

Abstract
Herpes simplex virus (HSV) type 1 capsids contain a single portal vertex that is composed of 12 copies of the UL6 gene product (pUL6), which forms a pore through which DNA is inserted during packaging. This unique vertex is also believed to comprise the site with which a molecular motor, termed the terminase, associates during the DNA packaging reaction. In HSV, the terminase likely comprises the UL15, UL28, and UL33 proteins (pUL15, pUL28, and pUL33, respectively). The current study was undertaken to identify portal domains required for interaction with the terminase. Both the amino and carboxyl termini, as well as amino acids 422 to 443 of pUL6 forming a putative leucine zipper motif, were critical for coimmunoprecipitation with pUL15 in the absence of other viral proteins. Amino acids 422 to 443 were also necessary for interaction with pUL28 in the absence of other viral proteins. By using an engineered recombinant virus, it was further determined that although amino acids 422 to 443 were dispensable for interaction with scaffold protein and incorporation of portal protein into capsids, they were necessary for coimmunoprecipitation of pUL6 and pUL15 from infected cell lysates, association of optimal levels of pUL15, pUL28, and pUL33 with capsids, and DNA cleavage and packaging. These data identify a portal protein domain critical for terminase association with the capsid and suggest that both the pUL15- and pUL28-bearing terminase subunits mediate docking of the terminase with the portal vertex.
The DNA replication machinery of herpes simplex virus (HSV) produces concatameric viral DNA in the nuclei of infected cells that is cleaved and packaged into preformed capsids (reviewed in references 2 and 4). These icosahedral capsids contain a unique pore, termed the portal vertex, through which viral DNA is inserted (15). The portal vertex comprises 12 copies of the UL6 gene (23). The portal also likely functions as the docking site of the terminase enzyme (24, 27, 30), which is responsible for cleavage of viral DNA and is a key part of the molecular motor that drives DNA through the portal during the packaging reaction (2, 19). In HSV, the terminase likely comprises the UL15, UL28, and UL33 gene products (5, 10, 26). The UL15 protein (pUL15) is believed to provide the energy for the packaging reaction through hydrolysis of ATP because it contains a highly conserved ATP binding motif that is essential for DNA packaging (7, 29); pUL28 likely provides sequence-specific DNA binding activity (1), and pUL33 enhances the pUL15/pUL28 interaction, primarily through its interactions with UL28 (26).
Incorporation of the portal into the capsid is mediated by its interaction with amino acids 143 to 151 of ICP35, the major component of the internal shell of the two-shelled capsid (9, 18, 28). In the absence of this scaffold protein sequence, the portal protein fails to interact efficiently with ICP35 in vitro and is not incorporated into the capsids.
Like those of all herpesviruses, the HSV type 1 (HSV-1) portal protein contains a potential leucine zipper between amino acids 422 and 443, in which three invariant leucines are separated by 6 amino acids, thus potentially placing them on one side of an α-helix (data not shown). Because such motifs have been implicated in a number of protein-protein interactions, and protein interactions represent critical functions of the portal vertex, this motif has garnered experimental interest. Specifically, deletion of codons 409 to 473, or changing leucines at positions 429 and 436 to glutamic acid, reduced incorporation of the portal into capsids and precluded normal formation of portal rings in vitro (14).
One goal of the current work was to determine how the terminase docks with the capsid. The portal protein pUL6 coimmunoprecipitates with both pUL15 and pUL28, suggesting these proteins interact in vivo (27). This hypothesis is also supported by observations that pUL6 (i) coimmunoprecipitates with either pUL15 or pUL28 overexpressed in insect cells and (ii) can alter the localization of pUL28 and pUL15 in mammalian cells when these proteins are coexpressed with pUL6 in the absence of other viral proteins (24).
Because our previous observations (27) indicated that the coimmunoprecipitation of pUL15 and pUL6 from infected cells is more robust than that of pUL28 and pUL6, we focused primarily on the pUL6/pUL15 interaction in the current study. The data indicated that although codons 422 to 443 of UL6 were dispensable for interaction with scaffold protein and incorporation of the portal into the capsid, they were critical for (i) DNA cleavage and packaging, (ii) interaction between pUL15 and pUL6 in lysates of both uninfected and infected cells, (iii) coimmunoprecipitation with transiently expressed pUL6 and pUL28, and (iv) association of normal levels of pUL15, pUL28, and pUL33 with the capsid. These data suggest that docking of the terminase with the capsid involves interactions between the portal protein and both the UL15-encoded and pUL28-encoded terminase subunits and shed new light on the importance of the putative leucine zipper of the portal in HSV DNA packaging.
MATERIALS AND METHODS
Viruses and cells.
CV1, Vero, and rabbit skin cells were obtained from the American Type Culture Collection (ATCC) and maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% newborn calf serum, 100 U penicillin per ml, and 100 μg of streptomycin per ml. The Flp-In-CV1 cell line was purchased from Invitrogen and was grown in DMEM supplemented with 10% newborn bovine serum, 100 U/ml penicillin, 100 μg/ml streptomycin, and 100 μg/ml Zeocin. The CV6 and CV6M cell lines described in this paper were cultured in DMEM supplemented with 10% newborn bovine serum, 100 U/ml of penicillin, 100 μg/ml of streptomycin, and 200 μg/ml of hygromycin B. HSV-1 strain F [HSV-1(F)] and UL6 null virus derived from HSV-1 strain 17 were described previously (8, 17). Recombinant virus vJB10 and its restored virus vJB10R are described in this paper.
Plasmids.
The full-length UL15 gene with a hemagglutinin (HA) tag inserted in-frame at the C terminus was amplified by PCR and cloned into the HindIII and EcoRV sites located in the multiple cloning site of expression vector pCDNA3. The resulting plasmid was designated pJB517. Plasmid pJB437 containing the entire UL6 coding sequence was described previously (27). UL6 genes fused at the 5′ or 3′ end to DNA encoding a Flag epitopic tag were amplified by PCR and cloned into the BamHI and EcoRI sites located in the multiple cloning site of expression vector pcDNA3. The resulting constructs were designated pJB444 and pJB445, respectively. To delete codons 422 to 443 from UL6, encoding a putative leucine zipper, two-step PCR was performed. The resultant PCR amplicons were cloned into pcDNA3 at the BamHI and EcoRI sites, and the resulting plasmid was designated pJB454. Similarly, amino-terminally truncated UL6 (as shown in the diagram of Fig. 1) was amplified by PCR, using pJB437 or pJB454 as a template. PCR products were cloned into the BamHI and EcoRI sites of pCDNA3, and the resulting plasmids were designated pJB507 and pJB645, respectively. To construct pJB456 and pJB557, plasmids pJB347 and pJB454 were digested with BamHI and NotI, and the inserts were purified and cloned into pcDNA5/FRT at the BamHI and NotI sites. All plasmid constructs were confirmed by immunoblotting after transient expression in mammalian cells and DNA sequencing by the Cornell University DNA sequencing and genotyping core facility (data not shown). To replace the sequence for the putative leucine zipper of pUL6 with the amino acid sequence of the GCN4 leucine zipper (LEDKVEELLSKNYHLENEVARL), two-step PCR was performed, the final PCR amplicons were cloned into pCDNA3 at the BamHI and EcoRI sites, and the resulting plasmid was designated pJB584.
