

Abstract
Mutation sites that arise in human mitochondrial DNA as a result of oxidation by a rhodium photooxidant have been identified. HeLa cells were incubated with [Rh(phi)2bpy]Cl3 (phi = 9,10-phenanthrenequinone diimine), an intercalating photooxidant, to allow the complex to enter the cell and bind mitochondrial DNA. Photoexcitation of DNA-bound [Rh(phi)2bpy]3+ can promote the oxidation of guanine from a distance through DNA-mediated charge transport. After two rounds of photolysis and growth of cells incubated with the rhodium complex, DNA mutations in a portion of the mitochondrial genome were assessed via manual sequencing. The mutational pattern is consistent with dG to dT transversions in the repetitive guanine tracts. Significantly, the mutational pattern found overlaps oxidative damage hot spots seen previously. These mutations are found within conserved sequence block II, a critical regulatory element involved in DNA replication, and these have been identified as sites of low oxidation potential to which oxidative damage is funneled. Based upon this mutational analysis and its correspondence to sites of long range oxidative damage, we infer a critical role for DNA charge transport in generating these mutations and, thus, in regulating mitochondrial DNA replication under oxidative stress.
INTRODUCTION
To protect cells from oxidative damage, a variety of repair processes and pathways must be activated (1-3). Mitochondria are particularly susceptible because there may be increased levels of toxic oxidants that are byproducts of oxidative phosphorylation within the mitochondrion (4,5). These oxygen species react with mitochondrial DNA and promote mutations (6). The most well studied modification to DNA is the oxidation of deoxyguanosine to 8-oxodeoxyguanosine (8-oxodG), but 23 other modified bases have been characterized (7). It has been observed that the 8-oxodG lesion leads to dG to dT transversions (8); base oxidation can also direct insertions and deletions, especially at microsatellites (9, 10).
Here we examine mutations generated in mitochondria using the one-electron photooxidant [Rh(phi)2bpy]3+ (phi = 9,10-phenanthrenequinone diimine). This complex binds to DNA by intercalation with little sequence-specificity, and, when photoexcited at high energy (313-325 nm), direct strand cleavage by the DNA-bound Rh occurs, marking the site of intercalator binding. Irradiation at longer wavelength (≥ 350 nm), however, promotes the oxidation of guanines to yield common irreversible products including 8-oxodG (11). The guanines oxidized need not, however, be localized to the Rh binding site, but instead oxidative damage to guanine doublets and triplets, sites of low oxidation potential in DNA, can be promoted from a distance through DNA-mediated charge transport. When the Rh complex is tethered to the end of a synthetic DNA oligonucleotide, oxidative damage has been observed ~200 Å from the site of Rh intercalation (12,13). Indeed, varied photooxidants have been found to react with DNA to promote oxidative damage to guanines from a distance (14). Holes injected into the DNA base pair stack can migrate through the DNA base stack to low oxidation potential sites, like the 5’-G of guanine doublets, to form guanine radicals that react with oxygen and water to form permanent DNA lesions that require repair (15,16).
Figure 1 illustrates the strategy we have utilized to examine mutations derived through oxidative damage. To promote oxidative stress in cells and specifically oxidative damage to DNA, we incubate cells with [Rh(phi)2bpy]3+ and irradiate. Under these oxidative conditions, some cells die, others undergo mutation, and many are left undamaged. Several rounds of Rh incubation, irradiation, and regrowth can, however, lead to enrichment of the cell population in mutants. These mutations can then be assayed by sequencing. It should be noted that the tris(chelate) metal complexes are readily taken up inside cells, with the more lipophilic cationic complexes taken up most efficiently (17). Rh complexes have been utilized to promote 1-electron DNA oxidation with photoactivation in a variety of cell studies (16, 18, 19).
Figure 1.
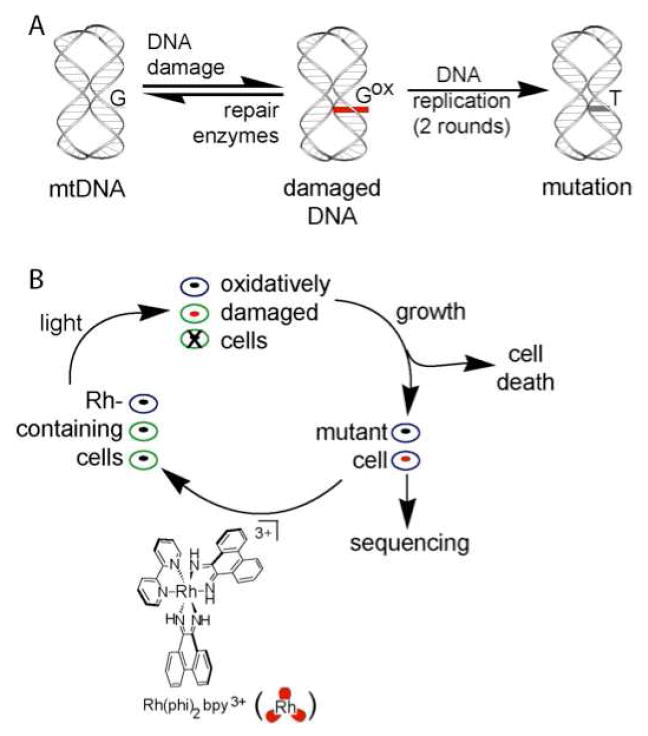
Strategy for inducing mutations in HeLa cell mitochondria. (A) Mitochondrial DNA under elevated oxidative stress yields some oxidative damage to the mitochondrial genomes (red). If unrepaired, modification of a dG leads to a mutation, here a dG to dT conversion. (B) In this study HeLa cells are incubated with [Rh(phi)2bpy]3+ (bottom), a non-specific DNA intercalator that, upon irradiation, induces DNA oxidative damage. Cells are either unmodified (blue), modified (green and red), or dead (x on cell) after exposure to Rh and light. After growing the cells, mutations can arise in the modified cells. Several cycles of oxidative damage and regrowth lead to the accumulation of mutations that can be identified by sequencing.
