMed] [Google Scholar]
- 15.Murphy PM, Baggiolini M, Charo IF, Hebert CA, Horuk R, Matsushima K, Miller LH, Oppenheim JJ, Power CA. International union of pharmacology. XXII. Nomenclature for chemokine receptors. Pharmacol Rev. 2000;52:145–176. [PubMed] [Google Scholar]
- 16.Arai H, I, Charo F. Differential regulation of G-protein-mediated signaling by chemokine receptors. J Biol Chem. 1996;271:21814–21819. doi: 10.1074/jbc.271.36.21814. [DOI] [PubMed] [Google Scholar]
- 17.Smith CD, Cox CC, Snyderman R. Receptor-coupled activation of phosphoinositide-specific phospholipase C by an N protein. Science. 1986;232:97–100. doi: 10.1126/science.3006254. [DOI] [PubMed] [Google Scholar]
- 18.Luttrell LM, Lefkowitz RJ. The role of β-arrestins in the termination and transduction of G-protein-coupled receptor signals. J Cell Sci. 2002;115:455–465. doi: 10.1242/jcs.115.3.455. [DOI] [PubMed] [Google Scholar]
- 19.Barlic J, Andrews JD, Kelvin AA, Bosinger SE, DeVries ME, Xu L, Dobransky T, Feldman RD, Ferguson SS, Kelvin DJ. Regulation of tyrosine kinase activation and granule release through β-arrestin by CXCRI. Nat Immunol. 2000;1:227–233. doi: 10.1038/79767. [DOI] [PubMed] [Google Scholar]
- 20.Richardson RM, Marjoram RJ, Barak LS, Snyderman R. Role of the cytoplasmic tails of CXCR1 and CXCR2 in mediating leukocyte migration, activation, and regulation. J Immunol. 2003;170:2904–2911. doi: 10.4049/jimmunol.170.6.2904. [DOI] [PubMed] [Google Scholar]
- 21.Yang W, Wang D, Richmond A. Role of clathrin-mediated endocytosis in CXCR2 sequestration, resensitization, and signal transduction. J Biol Chem. 1999;274:11328–11333. doi: 10.1074/jbc.274.16.11328. [DOI] [PubMed] [Google Scholar]
- 22.Fan GH, Lapierre LA, Goldenring JR, Sai J, Richmond A. Rab11-family interacting protein 2 and myosin Vb are required for CXCR2 recycling and receptor-mediated chemotaxis. Mol Biol Cell. 2004;15:2456–2469. doi: 10.1091/mbc.E03-09-0706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fan GH, Yang W, Sai J, Richmond A. Phosphorylation-independent association of CXCR2 with the protein phosphatase 2A core enzyme. J Biol Chem. 2001;276:16960–16968. doi: 10.1074/jbc.M009292200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fan GH, Yang W, Wang XJ, Qian Q, Richmond A. Identification of a motif in the carboxyl terminus of CXCR2 that is involved in adaptin 2 binding and receptor internalization. Biochemistry. 2001;40:791–800. doi: 10.1021/bi001661b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fan GH, Yang W, Sai J, Richmond A. Hsc/Hsp70 interacting protein (hip) associates with CXCR2 and regulates the receptor signaling and trafficking. J Biol Chem. 2002;277:6590–6597. doi: 10.1074/jbc.M110588200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bohn LM, Lefkowitz RJ, Gainetdinov RR, Peppel K, Caron MG, Lin FT. Enhanced morphine analgesia in mice lacking β-arrestin 2. Science. 1999;286:2495–2498. doi: 10.1126/science.286.5449.2495. [DOI] [PubMed] [Google Scholar]
- 27.Bohn LM, Gainetdinov RR, Lin FT, Lefkowitz RJ, Caron MG. Muopioid receptor desensitization by β-arrestin-2 determines morphine tolerance but not dependence. Nature. 2000;408:720–723. doi: 10.1038/35047086. [DOI] [PubMed] [Google Scholar]
