

Abstract
The extracellular signal-regulated kinase (ERK) pathway is generally mitogenic, but, upon strong activation, it causes cell cycle arrest by a not-yet fully understood mechanism. In response to genotoxic stress, Chk1 hyperphosphorylates Cdc25A, a positive cell cycle regulator, and targets it for Skp1/Cullin1/F-box protein (SCF)β-TrCP ubiquitin ligase-dependent degradation, thereby leading to cell cycle arrest. Here, we show that strong ERK activation can also phosphorylate and target Cdc25A for SCFβ-TrCP-dependent degradation. When strongly activated in Xenopus eggs, the ERK pathway induces prominent phosphorylation and SCFβ-TrCP-dependent degradation of Cdc25A. p90rsk, the kinase downstream of ERK, directly phosphorylates Cdc25A on multiple sites, which, interestingly, overlap with Chk1 phosphorylation sites. Furthermore, ERK itself phosphorylates Cdc25A on multiple sites, a major site of which apparently is phosphorylated by cyclin-dependent kinase (Cdk) in Chk1-induced degradation. p90rsk phosphorylation and ERK phosphorylation contribute, roughly equally and additively, to the degradation of Cdc25A, and such Cdc25A degradation occurs during oocyte maturation in which the endogenous ERK pathway is fully activated. Finally, and importantly, ERK-induced Cdc25A degradation can elicit cell cycle arrest in early embryos. These results suggest that strong ERK activation can target Cdc25A for degradation in a manner similar to, but independent of, Chk1 for cell cycle arrest.
INTRODUCTION
The Cdc25A phosphatase, a key cell cycle regulator in vertebrates, dephosphorylates and activates cyclin-dependent kinases (Cdks), thereby promoting cell cycle progression (Donzelli and Draetta, 2003). On genotoxic stress, however, the checkpoint kinases Chk1 and Chk2 hyperphosphorylate Cdc25A and target it for rapid degradation, thereby inducing cell cycle arrest for DNA repair (Bartek and Lukas, 2003). In human cells, Chk1 phosphorylation promotes the binding of β-transducing repeat-containing protein (β-TrCP), the F-box protein of the Skp1/Cullin1/F-box protein (SCF)β-TrCP ubiquitin ligase (Frescas and Pagano, 2008), to the doubly phosphorylated DSG motif (DpSGX2–4pS) of Cdc25A and thereby targets the phosphatase for degradation (Busino et al., 2003; Jin et al., 2003). In Xenopus eggs and embryos, activated Chk1 also phosphorylates and targets Xenopus Cdc25A for SCFβ-TrCP-dependent degradation, but, in this case, by promoting the binding of β-TrCP to the nonphosphorylated DDG motif (DDGX2D) (which also exists and functions in human Cdc25A) (Shimuta et al., 2002; Kanemori et al., 2005). In both human and Xenopus Cdc25A proteins, however, Chk1 phosphorylates multiple conserved Arg-X-X-Ser motifs in the N-terminal regulatory domain (Busino et al., 2003; Uto et al., 2004). Although phosphorylation of a conserved Ser-Pro motif is also required for the Chk1-induced degradation of human Cdc25A, the responsible kinase is not known (Busino et al., 2003).
Mitogen-activated protein kinase (MAPK) pathways regulate diverse cellular processes ranging from proliferation and differentiation to apoptosis. In vertebrates, there are three major MAPK signaling pathways: extracellular signal-regulated kinase (ERK), Jun NH2-terminal kinase (JNK), and p38 MAPK pathways (Raman et al., 2007). The ERK pathway is mainly activated by mitogenic stimuli, such as growth factors and phorbol esters (Meloche and Pouyssegur, 2007). In contrast, the JNK pathway is activated by radiation and other environmental stresses, whereas the p38 pathway is activated by numerous stresses, such as UV irradiation, cytokines, and osmotic shock (Raman et al., 2007). Interestingly, the p38 pathway is involved in the degradation of Cdc25A that occurs in response to UV irradiation and interleukin withdrawal in certain mammalian cells (Khaled et al., 2005; Reinhardt et al., 2007). This Cdc25A degradation is important for cell cycle arrest that follows, but its mechanism is poorly understood (Reinhardt et al., 2007).
Whereas weak or sustained activation of the ERK pathway is generally mitogenic, its strong activation by oncogenic Ras or activated Raf causes differentiation in certain cell types and cell cycle arrest (and senescence) in many cell types (Ebisuya et al., 2005; Meloche and Pouyssegur, 2007). This cell cycle arrest has been suggested to be elicited by the ERK-mediated up-regulation of negative cell cycle regulators, such as p53 and Cdk inhibitors (such as p21Cip1 and p16Ink4a) (Pumiglia and Decker, 1997; Serrano et al., 1997; Zhu et al., 1998; Meloche and Pouyssegur, 2007). Although hitherto not recognized, however, such cell cycle arrest might also be contributed to by an ERK-dependent down-regulation of some positive cell cycle regulator(s).