Most studies have shown that mutations within the mitochondria of tumors are not randomly distributed but occur predominately in a sequence of the genome termed the control region or d-loop (20). Nucleotides 303-315 within the control region (sequence 3’-G7AG5) are especially prone to insertions and deletions as well as dG to dT mutations (21). Nucleotides 303-315 are termed conserved sequence block II, a regulatory element that is involved in primer formation for mitochondrial DNA replication (22,23). Importantly, base oxidation arising from DNA charge transport has been shown both in vitro and in HeLa cells to damage predominately this conserved sequence block II (24,25). We hypothesize that, through DNA charge transport, damage is funneled to the regulatory element, so as to hinder replication of a damaged mitochondrion genome. While oxidative damage within conserved sequence block II, generated through DNA charge transport, has been documented, here we shown that this damage can lead to mitochondrial mutations. Moreover, we show that these mutations overlap with known mutations associated with cancers.
EXPERIMENTAL
Methods
All experiments were carried out using newly thawed HeLa cells. HeLa cells (Sigma, USA) were raised to ~75% confluence. Total DNA was purified using the DNAeasy kit (Qiagen, U.S.A.) and eluted in 2.5 mM Tris, pH 8.5. PCR was used to amplify a 1.5 kb DNA oligonucleotide that contained the control region using appropriate primers (24). Correct amplification was verified using restriction digest.
Damage to a PCR product with [Rh(phi)2bpy]3+
Damage to a PCR product containing the control region by [Rh(phi)2bpy]3+ was accomplished using 50 ng/μL of DNA in 50 μL (5 mM Tris, 25 mM NaCl, pH 8) and addition of various concentrations of [Rh(phi)2bpy]Cl3. Samples were irradiated at 365 nm for 15 min with a 1000 W Hg/Xe lamp outfitted with a 320 long-pass filter, then incubated with 99% piperidine [5 μL, 10 min, 95 °C], cooled, and 5 μL glacial acetic acid was added. The sample was prepared for primer extension by purification with a PCR purification kit (Qiaquick, Qiagen) to give 40 μL samples. A portion (30 μL) of the sample was then mixed with 2X reaction mix [2X Taq Buffer, 4 mM MgCl2, 200 μM dNTP, 0.25 μM 32P-endlabelled primer, and 0.3 U/μL Platinum Taq]. The sample was heated to 95 °C for 2.5 min. and cycled 40 times for primer extension [55 °C for 40 sec, 72 °C for 40 sec, 95 °C for 30 sec]. Samples were precipitated, and 6% denaturing PAGE was performed. Gels were visualized by overnight exposure to a phosphorimager screen and scanned. Data were quantified using the line function in ImageQuant (Amersham) and normalized in Excel.
Damage to DNA in HeLa cells to generate mutations with [Rh(phi)2bpy]3+
Fresh HeLa cells were raised to ~75% confluence, 2 million total cells. Cells were trypsinized, washed once with minimal media, pelleted, then resuspended in cold PBS and plated. Various [Rh(phi)2bpy]Cl3 concentrations in 2 mL PBS were added to the cells that were adherent to the bottom of a 75 mL (25 cm2 surface area) cell culture flask. Cells were irradiated at 37 °C for 20 minutes with a 1000 W Orion Solar Simulator (with a UVB/UVC blocking filter and a glass filter that eliminates wavelengths <345 nm) for 20 minutes. The Rh complex was then removed by washing with PBS, and 50,000 cells were plated and allowed to grow. The procedure was repeated four days later when cells had reached ~50% confluence. Following the second round of photolysis, cells were again allowed to grow to ~50% confluence, and total DNA was purified using the DNeasy kit (Qiagen, U.S.A.) and eluted from the column in 2.5 mM Tris, pH 8.5. The DNA was amplified by the addition of 20 μL of 2X reaction mix [2X Taq buffer, 4 mM MgCl2, 400 μM dNTP, 1 μM primer, and 10 U Taq] to 20 μL each DNA sample. The samples were heated to 95 °C for 2.5 minutes and cycled 40 times for primer extension [55 °C for 40 seconds, 72 °C for 40 seconds, 95 °C for 30 seconds]. After purification with the PCR Purification Kit (Qiagen, USA), sequencing reactions were accomplished by addition of a terminator dNTP [1X Vent Buffer, 400 μM dNTP, 150 μM acyclonucleotide (New England Biolabs), 5 μM 32P-endlabelled primer, 10 U Vent(exo-)] and cycled as listed above. Samples were precipitated and electrophoresed on a 6% polyacrylamide gel. Gels were visualized by overnight exposure to a phosphorimager screen and scanned. To visualize DNA damage in an agarose gel, DNA was collected as above, loading dye was added (20% glycerol, bromophenol blue, 1/10 volume), and electrophoresis was accomplished using ethiduim bromide under standard conditions. Fluorescence was captured by a digital camera outfitted with a uv-blocking filter and saved as tiff files. These files were quantified using Imagequant as previously described (24,25). All experiments have been carried out at least in duplicate.
RESULTS
[Rh(phi)2bpy]3+-induced oxidative DNA damage
Experiments to identify oxidative damage sites near conserved sequence block II were conducted. Total DNA was collected from HeLa cells and amplified by PCR to give a 1.5 kilobase product that corresponds to the control region in human mitochondria. Primer extension was carried out using a primer that begins at nucleotide 191 of the mitochondrial genome, allowing for visualization of conserved sequence block II (24). It should be noted that extension of a PCR copy of the control region leads to a slightly different set of primer extension products than with mitochondrial DNA purified from cells, examined previously (25). Notably, no primer extension stops are visualized within conserved sequence block II (Figure 2). These extension stops in purified mitochondrial DNA are replication intermediates (26), triplex in nature, and are removed during PCR. Primer extension from the control region PCR product yields ~7-fold higher signal.