FIG. 1.

(A) Schematic diagram of the full-length pUL15 (top line), pUL6 (second line from top), and UL6 mutant (other lines) proteins. The designation of the plasmid and genotype are indicated on the right. Numbers in the rightmost column followed by aa indicate the amino acids present in the corresponding construct. (B) Schematic diagram of the UL6 gene of the deletion mutant virus vJB10 or its corresponding genetically restored virus vJB10R.
Plasmid pJB112, containing the UL28 coding sequence cloned into pCDNA3, was described previously (22). Plasmid pCAGGS-nlsCre, expressing Cre recombinase, was a gift from Michael Kotlikoff, Cornell University. Plasmids pBAD-I-SceI, containing the gene encoding the Saccharomyces cerevisiae I-SceI endonuclease, and pEPkan-S, containing aphAI (encoding kanamycin resistance) adjacent to an I-SceI restriction site, were gifts from Nikolaus Osterrieder, Cornell University.
Construction of complementing cell lines.
CV6 and CV6M cell lines were made by using the Flp-In-CV1 system (Invitrogen) according to the manufacturer's protocol and as described previously (28). Briefly, either pJB456 or pJB557 was cotransfected with plasmid pOG44, encoding Flp recombinase under the constitutive cytomegalovirus promoter/enhancer, into an engineered cell line (Flp-In-CV1). Correct insertion of the shuttle vector caused simultaneous loss of Zeocin resistance and gain of hygromycin resistance. After recombination, cells resistant to hygromycin were selected by growth in DMEM supplemented with 10% newborn bovine serum and 200 μg/ml hygromycin B.
Construction of recombinant viruses.
Production and characterization of a bacterial artificial chromosome (BAC) containing the entire HSV-1(F) genome was described previously (20). Recombinant viruses were constructed by en passant mutagenesis, a two-step Red-mediated recombinant system described by Tischer et al. (22). The details of the procedure and their use in construction of recombinant HSV-1 genomes were also described previously (27). The primers for the production of a PCR amplicon for eventual deletion of codons 422 to 443 from the UL6 gene in the HSV-1(F)-containing BAC were as follows: forward, TTCCGCACGGCCGTGGTTAACAACATCAACGGCGTGGCGACCCAATTGCAGGAGTAGGGATAACAGGGTAATCGATTT; reverse, CCGGAGCTCGCGGTCGCGCTCCTGCAATTGGGTCGCCACGCCGTTGATGTTGTTGCCAGTGTTACAACCAATTAACC.
The expected mutation in the BAC DNA was confirmed by DNA sequencing, and the resulting recombinant BAC was designated bJB10. Purified bJB10 BAC DNA was cotransfected with a Cre expression plasmid (see above) into CV6 cells expressing pUL6. The presence of viable recombinant virus was confirmed by plaque formation on complementing CV6 cells, and the resulting virus was subjected to two further rounds of plaque purification. The genotype of the recombinant virus, designated vJB10, was confirmed by PCR and DNA sequencing, whereas the viral phenotype was characterized as described below in Results. To repair the mutated UL6 gene, rabbit skin cells were cotransfected with vJB10 viral DNA and linearized pRB132, which contains the gene fragment from bases 11820 to 21655 of the HSV-1 genome, according to the numbering scheme of McGeoch et al. (13). The virus arising from homologous recombination was able to form plaques on rabbit skin cells and was designated vJB10R. The genotype of vJB10R was confirmed by DNA sequencing, immunoblotting, and Southern blot analyses (data not shown).
Immunoprecipitation and immunoblotting.
These procedures were performed essentially as described previously (23, 24). Briefly, CV1 cells were either transfected with expression plasmids containing UL15, UL28, and UL6 or its derivatives or infected with wild-type or recombinant viruses. At 24 h after transfection or 18 h after infection, the cells were washed with cold phosphate-buffered saline (PBS) and lysed in cold radioimmunoprecipitation assay (RIPA) buffer containing 50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 1% NP-40, 0.25% sodium deoxycholate, 1 mM EDTA, and 1× protease inhibitor cocktail (Roche). Rabbit anti-HA polyclonal antibody (HA-probe, Y-11, SC-805 diluted 1:200; Santa Cruz Biotechnology) was used for pUL15 immunoprecipitation, whereas pUL6-specific rabbit polyclonal serum (diluted 1:100) was used to immunoprecipitate pUL6. Anti-ICP35 monoclonal antibodies (MCA 406; AbD Serotec) were used for immunoprecipitation at a dilution of 1:200. pUL28-specific rabbit polyclonal antisera were diluted 1:100 to immunoprecipitate pUL28. Immune complexes, RIPA buffer-solubilized clarified lysates, total lysates solubilized in 1% sodium dodecyl sulfate (SDS) and beta-mercaptoethanol, and in some experiments, SDS-denatured purified B capsids were separated on SDS-polyacrylamide gels and transferred to nitrocellulose membranes for immunoblotting.
Immunoblots were probed with anti-HA antibodies diluted 1:1,000, anti-pUL15C diluted 1:1,000, anti-pUL28 diluted 1:1,000, anti-pUL33 diluted 1:400, anti-ICP35 (MCA 406; AbD Serotec) diluted 1:2,000, anti-VP5 (HA018-100; Virusys Corporation) diluted 1:1,000, and/or anti-pUL6 polyclonal antiserum diluted 1:1,000. The bound immunoglobulins were revealed by reaction with horseradish peroxidase-conjugated anti-immunoglobulins and visualized by enhanced chemiluminescence (Amersham Pharmacia Biotech). Where applicable, the image intensities of specific bands on the immunoblots were quantified with an LAS-3000 mini Fujifilm imaging system (Fuji Photo Film Co., Ltd.).
Capsid purification.
CV1 cell monolayers from two 850-cm2 roller bottles were infected with either HSV-1(F) or mutant viruses at a multiplicity of infection (MOI) of 5 PFU/cell. The cells were harvested 20 hours later and washed with cold PBS. Cell pellets were suspended in 25 ml of lysis buffer (1 mM dithiothreitol, 1 mM EDTA, 20 mM Tris [pH 7.6], 500 mM NaCl, 1% Triton X-100, and protease inhibitor), sonicated briefly, and precleared by spinning at 10,000 × g for 15 min. The precleared lysates were pelleted through a 5-ml 35% sucrose cushion in TNE buffer (20 mM Tris-HCl [pH 7.6], 500 mM NaCl, 1 mM EDTA), in an SW28 ultracentrifuge tube at 24,000 rpm for 1 h. The pellets were resuspended in TNE buffer and applied to 20% to 50% sucrose gradients in SW41 ultracentrifuge tubes followed by centrifugation at 24,500 rpm for 1 h. After centrifugation, the light-refracting B capsid band was removed with a Pasteur pipette. Purity of the capsid preparations was evaluated by transmission electron microscopy and negative staining (data not shown).