- 28.Fong AM, Premont RT, Richardson RM, Yu YR, Lefkowitz RJ, Patel DD. Defective lymphocyte chemotaxis in β-arrestin 2- and GRK6-deficient mice. Proc Natl Acad Sci USA. 2002;99:7478–7483. doi: 10.1073/pnas.112198299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Edwards JC, Sedgwick AD, Willoughby DA. The formation of a structure with the features of synovial lining by subcutaneous injection of air: an in vivo tissue culture system. J Pathol. 1981;134:147–156. doi: 10.1002/path.1711340205. [DOI] [PubMed] [Google Scholar]
- 30.Sin YM, Sedgwick AD, Chea EP, Willoughby DA. Mast cells in newly formed lining tissue during acute inflammation: a six day air pouch model in the mouse. Ann Rheum Dis. 1986;45:873–877. doi: 10.1136/ard.45.10.873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Clish CB, O’Brien JA, Gronert K, Stahl GL, Petasis NA, Serhan CN. Local and systemic delivery of a stable aspirin-triggered lipoxin prevents neutrophil recruitment in vivo. Proc Natl Acad Sci USA. 1999;96:8247–8252. doi: 10.1073/pnas.96.14.8247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tomhave ED, Richardson RM, Didsbury JR, Menard L, Snyderman R, Ali H. Cross-desensitization of receptors for peptide chemoattractants: characterization of a new form of leukocyte regulation. J Immunol. 1994;153:3267–3275. [PubMed] [Google Scholar]
- 33.Chuntharapai A, Kim KJ. Regulation of the expression of IL-8 receptor A/B by IL-8: possible functions of each receptor. J Immunol. 1995;155:2587–2594. [PubMed] [Google Scholar]
- 34.Haribabu B, Richardson RM, Fisher I, Sozzani S, Peiper SC, Horuk R, Ali H, Snyderman R. Regulation of human chemokine receptors CXCR4: role of phosphorylation in desensitization and internalization. J Biol Chem. 1997;272:28726–28731. doi: 10.1074/jbc.272.45.28726. [DOI] [PubMed] [Google Scholar]
- 35.Meller N, Elitzur Y, Isakov N. Protein kinase C-θ (PKCθ) distribution analysis in hematopoietic cells: proliferating T cells exhibit high proportions of PKCθ in the particulate fraction. Cell Immunol. 1999;193:185–193. doi: 10.1006/cimm.1999.1478. [DOI] [PubMed] [Google Scholar]
- 36.Devalaraja RM, Nanney LB, Du J, Qian Q, Yu Y, Devalaraja MN, Richmond A. Delayed wound healing in CXCR2 knockout mice. J Invest Dermatol. 2000;115:234–244. doi: 10.1046/j.1523-1747.2000.00034.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nanney LB. Epidermal and dermal effects of epidermal growth factor during wound repair. J Invest Dermatol. 1990;94:624–629. doi: 10.1111/1523-1747.ep12876204. [DOI] [PubMed] [Google Scholar]
- 38.Thorey IS, Hinz B, Hoeflich A, Kaesler S, Bugnon P, Elmlinger M, Wanke R. Transgenic mice reveal novel activities of growth hormone in wound repair, angiogenesis and myofibroblast differentiation. J Biol Chem. 2004;279:26674–26684. doi: 10.1074/jbc.M311467200. [DOI] [PubMed] [Google Scholar]