Figure 2.
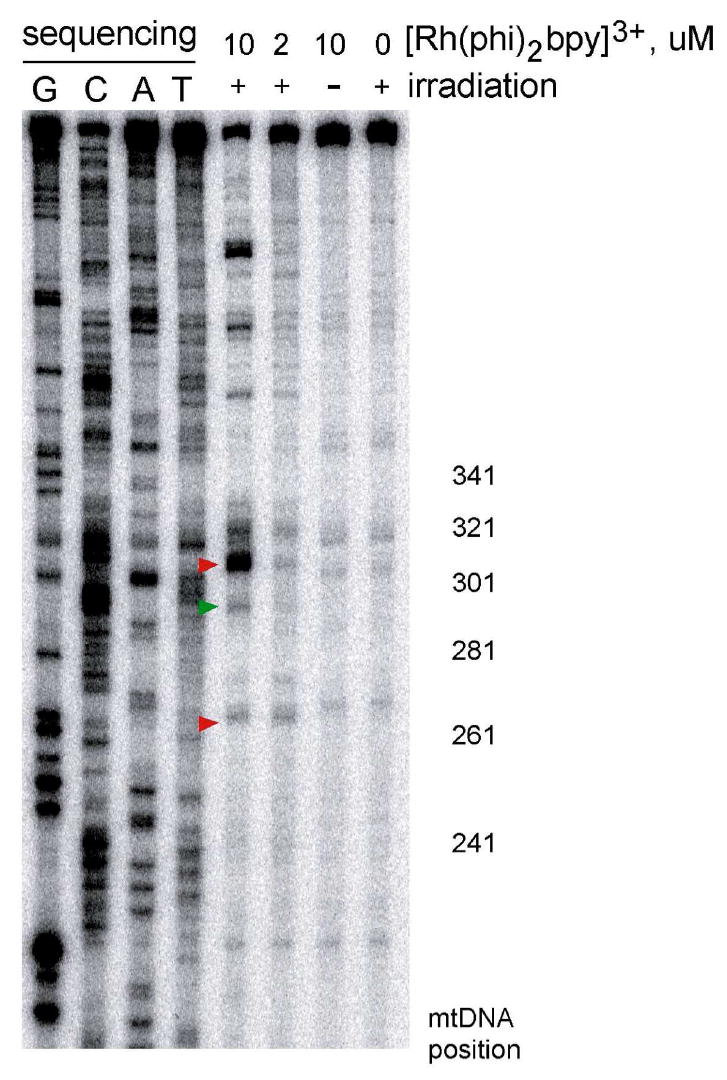
[Rh(phi)2bpy]3+-induced oxidative damage to the PCR-amplified mitochondrial DNA control region. A 1.5 kilobase copy of the control region of mitochondrial DNA from HeLa cells was prepared using PCR. After treatment with the [Rh(phi)2bpy]3+ photooxidant (10 or 2 μM) and incubation with piperidine, a primer extension reaction was used to visualize the DNA damage via PAGE. Shown are sequencing lanes along with DNA treated with 10, 2 μM, or 0 Rh with or without irradiation as indicated. The nucleotide positions within the control region are shown to the right. Addition of [Rh(phi)2bpy]3+ or irradiation alone do not produce oxidative damage as only large, unmodified DNA is observed. Addition of [Rh(phi)2bpy]3+, irradiation, and piperidine treatment leads to oxidative damage at nucleotides 303, 290, and 263. The most intense damage is at position 303, within conserved sequence block II. Modification at positions 303 and 263 are the result of DNA charge transport (red), damage at position 290 is at a chemically accessible site of high AT content (green).
Sites of oxidation on the control region PCR product were identified using 2.5 μg samples to which 10 or 2 μM [Rh(phi)2bpy]3+ was added. Irradiation at ~365 nm in the presence of DNA-bound Rh leads to base oxidation, which can be revealed as a strand break by treatment of the DNA with piperidine (11,12); after purification, a primer extension reaction is used to visualize the DNA damage pattern. Irradiation at higher energy in the presence of Rh but no piperidine treatment marks sites of Rh binding through direct strand cleavage. Sequencing is then used to identify the location of damage. With high energy irradiation and no piperidine treatment, low level damage is obtained across the DNA, reflecting non-specific binding of the Rh intercalator, and consistent with that seen earlier on mitochondrial DNA irradiated with Rh or after irradiation of mitochondria in the presence of Rh (24,25). Addition of [Rh(phi)2bpy]3+ without irradiation or irradiation of the DNA without [Rh(phi)2bpy]3+ and piperidine treatment shows a large, slowly migrating band that corresponds to the full length primer extension product since no base oxidation occurs (Figure 2). Addition of [Rh(phi)2bpy]3+ and irradiation leads to a series of stops at locations of base oxidation. With piperidine treatment, the strongest stop is located at nucleotides 303-315, reflecting base oxidation at this site (Figure 2). These data are fully consistent with previous studies on mitochondrial DNA purified from cells that indicate conserved sequence block II is a particularly low oxidation potential site within the mitochondrial genome (24,25). Though Rh binding is evident across the DNA (based upon higher energy direct strand cleavage), base oxidation promoted by Rh is evident preferentially in conserved sequence block II. Oxidative damage may thus be funneled to this site by DNA charge transport. Position 263 is also observed as a low oxidation potential site and has been previously identified as a site in purified mitochondrial DNA to which damage may also be funneled (25). Damage at positions 286-291 was identified previously as a site that is preferentially reactive to various chemical modifications (although direct strand cleavage with Rh is not observed) (24). Some Rh-specific damage is evident also at positions above 340, but the exact locations have not been sequenced.