Southern blotting.
Approximately 2 × 106 CV1 cells were infected with HSV-1(F), vJB10, or vJB10R. At 18 h postinfection, viral DNA was extracted, digested with BamHI, separated on 0.8% agarose gels, and transferred to nylon membranes as described previously (24). The bound DNA was UV cross-linked to the membrane and hybridized with a denatured [32P]dCTP-labeled BamHI P fragment of HSV-1(F) DNA at 42°C for 24 h. The membrane was washed twice with 1× SSC (0.15 M NaCl plus 0.015 M sodium citrate) at 42°C for 15 min each time and once with 0.1× SSC-0.1% SDS for 1 h at 64°C and then fluorographed by exposure to X-ray film at −80°C in the presence of intensifying screens.
Virus replication assay.
Approximately 2 × 106 cells in 25-cm2 flasks were infected with the viruses indicated in Table 1 at an MOI of 0.1 PFU/cell. After adsorption for 2 hours at 37°C with shaking, the inoculates were removed, and the cells were washed with CBS buffer (40 mM citric acid, 10 mM KCl, 135 mM NaCl [pH 3.0]) to remove residual infectivity. The cells were then washed with PBS once and overlaid with 5 ml of DMEM supplemented with 5% newborn calf serum. Twenty-four hours after infection, virus was harvested by three cycles of freezing and thawing, and the infectious yields were determined by plaque assay on the cell monolayers indicated in Table 1.
TABLE 1.
Virus replication assay
| Virus | Genotype of UL6 | Cell type infected/cell type used for plaque assay | Titer (PFU/ml)a |
|---|---|---|---|
| vJB10 | Deletion of codons 422 to 443 | CV1/CV6 | <102 |
| vJB10 | Deletion of codons 422 to 443 | CV6/CV6 | 1.2 × 107 ± 0.5 × 107 |
| vJB10 | Deletion of codons 422 to 443 | CV6M/CV6 | <102 |
| vJB10R | Deleted codons restored | CV1/Vero | 2.0 × 107 ± 0.76 × 107 |
| HSV-1(F) | Wild type | CV1/Vero | 2.5 × 107 ± 0.2 × 107 |
| HSV-1(F) | Wild type | CV6/Vero | 2.6 × 107 ± 0.5 × 107 |
| HSV-1(F) | Wild type | CV6M/Vero | 6.5 × 104 ± 0.8 × 104 |
Virus titers were determined as described in Materials and Methods. The data represent means ± standard deviations for three independent experiments.
RESULTS
To identify domains of pUL6 necessary for interaction with pUL15, we cloned pUL15 in-frame with an HA tag at its C terminus and wild-type UL6 into plasmids such that their expression was under the control of the human cytomegalovirus early promoter/enhancer. We chose this site in pUL15 to insert the HA tag because other insertions (cytomegalovirus glycoprotein B or Flag epitopes) at the C terminus of pUL15 have been tolerated with little effect on virus replication (3). The engineered plasmids were transfected into CV1 cells. Lysates of the transfected cells were then subjected to immunoprecipitation with HA-specific or pUL6-specific antibodies, and the presence of pUL6 and pUL15-HA in immunoprecipitated material was assessed by immunoblotting.
As shown in Fig. 2, lane 2, pUL15 and pUL6 were expressed to readily detectable levels in lysates of the transfected cells, but not in mock-transfected cells. Moreover, the HA-specific antibody readily immunoprecipitated HA-tagged pUL15, and the pUL6-specific antibody successfully immunoprecipitated pUL6. Most importantly for the purposes of this report, material immunoprecipitated with the HA-specific antibody also contained pUL6, and reaction with the pUL6 antibody caused coimmunoprecipitation of pUL15. We conclude that transiently expressed pUL15 and pUL6 interact as assessed by immunoprecipitation.
FIG. 2.
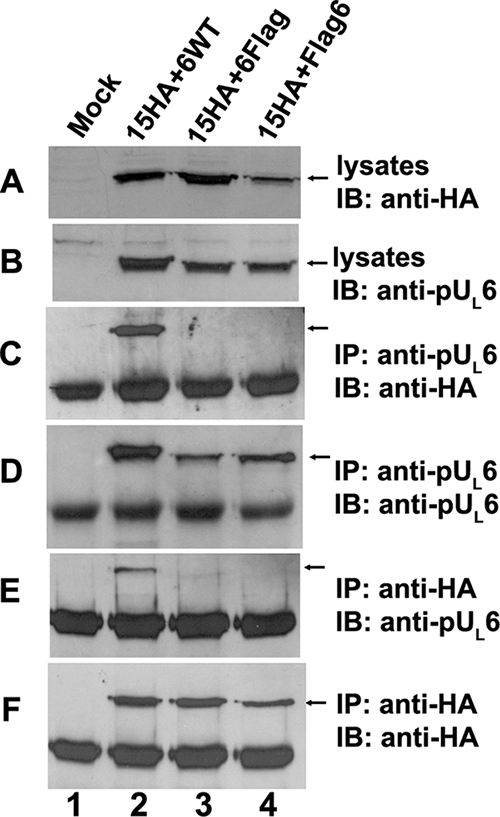
Insertions of Flag epitopic tags at the N or C terminus of pUL6 preclude interaction with transiently expressed pUL15. CV1 cells (2 × 106) were cotransfected with plasmids encoding pUL15-HA and wild-type UL6 (lane 2) or Flag-tagged pUL6 (lanes 3 and 4) or were mock transfected (lane 1). Twenty-four hours after transfection, reciprocal coimmunoprecipitation with either anti-pUL6 or anti-HA antibodies was performed. Cell lysates and immunoprecipitated proteins were electrophoretically separated in denaturing 12% polyacrylamide gels and transferred onto a nitrocellulose membrane. The transferred proteins were probed with anti-pUL6 (B, D, and E) or anti-HA (A, C, and F) antibodies. Bound immunoglobulins were revealed by enhanced chemiluminescence. IP, immunoprecipitation with the indicated antibody; IB, immunoblotting with the indicated antibody. The arrows indicate the positions of proteins of interest.