- 39.Milatovic S, Nanney LB, Yu Y, White JR, Richmond A. Impaired healing of nitrogen mustard wounds in CXCR2 null mice. Wound Repair Regen. 2003;11:213–219. doi: 10.1046/j.1524-475x.2003.11310.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tohgo A, Pierce KL, Choy EW, Lefkowitz RJ, Luttrell LM. β-Arrestin scaffolding of the ERK cascade enhances cytosolic ERK activity but inhibits ERK-mediated transcription following angiotensin AT1a receptor stimulation. J Biol Chem. 2002;277:9429–9436. doi: 10.1074/jbc.M106457200. [DOI] [PubMed] [Google Scholar]
- 41.Tohgo A, Choy EW, Gesty-Palmer D, Pierce KL, Laporte S, Oakley RH, Caron MG, Lefkowitz RJ, Luttrell LM. The stability of the G protein-coupled receptor-β-arrestin interaction determines the mechanism and functional consequence of ERK activation. J Biol Chem. 2003;278:6258–6267. doi: 10.1074/jbc.M212231200. [DOI] [PubMed] [Google Scholar]
- 42.Sun Y, Cheng Z, Ma L, Pei G. β-Arrestin 2 is critically involved in CXCR4-mediated chemotaxis, and this is mediated by its enhancement of p38 MAPK activation. J Biol Chem. 2002;277:49212–49219. doi: 10.1074/jbc.M207294200. [DOI] [PubMed] [Google Scholar]
- 43.Richardson RM, Pridgen BC, Haribabu B, Snyderman R. Regulation of the human chemokine receptor CCR1: cross-regulation by CXCR1 and CXCR2. J Biol Chem. 2000;275:9201–9208. doi: 10.1074/jbc.275.13.9201. [DOI] [PubMed] [Google Scholar]
- 44.Zhao M, Wimmer A, Trieu K, Discipio RG, Schraufstatter IU. Arrestin regulates MAPK activation and prevents NADPH oxidase-dependent death of cells expressing CXCR2. J Biol Chem. 2004;279:49259–49267. doi: 10.1074/jbc.M405118200. [DOI] [PubMed] [Google Scholar]
- 45.Springer TA. Traffic signals on endothelium for lymphocyte recirculation and leukocyte emigration. Annu Rev Physiol. 1995;57:827–872. doi: 10.1146/annurev.ph.57.030195.004143. [DOI] [PubMed] [Google Scholar]
- 46.Smith ML, Olson TS, Ley K. CXCR2- and E-selectin-induced neutrophil arrest during inflammation in vivo. J Exp Med. 2004;200:935–939. doi: 10.1084/jem.20040424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pierce KL, Lefkowitz RJ. Classical and new roles of β-arrestins in the regulation of G-protein-coupled receptors. Nat Rev Neurosci. 2001;2:727–733. doi: 10.1038/35094577. [DOI] [PubMed] [Google Scholar]
- 48.Goodman OB, Jr, Krupnick JG, Santini F, Gurevich VV, Penn RB, Gagnon AW, Keen JH, Benovic JL. β-Arrestin acts as a clathrin adaptor in endocytosis of the β2-adrenergic receptor. Nature. 1996;383:447–450. doi: 10.1038/383447a0. [DOI] [PubMed] [Google Scholar]
- 49.Laporte SA, Oakley RH, Zhang J, Holt JA, Ferguson SS, Caron MG, Barak LS. The β2-adrenergic receptor/βarrestin complex recruits the clathrin adaptor AP-2 during endocytosis. Proc Natl Acad Sci USA. 1999;96:3712–3717. doi: 10.1073/pnas.96.7.3712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Oakley RH, Laporte SA, Holt JA, Caron MG, Barak LS. Differential affinities of visual arrestin, β arrestin 1, and β arrestin 2 for G protein-coupled receptors delineate two major classes of receptors. J Biol Chem. 2000;275:17201–17210. doi: 10.1074/jbc.M910348199. [DOI] [PubMed] [Google Scholar]
- 51.Conner DA, Mathier MA, Mortensen RM, Christe M, Vatner SF, Seidman CE, Seidman JG. β-Arrestin1 knockout mice appear normal but demonstrate altered cardiac responses to β-adrenergic stimulation. Circ Res. 1997;81:1021–1026. doi: 10.1161/01.res.81.6.1021. [DOI] [PubMed] [Google Scholar]