Assessing mitochondrial DNA damage in HeLa cells by [Rh(phi)2bpy]3+
HeLa cells (approximately 2×106 in each cell culture flask) were grown to confluence, dosed with various concentrations of [Rh(phi)2bpy]3+ for 1 h, and then irradiated. After irradiation, total DNA was collected from 1×106 cells and visualized on a 0.75% agarose gel stained with ethidium bromide. As illustrated in Figure 3, when oxidation occurs, DNA is fragmented into smaller random sizes. These smaller DNA fragments are below the detection limit of ethidium bromide staining and, therefore, only a loss in the slow-moving, undamaged DNA band can be observed as an indicator of the extent of oxidative damage in the HeLa cells. A 20 minute irradiation at ~365 nm with no Rh added leads to less than 5% change in the DNA band. At 7 μM [Rh(phi)2bpy]3+, the high molecular weight DNA band loses 36% of its intensity, while at 30 μM Rh, 75% is lost. Therefore, extensive DNA damage occurs with incubation of [Rh(phi)2bpy]3+and irradiation.
Figure 3.
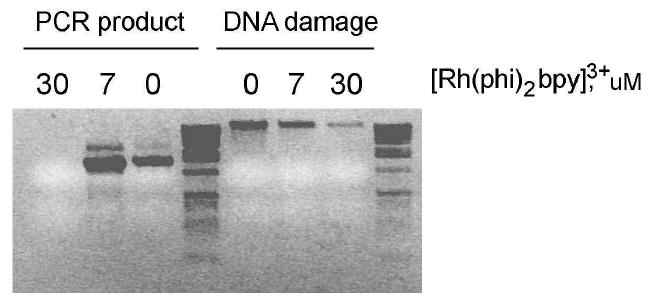
Agarose gel detection of global DNA damage in HeLa cells. Addition of [Rh(phi)2bpy]3+ to HeLa cells and irradiation leads to DNA damage that is visualized as a loss of double-stranded, high molecular weight DNA. Shown are amplified DNA samples after incubation of cells with or without Rh (0, 7, 30 μM) and irradiation for 20 min. The amount of DNA damage is sensitive to the amount of photooxidant; 30 μM [Rh(phi)2bpy]3+ causes more loss of unmodified DNA than 7 μM [Rh(phi)2bpy]3+. Cells from these samples were grown for another 24 hours and the mitochondrial control region was PCR-amplified. At 30 μM [Rh(phi)2bpy]3+, no amplifiable DNA is seen.
The viability of cells after [Rh(phi)2bpy]3+-induced oxidation was also examined. From the samples above, 50,000 cells were plated and grown for an additional day. Cells irradiated without Rh or with 7 μM Rh yielded significant numbers of adherent cells, and new cells could be seen in the microscope. In contrast, treatment with 30 μM Rh and irradiation showed limited numbers of adherent cells, ruptured cellular debris, and few new cells visible. DNA from the adherent cells in all three cases was extracted, the control region of mitochondrial DNA was amplified using PCR, and the product also visualized by agarose gel electrophoresis (Figure 3). With 30 μM Rh irradiation, no DNA was detectable with PCR amplification. We therefore utilized 7 μM Rh and 20 minutes of irradiation in subsequent experiments; limited cell growth occurred after too much DNA damage.
Inducing mitochondrial DNA mutations in HeLa cells
HeLa cells from a single thawed stock were then grown in three cell culture flasks. Once 50% confluence was reached, cells from each flask were split into 50,000 and one million cell samples. The one million cells were used for sequencing by extracting total DNA, PCR-amplifying the control region, and nucleotides surrounding conserved sequence block II were sequenced using an appropriate 32P-labeled primer. The sequencing results are labeled as 0 in Figure 4. Each set of 50,000 cells were plated and allowed to grow to 50% confluence. In one cell culture flask, 7 μM Rh was added but not irradiated, the second flask was irradiated with ~365 nm light but no Rh, and, in the third flask, 7 μM Rh was added and the flask was irradiated to promote DNA damage. Each flask was then split into two flasks by adding 50,000 cells to fresh media and grown to 50% confluence. One flask for each sample was used for sequencing (this set is labeled 1 in Figure 4), while a second flask of cells was used to repeat the experiment (labeled 2 in Figure 4, i.e. these cells have been oxidized twice). An important consideration in these experiments is the number of cells to be plated after Rh-induced DNA damage. A condition with too many cells leads to mutants not present in a high enough percentage for definitive assignment, while with too few cells, the chance of finding a mutant is low.
Figure 4.