To identify domains of pUL6 involved in interactions with the terminase subunit pUL15, a series of transient expression constructs were made; their relevant sequences are indicated in Fig. 1A. Expression plasmids bearing these constructs were contransfected with plasmids expressing full-length pUL15-HA into CV1 cells, and lysates of the cells were immunoprecipitated with the HA- and pUL6-specific antibodies. As shown in Fig. 2, lanes 3 and 4, placement of a Flag epitopic tag at either the C terminus or the N terminus of pUL6 did not interfere with the expression or immunoprecipitation of pUL6 with its cognate antibody, but it completely precluded coimmunoprecipitation with pUL15. These data suggest that both the N and C termini of pUL6 are important for the interaction with pUL15. To confirm that the N and C termini of pUL6 were necessary for the coimmunoprecipitation with pUL15, a plasmid construct encoding pUL6 amino acids 314 to 676 (Fig. 3) and a construct encoding only the first 321 amino acids of pUL6 (not shown) were coexpressed with pUL15 and were subjected to immunoprecipitation with HA antibodies. As shown in Fig. 3, the C-terminal amino acids 314 to 676 of pUL6 were expressed and were recognized by the pUL6 antibody, but they did not coimmunoprecipitate with pUL15-HA. Similar negative results were obtained upon expression of the first 321 codons encoding the sequence of pUL6 followed by attempted coimmunoprecipitation of pUL15-HA with the HA antibody (not shown). We conclude that both N and C termini are necessary for interaction with pUL15 in this transient immunoprecipitation assay.
FIG. 3.
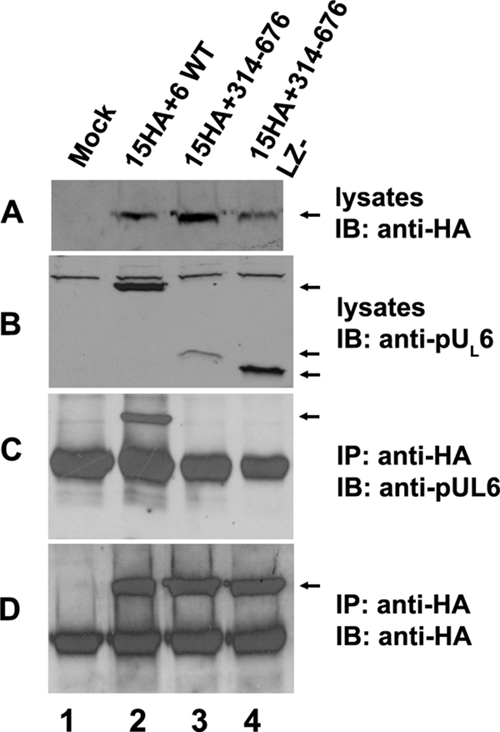
Only the full-length pUL6 interacts with pUL15 in transiently transfected cells. CV1 cells (2 × 106) were mock transfected (lane 1) or cotransfected with plasmids encoding pUL15-HA and full-length pUL6 (lane 2), N-terminally truncated pUL6 (lane 3), or N-terminally truncated pUL6 lacking the putative leucine zipper (lane 4). Twenty-four hours after transfection, coimmunoprecipitation was performed with anti-pUL6 antibodies. Cell lysates and immunoprecipitated material were separated on 12% SDS-polyacrylamide gels and transferred to a nitrocellulose membrane. The transferred proteins were probed with anti-pUL6 (B and C) or anti-HA (A and D) antibodies. Bound immunoglobulins were revealed by reaction with appropriately conjugated anti-immunoglobulins followed by enhanced chemiluminescence. IP, immunoprecipitation with the indicated antibody; IB, immunoblotting with the indicated antibody. The arrows indicate the positions of proteins of interest.
To investigate the role of the putative leucine zipper in coimmunoprecipitation of transiently expressed pUL6 and pUL15, codons 422 to 443 of UL6 were deleted, the truncated protein was coexpressed with pUL15-HA, and the lysates were subjected to immunoprecipitation with pUL6- and HA-specific antibodies. As shown in Fig. 4, lane 2, the absence of UL6 codons 422 to 443 precluded coimmunoprecipitation of the truncated pUL6 and full-length pUL15, despite the presence of ample levels of soluble pUL6 and pUL15 proteins in the starting lysates, and the successful coimmunoprecipitation of both proteins with their cognate antibodies. These data indicate that the putative leucine zipper in pUL6 is required for the interaction with pUL15. To determine if a known leucine zipper could complement the function of codons 422 to 443, these codons were replaced with DNA encoding the leucine zipper of GCN4 (16) and the protein was coexpressed with pUL15 and subjected to the coimmunoprecipitation reaction. As shown in Fig. 4, lane 3, the GCN4 motif was not sufficient to restore the ability of truncated pUL6 to interact with pUL15. Thus, either the GCN4 motif is unable to form a leucine zipper when expressed in the context of pUL6, or codons 422 to 443 mediate the interaction with pUL15 through means other than as a leucine zipper.
FIG. 4.
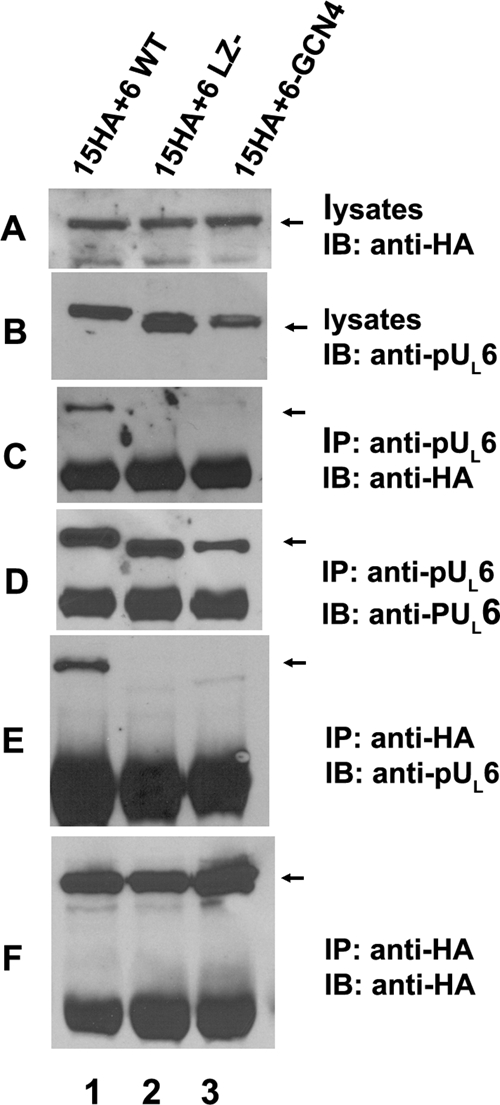
The putative leucine zipper of pUL6 of HSV-1(F) is required for pUL6 and pUL15 interaction in the absence of other viral proteins. CV1 cells were cotransfected with plasmids encoding full-length pUL15 with a C-terminal HA tag and wild-type pUL6 (lane 1), pUL6 lacking amino acids 422 to 443 (lane 2), or a pUL6-GCN4 chimera (lane 3) in which codons 422 to 443 of UL6 were replaced with the sequence encoding the GCN4 leucine zipper. Twenty-four hours after transfection, reciprocal coimmunoprecipitations were performed with anti-pUL6 or anti-HA antibodies. Cell lysates and immunoprecipitated proteins were separated on denaturing 12% polyacrylamide gels and transferred to a nitrocellulose membrane. The transferred proteins were probed with anti-pUL6 (B, D, and E) or anti-HA (A, C, and F) antibodies. Bound immunoglobulins were detected as indicated in the legend to Fig. 3. IP, immunoprecipitation; IB, immunoblotting. The arrows indicate the positions of proteins of interest.