In this study, we have investigated whether strong ERK activation can induce down-regulation (or degradation) of the positive cell cycle regulator Cdc25A. We show that the ERK pathway, when strongly activated in Xenopus eggs, phosphorylates and targets Cdc25A for degradation. This degradation involves the SCFβ-TrCP ubiquitin ligase and requires phosphorylations by both ERK and its downstream kinase p90rsk. Furthermore, strong ERK activation can cause cell cycle arrest in early embryos by targeting Cdc25A for degradation. These results, together with other results, suggest that, unexpectedly, the ERK pathway can target Cdc25A for degradation in a manner very similar to Chk1 and that Cdc25A degradation contributes to cell cycle arrest caused by strong ERK activation.
MATERIALS AND METHODS
Oocytes, Eggs, and Embryos
Oocytes, unfertilized eggs, and embryos were prepared, microinjected, and cultured as described previously (Kanemori et al., 2005; Inoue et al., 2007). Oocyte maturation and artificial egg activation were induced by progesterone (5 mg/ml) and the calcium ionophore A23187 (1 mg/ml), respectively, as described previously (Kanemori et al., 2005; Inoue et al., 2007). Artificially activated eggs are a very good system (for their single-cell nature) to express exogenous proteins (by mRNA injection) and to analyze regulation of cell cycle regulators, such as Cdc25A and Wee1 (Kanemori et al., 2005; Okamoto and Sagata, 2007). We usually used the eggs after 40 min of artificial activation.
cDNAs and In Vitro Transcription
cDNAs encoding Xenopus Cdc25A, a constitutively active form of Xenopus p90rsk, and a dominant-negative mutant of Xenopus β-TrCP were described previously (Shimuta et al., 2002; Kanemori et al., 2005; Inoue et al., 2007). cDNAs encoding mouse mitogen-activated protein kinase kinase (MKK) 1 or MAP kinase-ERK kinase (MEK) 1 (accession number NM 008927), human MKK6 (NM 002758), Xenopus MKK7 (NM 001087648), Xenopus JNKa (AB073999), human p21Cip1 (NM 000389), and human MKP3 (NM 001946) were isolated by polymerase chain reaction (PCR) from appropriate cDNA libraries. A constitutively active form of MEK1 was made by deleting the region encompassing amino acids 32–51 and by mutating two serine residues (Ser-218→Asp, Ser-222→Glu) (Mansour et al., 1994). A constitutively active form of MKK6 was made by mutating two residues (Ser-207→Asp, Thr-211→Asp) (Alonso et al., 2000), whereas that of MKK7 by mutating three residues (Ser-268→Asp, Thr-272→Glu, Ser-274→Asp) (Yamanaka et al., 2002). All the cDNA constructs were subcloned into either the N-terminally Myc-tagged or glutathione S-transferase (GST)-tagged pT7-G (UKII-) transcription vectors (Uto et al., 2004; Kanemori et al., 2005). In vitro mutagenesis and transcription of the cDNAs were performed as described previously (Shimuta et al., 2002).
In Vitro Translation
In vitro translation of Xe-Cdc25A mRNA was performed using wheat germ extracts (L4380; Promega, Madison, WI) as described previously (Inoue et al., 2007).
GST Fusion Proteins
cDNAs encoding Xenopus Cdc25A peptides (S36, residues 16-56; S85, 58-102; S120, 91-136; S129, 104-154; S137, 120-168; S190, 166-215; S198, 175-213; and S295, 265-316) and their Ser→Ala mutant peptides were subcloned into the pGEX-3X plasmid vector, and the GST-fused peptides were bacterially expressed and purified by standard methods.
Morpholino Oligonucleotides (Oligos)
Antisense morphlino oligos against Xenopus Erp1 mRNA were prepared and injected into one-cell embryos as described previously (Inoue et al., 2007).
In Vitro Kinase Assays
For in vitro p90rsk and ERK kinase assays, GST-fused Xe-Cdc25A peptides were incubated with [γ-32P]ATP and either p90rsk2 protein (14–480; Millipore, Billerica, MA) or ERK2 protein (P6080; New England Biolabs, Ipswich, MA) and analyzed essentially as described previously (Inoue et al., 2007). In vitro translated Xe-Cdc25A protein was incubated with ERK2 protein in the absence of [γ-32P]ATP as described previously (Inoue et al., 2007).