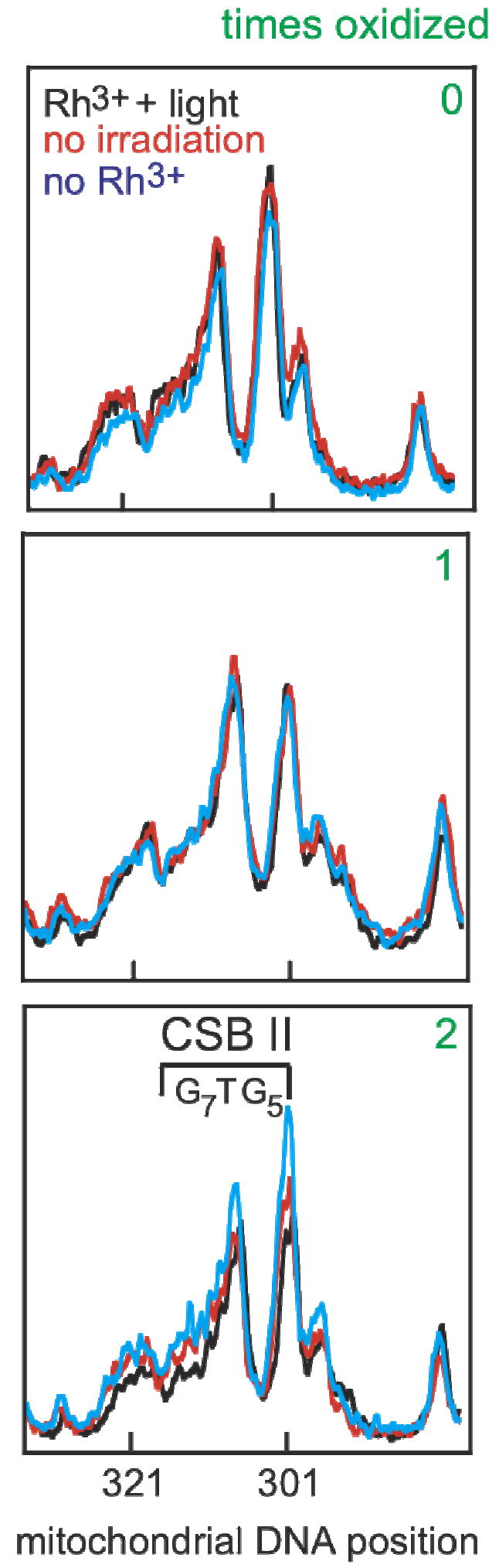
Guanine sequencing of conserved sequence block II over several cycles of oxidation and regrowth. Cellular DNA was extracted, the mitochondrial control region was amplified, and sequencing was accomplished using a 32P-labeled primer and dC terminator nucleotide. The results shown are line plots of the PAGE gels from nucleotides 284-330. Conserved sequence block II (303-315, 3’-G7AG5, marked as CSB II) is a region comprised of many guanosine residues and is prone to oxidation. A reference dG, that is not within conserved sequence block II, at position 285 is on the right of each plot. At first, cells are genetically identical, and sequencing lanes overlap (0). Cells were treated with [Rh(phi)2bpy]3+ only (no irradiation, red), only irradiated with ~365 nm light (no Rh, blue), or given Rh and irradiated (Rh+light, black) and allowed to grow. Little, if any, change in the dG content occurs in conserved sequence block II in all three cases after one cycle (labeled as 1). The same cells were treated again (labeled as 2). After two cycles, cells treated with [Rh(phi)2bpy]3+ only (red) and only irradiated (blue) have very similar dG content in conserved sequence block II. In contrast, incubation of cells with [Rh(phi)2bpy]3+ and irradiation (black) shows a decrease in the dG content in conserved sequence block II. The lack of change at position 285 with Rh and light indicates the loss of dG content is specific to conserved sequence block II.
Figure 4 shows line plots of guanosine sequencing results for nucleotides 284-330 from DNA amplified with or without oxidative damage. These plots show conserved sequence block II to the left and a nearby guanine 285 to the right for comparison (Figure 4). As expected, sequencing line plots overlap when nothing has been done to the HeLa cells (labeled 0 in Figure 4), as these HeLa cells are genetically identical. Cells that were irradiated without Rh show no changes in either position 285 or in positions 303-315, conserved sequence block II, after one and two irradiations/growth cycles. In contrast, cells treated with [Rh(phi)2bpy]3+ and irradiated show a decrease in guanine content within conserved sequence block II after two rounds of oxidation and growth (compare plots on Figure 4 bottom after 2 cycles). Importantly, position 285 shows little if any decrease after 0, 1, or 2 cycles of oxidation and growth, indicating that the decrease in guanosine content is localized to the oxidation-prone regions to which damage is funneled and does not reflect a global decrease in DNA yield. Analyzing guanosine content at various positions within conserved sequence block II gives a variable decrease in guanine content from as much a 42% to as little as 9% measured from peak heights. Cells given Rh but not irradiated show a much smaller decrease in the guanosine content, presumably, from ambient light absorbed during workup. The change with Rh addition and irradiation is therefore significant. Mutations in oxidation hot spots are detectable after two cycles of oxidation and growth.
Determining Specific Mitochondrial DNA Mutations
Another set of fresh cells was examined to identify the specific types of mutations that arise upon Rh-induced DNA damage (Figure 5). Sequencing using the four terminating nucleotides uncovers all types of mutation that occur. Cells were either treated with [Rh(phi)2bpy]3+ and irradiated or treated with Rh without light as a control. Again, mutations become apparent after two rounds of [Rh(phi)2bpy]3+ incubation and irradiation, while no mutations are seen with cells not irradiated but treated with Rh (no DNA damage, Figure 5). Note that the primer is the complement of the strand being interrogated. When visualizing 175 nucleotides of the control region, mutations are evident at only four sites. The locations of mutations found are at positions 303-315, 286-291, 263, and 208-220. Interestingly, all positions have been identified as nucleotides susceptible to Rh-induced DNA damage (24, 25 and Figure 2).
Figure 5.