To determine whether pUL6 and pUL28 can interact in the absence of other viral proteins in mammalian cells and whether codons 422 to 443 affected this interaction, CV1 cells were transfected with plasmids encoding full-length pUL28 and either wild-type pUL6, pUL6 lacking sequences corresponding to codons 422 to 443, or pUL6 in which the sequences corresponding to codons 422 to 443 was replaced by the GCN4 leucine zipper motif (16). Lysates of the cells were then reacted with pUL28-specific antibody, and the presence or absence of pUL6 and pUL28 in immunoprecipitated material was determined by immunoblotting. As shown in Fig. 5, lane 1, wild-type pUL6 was readily coimmunoprecipitated with pUL28-specific antibody. In contrast, the absence of the leucine zipper of pUL6 precluded this coimmunoprecipitation whether or not it was replaced with the GCN4 leucine zipper motif.
FIG. 5.
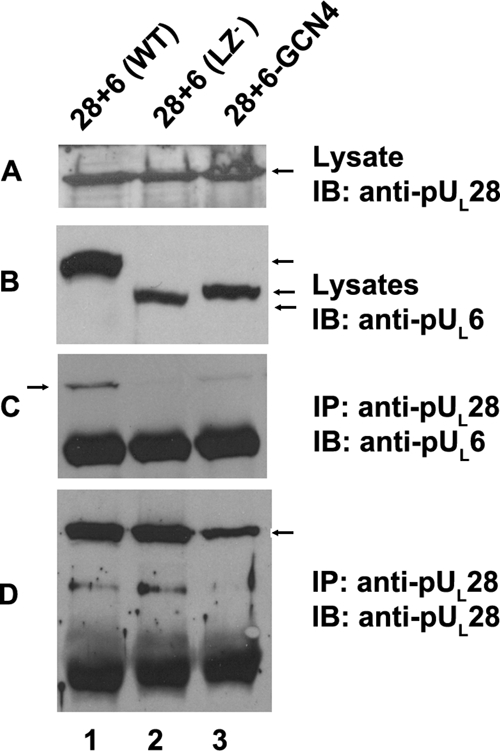
The putative leucine zipper of HSV-1(F) pUL6 is required for interaction with pUL28 in the absence of other viral proteins. Cells were transfected with plasmids encoding full-length pUL28 and wild-type pUL6 (lane 1), pUL6 lacking codons 422 to 443 (lane 2), or pUL6 in which codons 422 to 443 were replaced by a leucine zipper of GCN4 (lane 3). The cells were lysed 24 h later and either immunoblotted with antibodies directed against pUL6 or pUL28 (A and B) or subjected to immunoprecipitation with antibody directed against pUL28. The presence of pUL6 or pUL28 in immunoprecipitated material was determined by immunoblotting (C and D, respectively). IP, antibody used for immunoprecipitation in the corresponding panel; IB, antibody used for immunoblotting in the corresponding panel. The arrows indicate the positions of proteins of interest.
To determine the role of codons 422 to 443 of pUL6 in infected cells, a recombinant virus lacking these codons was constructed as detailed in Materials and Methods. The virus was designated vJB10. Initial experiments indicated that vJB10 was unable to propagate on CV1 cells, so viral stocks were generated on CV1 cells engineered to express UL6. To confirm the conclusion that the deletion within UL6 was lethal for viral replication, CV1 cells, CV6 cells (CV1 cells expressing pUL6), and CV6M cells (CV1 cells expressing UL6 lacking codons 422 to 443) were infected with wild-type HSV-1(F) and vJB10. After 24 h, the cells were frozen and thawed three times, and the titer of infectious virus was determined on CV6 or Vero cells as indicated in Table 1. The results indicated that the UL6 deletion virus was unable to produce substantial amounts of infectious virus upon infection of either CV1 or CV6M cells. In contrast, the virus was able to replicate to levels approaching those produced by HSV-1(F) in CV6 cells. Interestingly, CV6M cells were refractory to replication of HSV-1(F) such that titers were reduced approximately 380-fold compared to those of infectious virus obtained from CV1 or CV6 cells. These data suggest that the mutant pUL6 expressed in CV6M cells served in a dominant negative fashion to inhibit wild-type virus replication.
To determine the role of pUL6 amino acids 422 to 443 on the pUL6/pUL15 interaction in infected cells, CV1 cells were infected with HSV-1(F) or vJB10, and infected cell lysates were immunoprecipitated with pUL6- or pUL15-specific antisera. The presence of either pUL6 or pUL15 in immunoprecipitated material was then assessed by immunoblotting. As shown in Fig. 6, the absence of codons 422 to 443 did not preclude expression or solubility of pUL6 but abrogated coimmunoprecipitation with pUL15. This was despite the fact that wild-type pUL6 and pUL15 coexpressed in HSV-1(F)-infected cells readily coimmunoprecipitated. We therefore conclude that UL6 codons 422 to 443 are required for interaction with pUL15 in infected cell lysates.
FIG. 6.
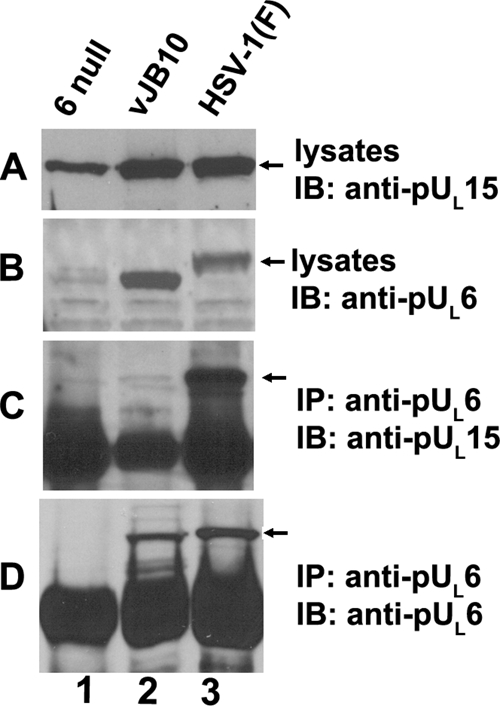
The putative leucine zipper of HSV-1(F) pUL6 is required for the interaction between pUL6 and pUL15 in virus-infected cells. CV1 (2 × 106) cells were infected with a UL6 null mutant, leucine zipper deletion virus (vJB10), or HSV-1(F) at an MOI of 5 PFU per cell. Eighteen hours after infection, cells were lysed in RIPA buffer with 1 M NaCl, and immunoprecipitation was performed with anti-pUL6 antibody. Cell lysates and immunoprecipitated proteins were electrophoretically separated on four separate polyacrylamide gels and immunoblotted with anti-pUL15C or pUL6-specific antibodies. Bound immunoglobulins were revealed as indicated in the legend to Fig. 3. The arrows indicate the positions of proteins of interest. Note that the gels were run for different periods of time, accounting for the different migrations of bands in the different panels. IP, antibody used for immunoprecipitation in the corresponding panel; IB, antibody used for immunoblotting in the corresponding panel.
To determine the effect of the deletion of codons 422 to 443 on viral DNA cleavage, cells were infected with HSV-1(F), vJB10, or vJB10R, a virus derived from vJB10 but containing a UL6 gene restored to wild-type sequences. DNA was purified from the infected cells, digested with BamHI, electrophoretically separated on an agarose gel, transferred to a nylon membrane, denatured, and reacted with a radiolabeled probe bearing end-specific sequences. As noted previously (3), this probe hybridizes to the BamHI junction S-P fragment and the S-terminal BamHI P fragment in BamHI-digested HSV-1(F) DNA; this was also the case in digested vJB10R DNA. In contrast, only the S-P fragment was recognized in digested vJB10 DNA (Fig. 7). Probing the Southern blot with an L-terminal probe produced similar results (data not shown), indicating that neither DNA cleavage event occurs in cells infected with vJB10. As might be expected in the absence of DNA cleavage, we also noted that only capsids lacking DNA were detected in cells infected with the virus lacking UL6 codons 422 to 443 (not shown). We conclude that UL6 codons 422 to 443 are necessary for cleavage of concatameric viral DNA.
FIG. 7.
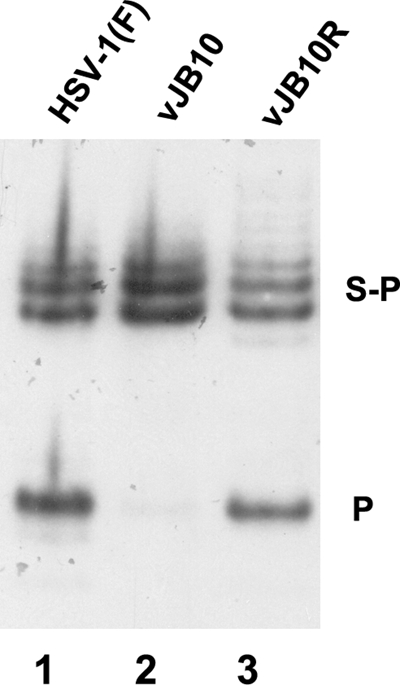
The virus with mutant UL6 lacking the putative leucine zipper is defective in viral DNA cleavage. Approximately 2 × 106 CV1 cells were infected with 5 PFU per cell of HSV-1(F), vJB10, or vJB10R, which was derived from vJB10 but contains a genetically restored UL6 gene. Viral DNA was extracted about 15 hours after infection, digested with BamHI, transferred to a nylon membrane (0.45 μM), and hybridized with a radiolabeled BamHI P fragment of HSV-1(F) DNA. Bound probe DNA was detected by fluorography.
To determine whether the deletion of codons 422 to 443 affected interaction with scaffold proteins, cells were mock infected or infected with the UL6 null virus, vJB10, or HSV-1(F), lysed, and subjected to immunoprecipitation with anti-ICP35 antibody. As shown in Fig. 8, ICP35 was efficiently immunoprecipitated with its cognate antibody from all three infected cell lysates. Immunoblotting of immunoprecipitated material with the pUL6-specific antibody revealed that pUL6 was coimmunoprecipitated with ICP35 from lysates of cells infected with HSV-1(F) and vJB10. We therefore conclude that the defect in DNA cleavage induced by the absence of UL6 codons 422 to 443 was not attributable to a failure of pUL6 to interact with scaffold protein.
FIG. 8.

Deletion of the putative leucine zipper of pUL6 does not block interaction with the scaffold protein ICP35. CV1 cells were infected with 5 PFU per cell of the UL6 null mutant, leucine zipper deletion virus (vJB10), or HSV-1(F). Eighteen hours after infection, the cells were lysed in RIPA buffer and reacted with ICP35-specific monoclonal antibody. Immunoprecipitated proteins were immunoblotted with anti-pUL6 (A) or anti-ICP35 (B) antibodies. Bound immunoglobulins were revealed by reaction with appropriately conjugated immunoglobulins followed by enhanced chemiluminescence.
To determine the effect of codons 422 to 443 on the association of portal protein and pUL15 with capsids, B capsids were purified from cells infected with HSV-1(F), the UL6 null virus, or vJB10. The capsids were then denatured, and associated proteins were electrophoretically separated on SDS-polyacrylamide gels, transferred to nitrocellulose, and subjected to immunoblotting with pUL6-, pUL15-, or VP5-specific antibodies, the latter serving as a loading control. The relative levels of the chemiluminescent signals on the immunoblots were then quantified by densitometry, and the level of signal was normalized to the levels obtained in the lanes containing HSV-1(F) capsid proteins. The results are shown in Fig. 9. Given that other terminases have been shown to interact with both the major capsid protein and portal protein (19), it was not unexpected that some terminase proteins were detected in capsids lacking portals (i.e., those from cells infected with the UL6 null mutant). The deletion in vJB10 did not reduce the incorporation of pUL6 into the capsid discernibly, whereas the level of pUL15 incorporated into vJB10 capsids was reduced by approximately 90%. This level was similar to the levels detected in pUL6 null capsids. We therefore conclude that the deletion of codons 422 to 443 precludes the association of pUL15 with the portal vertex.
FIG. 9.
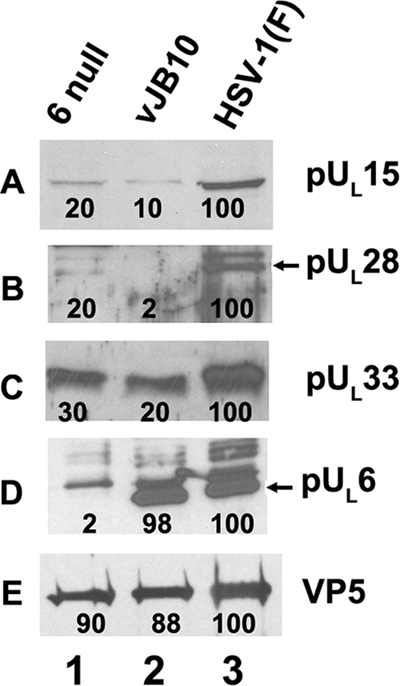
Immunoblot of B capsids probed with pUL15-, pUL6-, pUL33-, pUL28, or VP5-specific antibodies. Approximately 4 × 108 CV1 cells were infected with viruses UL6 null, vJB10, or HSV-1(F) at an MOI of 5 PFU per cell. Twenty hours after infection, capsids were purified, and B capsids were denatured, separated by SDS-polyacrylamide gel electrophoresis, and transferred onto a nitrocellulose membrane, followed by immunoblotting with anti-pUL15C, anti-pUL6, or anti-VP5 antibodies. Bound immunoglobulin was revealed by enhanced chemiluminescence as described in the legend of Fig. 3. The intensity of each band was determined using an LAS-3000 mini Fujifilm imaging system. The intensity of the band on each panel is reported as a percentage of the signal in the lane containing HSV-1(F) proteins and is shown below each band.