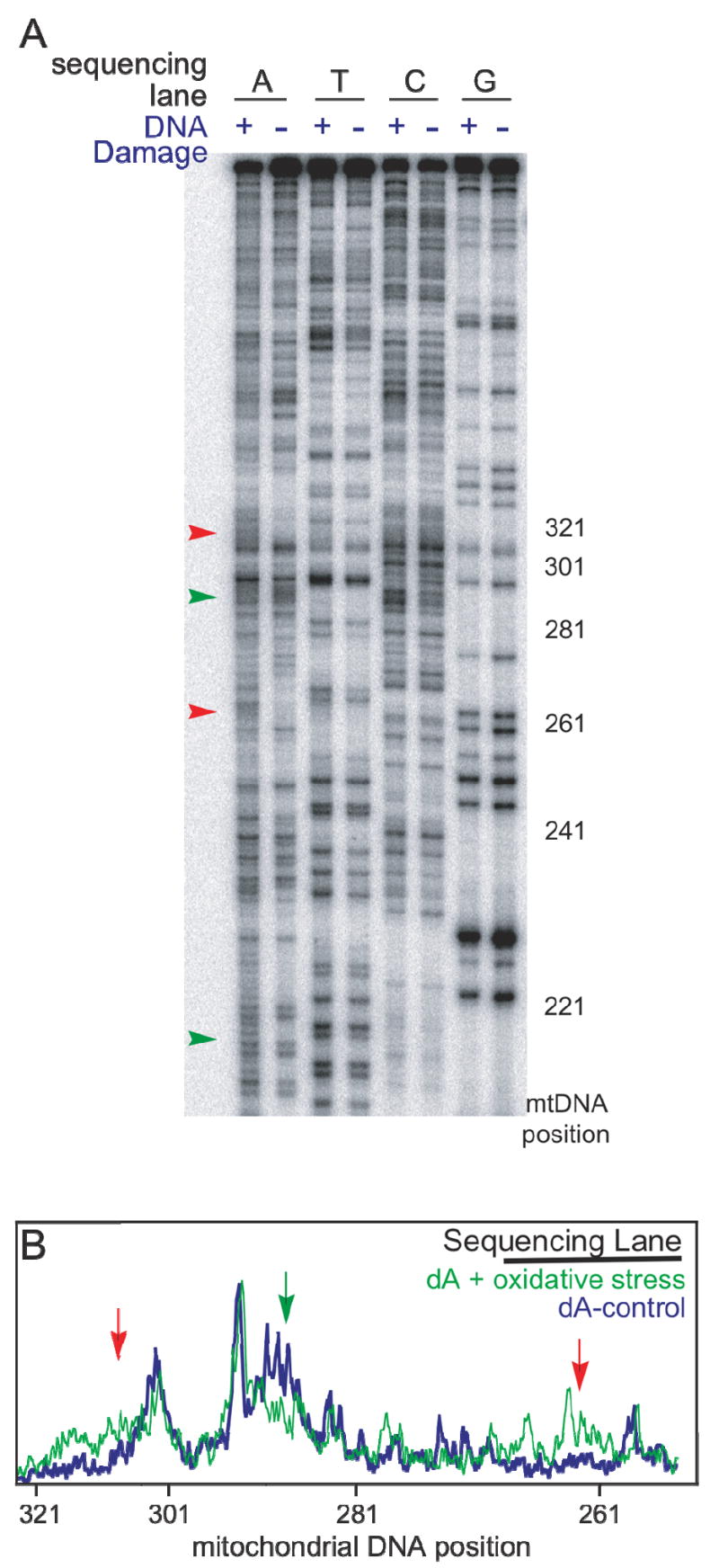
Mutations after Rh-induced DNA damage in HeLa cells. (A) Sequencing PAGE gel using all four terminator nucleotides after two cycles of Rh-induced oxidative DNA damage and growth. HeLa cells that were irradiated and treated with [Rh(phi)2bpy]3+ (DNA Damage plus) show mutations observed as changes in the sequencing lanes (arrows) as compared to cells that where incubated with Rh and not irradiated (DNA Damage minus). The mutations associated with DNA charge transport (red) are consistent with conversion of dG to dT or deletion of dG. Other mutations are also observed (green). (B) Line plot of the first two lanes of the same gel. Note that it is the complementary strand that is sequenced, so that damage associated with a G to T transversion yields an increase in the complementary A.
In particular, both positions 303-315 and 263 are prone to oxidative damage via DNA charge transport. Here, positions 303-315 show evidence of dG to dT transversions, seen as an increase of 28% in dA content calculated from integration in the amount of dA over the 12 bases. Notably the increase in dA content is specific to cells that have been through two rounds of Rh-induced DNA damage and growth. A corresponding loss of dC is also found, although more blurry, and as we would expect with either dG to dT transversions or deletions of dG (Figure 5). While the dG to dT transversions are supported by the increase in dA content, deletions are also suggested, since the increase in intensity (transversion mutations) observed in Figure 5B (green plot) is uniform while the loss of dG (Figure 4) is non-uniform. These observations would be consistent with deletions along with dG to dT transversions within conserved sequence block II. Changes are also evident at position 263, consistent with mutation from a dG to a dT. We also found mutations at nucleotides 286-291 and 208-220, which are flexible sequences (24) of T5 and (AATT)3 and are highly susceptible to mutation, particularly with irradiation. We find at position 286-291 deletions or mutation from a dT to a dG (green arrow, Figure 5), while positions 208-220 are mutated from a dA to a dT. These positions are damaged by a mechanism that is not associated with DNA oxidation. Importantly, nucleotides 263 and 303-315 are known as mutational hot spots in cancers (24). Oxidative DNA damage through DNA charge transport thus appears to lead to mutations that are characteristically found in tumors.
DISCUSSION
Hot spots for oxidative damage are present within the control region of the mitochondrial genome and specifically within the highly conserved sequence block II. We have seen earlier that damage is preferentially funneled to this region through DNA charge transport (24,25). Here we see that long range damage in conserved sequence block II can also promote mutations. Specifically, we find dG to dT transversions within the guanine-rich tracts of conserved sequence block II. These mutations would be expected with oxidation of guanines to 8-oxodG; other guanine oxidation products could also occur. In fact, we have identified 8-oxodG as one product of long range oxidative damage in Rh-tethered oligonucleotides (11,27). Importantly, these lesions are commonly found associated with oxidative stress (5-7). Perhaps even more interesting, however, is the role that DNA-mediated charge transport may play within the mitochondrion. We observe that damage to this region of the mitochondrial genome and therefore the mutations that occur as a result of this damage arise from a distance through DNA charge transport. Indeed, we may consider now that DNA charge transport actually serves to direct mutations to this region of the mitochondrial genome.
The preferential funneling of damage in the mitochondrion is seen to provide a checkpoint for damage and oxidative stress within the cell. How that checkpoint may effectively operate or become dysfunctional as a result of mutation is illustrated in Figure 6. Under oxidative stress, damage to the mitochondrial genome occurs, and it is directed to conserved sequence block II through DNA-mediated charge transport, leading to the formation of base lesions, like 8-oxodG. Each mitochondrion has several mitochondrial DNA genomes, some of which may be oxidatively modified while others remain undamaged. Though elements of mitochondrial DNA replication remain unknown and controversial (22,23), replication will either be inhibited or surely altered by DNA modification within the sequence block. We observe mutations consistent with the formation of the 8-oxodG lesion within this region, but other oxidative lesions associated with guanine oxidation have also been identified, and these too may inhibit and/or alter replication (28). Likely then, because conserved sequence block II represents both the hot spot for damage and the replication control site, damage in this region should lead to the selective replication of unmodified genomes.
Figure 6.
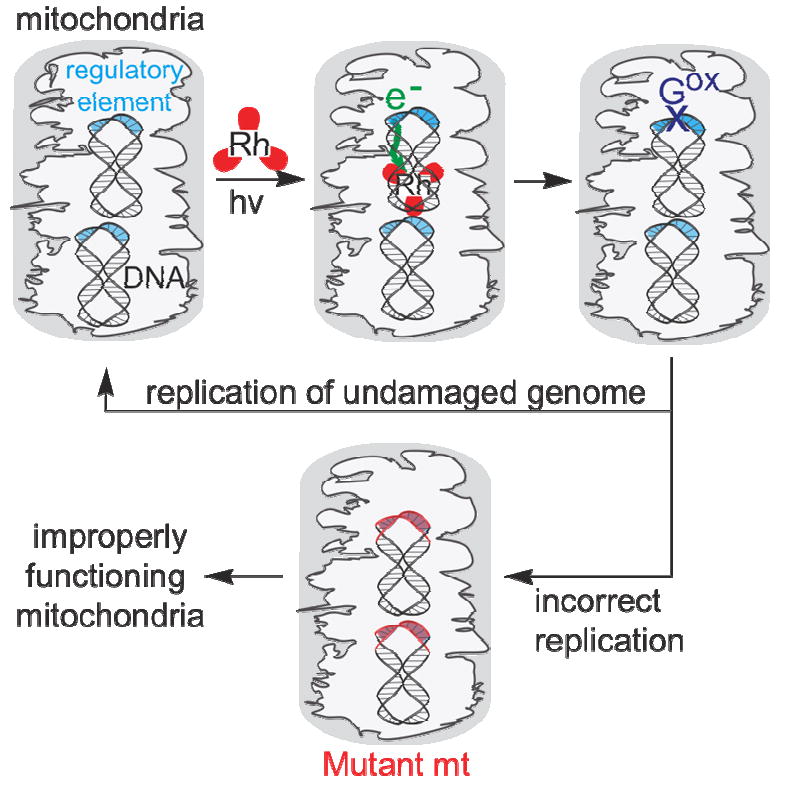
Schematic illustration how DNA charge transport may promote damage and mutations that regulate mitochondrial replication. Mitochondria contain several genomes. Under oxidative stress some genomes are damaged, and through DNA charge transport, this damage is preferentially funneled to conserved sequence block II (green arrow). This funneling of damage to conserved sequence block II can serve as a damage checkpoint, limiting the replication of damaged mitochondrial genomes. Some damaged mitochondria can survive, however, and here, mutations occur, as we have seen. These mutations may lead to improperly functioning mitochondria and cancerous transformation.
Nonetheless, replication of damaged DNA can occur, albeit with low probability, and replication of oxidized guanine lesions leads to mutation, specifically dG to dT transversions and insertions/deletions within the guanine tract. The results described here identify these specific mutations within conserved sequence block II and that these mutations arise through the funneling of oxidative damage to these sites. Incubation of cells with [Rh(phi)2bpy]3+, a potent one-electron photooxidant that intercalates in DNA, with two cycles of irradiation and cell growth, promotes specific mutation at the sites where Rh-induced damage on the mitochondrial DNA has been identified.
Perhaps more remarkable is the finding that these specific mutations correspond to those commonly associated with cancers. Such mutations certainly would hinder mitochondrial function. Moreover, the loss of guanine content within the conserved sequence block would mean that this region would no longer serve as a low oxidation potential checkpoint to limit replication of damaged genomes. Thus, mutations would further propogate, increasingly limiting mitochondrial function. In fact, such mutations would be expected to provide an advantage to tumor cells that utilize glycolysis preferentially for energy production. Therefore, while mutation of conserved sequence block II itself may not be pathogenic, it may impart to a tumor a higher chance to survive under hypoxic conditions.
These studies provide another example of how DNA-mediated charge transport may serve a useful role within the cell (16). By funneling damage to specific control sites, the signal for oxidative stress to the mitochondrial genome is transmitted. If replication proceeds despite this signaling, mutations arise. Indeed, these mutations are characteristically associated with cancerous transformations.
Footnotes
We are grateful to the NIH (GM49216) for their financial support of this research including a minority postdoctoral fellowship to E. J. M.