To determine whether capsid association of other putative terminase subunits requires UL6 codons 422 to 443, immunoblots of capsids purified from cells infected with HSV-1(F), vJB10, or the UL6 null virus were also probed with antibodies directed against pUL28 and pUL33. The results as shown in Fig. 9 indicated that levels of pUL28 and pUL33 were reduced to various extents in vJB10 mutant capsids compared to those in wild-type capsids.
DISCUSSION
The data presented herein indicate that codons 422 to 443 of UL6 are critical for (i) interaction with pUL15 and pUL28, as assessed by immunoprecipitation, and (ii) the association of all three putative terminase subunits with portal-bearing capsids. This is the first genetic evidence of a domain of pUL6 critical to interaction with putative terminase subunits in infected cells and capsids.
The observations that both pUL15 and pUL28 can interact with pUL6 independently suggest that docking of the terminase with the portal involves interactions between the two larger subunits of the terminase and the portal. This conclusion is consistent with the data of others showing that pUL28 and pUL15 can interact with pUL6 independently (24). Parenthetically, we did not detect an interaction between pUL6 and pUL33 in the absence of other viral proteins as assessed by immunoprecipitation (data not shown).
In the absence of structural data, it is unclear that a leucine zipper is actually encoded by codons 422 to 443 of UL6. Possibly the best arguments for such a motif are the conservation of aligned leucines in a number of herpesvirus portals and the observation that substitution of leucines 429 and 436 with glutamic acids precludes normal portal ring formation and incorporation of the portal into capsids (14). Nevertheless, replacement of the putative leucine zipper with a sequence known to form such motifs in other contexts was insufficient to restore pUL15 or pUL28 binding (Fig. 4 and 5).
Although the structure of pUL6 is unknown, alignment of herpesvirus portal proteins with the solved structures of bacteriophage φ29 and SPP1 portals potentially reveals a structurally conserved region consisting of several α-helices (12). The most C-terminal of these helices (helix 6) is predicted at amino acids 406 to 454 of pUL6 by using PSIPRED version 2.6 (6, 11). Helix 6 is a long, kinked α-helix that emanates roughly perpendicularly to the DNA channel, forming an external ridge or wing in the SPP1 portal (12). In the bacteriophage systems studied, this helix is proposed to interact with other portal subunits through a relatively sparse set of hydrogen bonds rather than through a leucine zipper.
The mutations described here and in previous studies are predicted to affect the stability, charge, register, and/or length of helix 6 as follows: (i) deletion of codons 409 to 473 should completely remove the helix, (ii) deletion of codons 422 to 443 should decrease its length by 7 amino acids and truncate and displace a hydrophobic patch by about 140 degrees relative to that of the wild type motif, (iii) replacement of codons 422 to 443 with the GCN4 leucine zipper should maintain the position of the leucines and hydrophobic patch and lengthen the helix by 9 amino acids, and (iv) changing leucines 429 and 436 to glutamic acids should disperse hydrophobic residues, precluding a prominent hydrophobic region.
Normal levels of portal proteins were incorporated into capsids purified from cells infected with vJB10 (Fig. 9) but were less efficiently incorporated into capsids when (i) codons 409 to 473 were deleted or (ii) leucines 429 and 436 were changed to glutamic acids (14). Given that vJB10 preserves a remnant (albeit displaced) of the hydrophobic region of helix 6, the data suggest that hydrophobic interactions are involved in intersubunit interactions within the HSV-1 portal. On the other hand, the failure of GCN4 to restore terminase binding of the deletion in helix 6 argues against the possibility that subunit interactions involve interdigitation of leucines as in a classic leucine zipper.
As might be expected given the structure of other portals, attempts to demonstrate an interaction between pUL6 amino acids 422 to 443 with either pUL6, pUL28, or pUL15 have not been successful (not shown). Taken together, the data support the idea that this region plays an indirect role in portal/terminase interactions, probably by ensuring proper portal morphology.
Our data also suggest that both amino and carboxyl termini of pUL6 are necessary for interaction with pUL15. Specifically, insertion of epitopic tags into the N or C terminus of pUL6 did not preclude protein solubility, expression of the recombinant proteins, or interaction with ICP35 (not shown), but precluded coimmunoprecipitation with pUL15 in transient expression assays. Moreover, deletion of either the N terminus or C terminus of pUL6 precluded coimmunoprecipitation with pUL15. Little is known about the orientation of pUL6 in the capsid other than the fact that epitopes within the C-terminal 298 amino acids of pUL6 are located at the external surface (15, 21, 25). Moreover, the termini are not homologous to bacteriophage portal proteins and so do not provide clues as to function. Thus, it is uncertain if the pUL6 termini interact directly with pUL15 or act to ensure proper conformation of pUL6 and, thereby, promote interaction with pUL15 indirectly.
The large number of UL6 mutations that can preclude the interaction with pUL15 is striking to us, and we speculate that terminase docking is exquisitely dependent on proper conformation of the portal in the capsid. This is supported by the observation that expression of the defective portal protein acts as a dominant negative inhibitor of wild-type virus (Table 1), presumably because optimal portal conformation cannot tolerate defective subunits. Because the terminase with bound DNA may have a choice of a number of potential capsids with which to dock, this structural constraint to at least the pUL15/pUL6 interaction may represent an important assembly checkpoint to ensure efficient viral DNA cleavage and packaging.
Acknowledgments
We thank Fred Homa for interesting discussions.
These studies were supported by R01 grant GM50741 from the National Institutes of Health.
Footnotes
Published ahead of print on 18 February 2009.
REFERENCES
- 1.Adelman, K., B. Salmon, and J. D. Baines. 2001. Herpes simplex DNA packaging sequences adopt novel structures that are specifically recognized by a component of the cleavage and packaging machinery. Proc. Natl. Acad. Sci. USA 983086-3091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baines, J. D., and C. Duffy. 2006. Nucleocapsid assembly and envelopment of herpes simplex virus, p. 175-204. In R. M. Sandri-Goldin (ed.), Alphaherpesviruses: pathogenesis, molecular biology and infection control. Caister Scientific Press, Norfolk, United Kingdom.
- 3.Baines, J. D., A. P. W. Poon, J. Rovnak, and B. Roizman. 1994. The UL15 gene of herpes simplex virus encodes two proteins and is required for cleavage of viral DNA. J. Virol. 688118-8124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baines, J. D., and S. K. Weller. 2004. Cleavage and packaging of herpes simplex virus 1 DNA, p. 1-16. In C. Catalano (ed.), Viral genome packaging. Kluwer Academic/Plenum, London, United Kingdom.