References
- 1.Hoeijmakers JHJ. Genome maintenance mechanisms for preventing cancer. Nature. 2001;411:366–374. doi: 10.1038/35077232. [DOI] [PubMed] [Google Scholar]
- 2.Jones PA, Laird PW. Cancer epigenetics comes of age. Nat Genet. 1999;21:163–167. doi: 10.1038/5947. [DOI] [PubMed] [Google Scholar]
- 3.Polyak K, Xia Y, Zweier JL, Kinzler KW, Vogelstein B. A model for p53-induced apoptosis. Nature. 1997;389:300–305. doi: 10.1038/38525. [DOI] [PubMed] [Google Scholar]
- 4.Wallace DC. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: A dawn for evolutionary medicine. Annu Rev Genet. 2005;39:359–407. doi: 10.1146/annurev.genet.39.110304.095751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cadenas E, Davies KJA. Mitochondrial free radical generation, oxidative stress, and aging. Free Radic Biol Med. 2000;29:222–230. doi: 10.1016/s0891-5849(00)00317-8. [DOI] [PubMed] [Google Scholar]
- 6.Nakamoto H, Kaneko T, Tahara S, Hayashi E, Naito H, Radak Z, Goto S. Regular exercise reduces 8-oxodG in the nuclear and mitochondrial DNA and modulates the DNA repair activity in the liver of old rats. Exp Gerontol. 2007;42:287–295. doi: 10.1016/j.exger.2006.11.006. [DOI] [PubMed] [Google Scholar]
- 7.Cooke MS, Evans MD, Dizdaroglu M, Lunec J. Oxidative DNA damage: mechanisms, mutation, and disease. FASEB J. 2003;17:1195–1214. doi: 10.1096/fj.02-0752rev. [DOI] [PubMed] [Google Scholar]
- 8.Hsu GW, Ober M, Carell T, Beese LS. Error-prone replication of oxidatively damaged DNA by a high-fidelity DNA polymerase. Nature. 2004;431:217–221. doi: 10.1038/nature02908. [DOI] [PubMed] [Google Scholar]
- 9.Slebos RJC, Li M, Vadivelu S, Burkey BB, Netterville JL, Sinard R, Gilbert J, Murphy B, Chung CH, Shyr Y, Yarbrough WG. Microsatellite mutations in buccal cells are associated with aging and head and neck carcinoma. Br J Cancer. 2008;98:619–626. doi: 10.1038/sj.bjc.6604198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jackson AL, Chen R, Loeb LA. Induction of microsatellite instability by oxidative DNA damage. Proc Natl Acad Sci U S A. 1998;95:12468–12473. doi: 10.1073/pnas.95.21.12468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hall DB, Holmlin RE, Barton JK. Oxidative DNA damage through long-range electron transfer. Nature. 1996;382:731–735. doi: 10.1038/382731a0. [DOI] [PubMed] [Google Scholar]
- 12.Nunez ME, Hall DB, Barton JK. Long-range oxidative damage to DNA: effects of distance and sequence. Chem Biol. 1998;6:85–97. doi: 10.1016/S1074-5521(99)80005-2. [DOI] [PubMed] [Google Scholar]
- 13.Henderson PT, Jones D, Hampikian G, Kan Y, Schuster GB. Long-distance charge transport in duplex DNA: The phonon-assisted polaron-like hopping mechanism. Proc Natl Acad Sci USA. 1999;96:8353–8358. doi: 10.1073/pnas.96.15.8353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Williams TT, Dohno C, Stemp EDA, Barton JK. Effects of the photooxidant on DNA-mediate charge transport. J Am Chem Soc. 2004;126:8148–8158. doi: 10.1021/ja049869y. [DOI] [PubMed] [Google Scholar]
- 15.Sugiyama H, Saito I. Theoretical studies of GG-specific photocleavage of DNA via electron transfer: Significant lowering of ionization potential and 5’-localization of HOMO of stacked GG bases in B-form DNA. J Am Chem Soc. 1996;118:7063–7068. [Google Scholar]
- 16.Merino EJ, Boal AK, Barton JK. Biological contexts for DNA charge transport chemistry. Curr Opin Chem Biol. 2008;12:229–237. doi: 10.1016/j.cbpa.2008.01.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Puckett CA, Barton JK. Methods to Explore Cellular Uptake of Ruthenium Complexes. J Am Chem Soc. 2007;129:46–47. doi: 10.1021/ja0677564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Augustyn KE, Merino EJ, Barton JK. A Role for DNA Charge Transport in Regulating p53: Oxidation of the DNA-bound Protein from a Distance. Proc Natl Acad Sci U S A. 2007;104:189107–18912. doi: 10.1073/pnas.0709326104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nunez ME, Holmquist GP, Barton JK. Evidence for DNA Charge Transport in the Nucleus. Biochemistry. 2001;40:12465–12471. doi: 10.1021/bi011560t. [DOI] [PubMed] [Google Scholar]
- 20.Tan DJ, Bai RK, Wong LJC. Comprehensive scanning of somatic mitochondrial DNA mutations in breast cancer. Cancer Res. 2002;62:972–976. [PubMed] [Google Scholar]
- 21.Lièvre A, Blons H, Houllier AM, Laccourreye O, Brasnu D, Beaune P, Laurent-Puig P. Clinicopathological significance of mitochondrial D-Loop mutations in head and neck carcinoma. Br J Cancer. 2006;94:692–697. doi: 10.1038/sj.bjc.6602993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xu B, Clayton DA. RNA-DNA hybrid formation at the human mitochondrial heavy-strand origin ceases at replication start sites: an implication for RNA-DNA hybrids serving as primers. EMBO J. 1996;15:3135–3143. [PMC free article] [PubMed] [Google Scholar]
- 23.Pham XH, Farge G, Shi YH, Gaspari M, Gustafsson CM, Falkenberg M. Conserved sequence box II directs transcription termination and primer formation in mitochondria. J Biol Chem. 2006;281:24647–24652. doi: 10.1074/jbc.M602429200. [DOI] [PubMed] [Google Scholar]
- 24.Merino EJ, Barton JK. Oxidation by DNA charge transport damages conserved sequence block II, a regulatory element in mitochondrial DNA. Biochemistry. 2007;46:2805–2811. doi: 10.1021/bi062024+. [DOI] [PubMed] [Google Scholar]
- 25.Merino EJ, Barton JK. DNA oxidation by charge transport in mitochondria. Biochemistry. 2008;47:1511–1517. doi: 10.1021/bi701775s. [DOI] [PubMed] [Google Scholar]
- 26.King TC, Low RL. Mitochondrial DNA displacement loop structure depends on growth state in bovine cells. J Biol Chem. 1987;262:6214–6220. [PubMed] [Google Scholar]
- 27.Hall DB. Ph.D. thesis. California Institute of Technology; 1997. [Google Scholar]
- 28.Henderson PT, Delaney JC, Muller JG, Neeley WL, Tannenbaum SR, Burrows CJ. The hydantoin lesions formed from oxidation of 7,8-dihydro-8-oxoguanine are potent sources of replication errors in vivo. Biochemistry. 2003;42:9257–9262. doi: 10.1021/bi0347252. [DOI] [PubMed] [Google Scholar]