- 5.Beard, P. M., N. S. Taus, and J. D. Baines. 2002. The DNA cleavage and packaging proteins encoded by genes UL28, UL15, and UL33 of herpes simplex virus 1 form a complex in infected cells. J. Virol. 764785-4791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bryson, K., L. J. McGuffin, R. L. Marsden, J. J. Ward, J. S. Sodhi, and D. T. Jones. 2005. Protein structure prediction servers at University College London. Nucleic Acids Res. 33W36-W38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Davison, A. J. 1992. Channel catfish virus: a new type of herpesvirus. Virology 1869-14. [DOI] [PubMed] [Google Scholar]
- 8.Ejercito, P. M., E. D. Kieff, and B. Roizman. 1968. Characterization of herpes simplex virus strains differing in their effects on social behavior of infected cells. J. Gen. Virol. 2357-364. [DOI] [PubMed] [Google Scholar]
- 9.Huffman, J. B., W. W. Newcomb, J. C. Brown, and F. L. Homa. 2008. Amino acids 143 to 150 of the herpes simplex virus type 1 scaffold protein are required for the formation of portal-containing capsids. J. Virol. 826778-6781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jacobson, J. G., K. Yang, J. D. Baines, and F. L. Homa. 2006. Linker insertion mutations in the herpes simplex virus type 1 UL28 gene: effects on UL28 interaction with UL15 and UL33 and identification of a second-site mutation in the UL15 gene that suppresses a lethal UL28 mutation. J. Virol. 8012312-12323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jones, D. T. 1999. Protein secondary structure prediction based on position-specific scoring matrices. J. Mol. Biol. 292195-202. [DOI] [PubMed] [Google Scholar]
- 12.Lebedev, A. A., M. H. Krause, A. L. Isidro, A. A. Vagin, E. V. Orlova, J. Turner, P. Tavares, and A. A. Antson. 2007. Structural framework for DNA translocation via the viral portal protein. EMBO J. 261984-1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McGeoch, D. J., M. A. Dalrymple, A. J. Davison, A. Dolan, M. C. Frame, D. McNab, L. J. Perry, J. E. Scott, and P. Taylor. 1988. The complete DNA sequence of the long unique region in the genome of herpes simplex virus type 1. J. Gen. Virol. 691531-1574. [DOI] [PubMed] [Google Scholar]
- 14.Nellissery, J. K., R. Szczepaniak, C. Lamberti, and S. K. Weller. 2007. A putative leucine zipper within the herpes simplex virus type 1 UL6 protein is required for portal ring formation. J. Virol. 818868-8877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Newcomb, W. W., R. M. Juhas, D. R. Thomsen, F. L. Homa, A. D. Burch, S. K. Weller, and J. C. Brown. 2001. The UL6 gene product forms the portal for entry of DNA into the herpes simplex virus capsid. J. Virol. 7510923-10932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.O'Shea, E. K., J. D. Klemm, P. S. Kim, and T. Alber. 1991. X-ray structure of the GCN4 leucine zipper, a two-stranded, parallel coiled coil. Science 254539-544. [DOI] [PubMed] [Google Scholar]
- 17.Patel, A. H., F. J. Rixon, C. Cunningham, and A. J. Davison. 1996. Isolation and characterization of herpes simplex virus type 1 mutants defective in the UL6 gene. Virology 217111-123. [DOI] [PubMed] [Google Scholar]
- 18.Singer, G. P., W. W. Newcomb, D. R. Thomsen, F. L. Homa, and J. C. Brown. 2005. Identification of a region in the herpes simplex virus scaffolding protein required for interaction with the portal. J. Virol. 79132-139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sun, S., K. Kondabagil, B. Draper, T. I. Alam, V. D. Bowman, Z. Zhang, S. Hegde, A. Fokine, M. G. Rossmann, and V. B. Rao. 2008. The structure of the phage T4 DNA packaging motor suggests a mechanism dependent on electrostatic forces. Cell 1351251-1262. [DOI] [PubMed] [Google Scholar]
- 20.Tanaka, M., H. Kagawa, Y. Yamanashi, T. Sata, and Y. Kawaguchi. 2003. Construction of an excisable bacterial artificial chromosome containing a full-length infectious clone of herpes simplex virus type 1: viruses reconstituted from the clone exhibit wild-type properties in vitro and in vivo. J. Virol. 771382-1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Taus, N. S., B. Salmon, and J. D. Baines. 1998. The herpes simplex virus 1 UL17 gene is required for localization of capsids and major and minor capsid proteins to intranuclear sites where viral DNA is cleaved and packaged. Virology 252115-125. [DOI] [PubMed] [Google Scholar]
- 22.Tischer, B. K., J. von Einem, B. Kaufer, and N. Osterrieder. 2006. Two-step RED-mediated recombination for versatile high-efficiency markerless DNA manipulation in Eschericia coli. BioTechniques 40191-197. [DOI] [PubMed] [Google Scholar]
- 23.Trus, B. L., N. Cheng, W. W. Newcomb, F. L. Homa, J. C. Brown, and A. C. Steven. 2004. Structure and polymorphism of the UL6 portal protein of herpes simplex virus type 1. J. Virol. 7812668-12671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.White, C. A., N. D. Stow, A. H. Patel, M. Hughes, and V. G. Preston. 2003. Herpes simplex virus type 1 portal protein UL6 interacts with the putative terminase subunits UL15 and UL28. J. Virol. 776351-6358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wills, E., L. Scholtes, and J. D. Baines. 2006. Herpes simplex virus 1 DNA packaging proteins encoded by UL6, UL15, UL17, UL28, and UL33 are located on the external surface of the viral capsid. J. Virol. 8010894-10899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang, K., and J. D. Baines. 2006. The putative terminase subunit of herpes simplex virus 1 encoded by UL28 is necessary and sufficient to mediate interaction between pUL15 and pUL33. J. Virol. 805733-5739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang, K., F. Homa, and J. D. Baines. 2007. Putative terminase subunits of herpes simplex virus 1 form a complex in the cytoplasm and interact with portal protein in the nucleus. J. Virol. 816419-6433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yang, K., and J. D. Baines. 2008. Domain within herpes simplex virus 1 scaffold proteins required for interaction with portal protein in infected cells and incorporation of the portal vertex into capsids. J. Virol. 825021-5030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yu, D., and S. K. Weller. 1998. Genetic analysis of the UL 15 gene locus for the putative terminase of herpes simplex virus type 1. Virology 24332-44. [DOI] [PubMed] [Google Scholar]
- 30.Yu, D., and S. K. Weller. 1998. Herpes simplex virus type 1 cleavage and packaging proteins UL15 and UL28 are associated with B but not C capsids during packaging. J. Virol. 727428-7439. [DOI] [PMC free article] [PubMed] [Google Scholar]