Antibodies and Immunoblotting
Anti-Xenopus Cdc25A phospho-Ser-85 antibody was raised in rabbits against peptides (LDpSPIKMDLRC) and affinity-purified by standard methods. Routinely, proteins equivalent to one oocyte or egg were analyzed by immunoblotting using anti-Xenopus Cdc25A antibody (Shimuta et al., 2002), anti-Myc antibody (A-14; Santa Cruz Biotechnology, Santa Cruz, CA), anti-GST antibody (Z-5; Santa Cruz Biotechnology), anti-phospho-p44/42 MAPK antibody (9106; Cell Signaling Technology, Danvers, MA), anti-phospho-JNK antibody (9251; Cell Signaling Technology), anti-phospho-p38 antibody (9211; Cell Signaling Technology), anti-Cdk1 phospho-Thr-14/Tyr-15 antibody (44–686G; BioSource International, Camarillo, CA), anti-Xenopus Erp1 antibody (Inoue et al., 2007), or anti-Xenopus Cdc25A phospho-Ser-85 antibody, essentially as described previously (Uto et al., 2004). In some experiments, oocyte or egg extracts before immunoblotting were treated with λ phosphatase as described previously (Inoue et al., 2007).
RESULTS
Activation of ERK Induces Degradation of Xe-Cdc25A in Xenopus Eggs
First, we examined whether strong activation of ERK, p38, or JNK MAPKs could induce degradation of Cdc25A. For this, we ectopically expressed, by mRNA injection, their immediate upstream activators (MKKs) in artificially activated Xenopus eggs, and then monitored activating phosphorylation of endogenous MAPKs and the levels of endogenous Cdc25A (Xe-Cdc25A) by immunoblotting. Activated eggs (which mimic fertilized eggs) are a very good in vivo system to reproducibly analyze regulatory mechanisms of cell cycle regulators, such as Cdc25A and Wee1 (Kanemori et al., 2005; Okamoto and Sagata, 2007; see Materials and Methods). Ectopic expression of a constitutively active (CA) form of MKK6, an activator of p38, efficiently induced activating phosphorylation of endogenous p38 but only very weakly induced the mobility shift and degradation of Xe-Cdc25A (Figure 1A, MKK6-CA) (see Discussion). Expression of a CA form of MKK7 (a JNK activator) (together with JNK; Lei et al., 2002) did not induce any appreciable mobility shift or degradation of Xe-Cdc25A (Figure 1A, MKK7-CA + JNK). In contrast to these, expression of a CA form of MEK1 (or MKK1), an activator of ERK, strongly induced both mobility upshifts (due to phosphorylation; Figure 1B) and degradation of Xe-Cdc25A (Figure 1A, MEK-CA). In this experiment, however, MEK-CA expression induced activating phosphorylation of not only ERK but also JNK (Figure 1A, MEK-CA). However, JNK activation after MEK-CA expression was far much weaker than that after MKK7-CA expression, which itself could not induce any mobility shift or degradation of Xe-Cdc25A (Figure 1A, MKK7-CA + JNK). Thus, these results suggest that strong activation of the MEK/ERK pathway can specifically induce prominent phosphorylation and degradation of endogenous Xe-Cdc25A in Xenopus eggs.
Figure 1.

Induction of SCFβ-TrCP-dependent degradation of Xe-Cdc25A by the ERK pathway. (A) Artificially activated eggs (or eggs after 40 min of calcium ionophore treatment) were injected or not with 9 ng of JNK mRNA, reinjected 2.5 h later (time 0) with 9 ng of either MEK-CA, MKK6-CA, or MKK7-CA mRNAs, and analyzed at 20-min intervals by immunoblotting (IB) with anti-Xe-Cdc25A, anti-phospho-JNK, anti-phospho-ERK, and anti-phospho-p38 antibodies. (B) Activated eggs were injected with 9 ng of GST mRNA (Control) or MEK-CA mRNA. Egg extracts were then treated (+λ) or not (−λ) with λ phosphatase and analyzed for Xe-Cdc25A by immunoblotting. (C) Activated eggs were injected with 18 ng of GST mRNA (Control) or dominant-negative β-TrCPΔF mRNA, reinjected 2.5 h later with 9 ng of MEK-CA mRNA, and then analyzed for Xe-Cdc25A by immunoblotting. (D) Activated eggs were injected or not with 9 ng of MKP3 mRNA, reinjected 40 min later with 2 ng of mRNA encoding Myc-tagged WT or D231A Xe-Cdc25A, further injected 2.5 h later with 9 ng of MEK-CA mRNA, and then analyzed for Myc-Xe-Cdc25A constructs and phospho-ERK by immunoblotting. Five, three, four, and four independent experiments were performed for A, B, C, and D, respectively, and, for each, a typical result is shown.
Involvement of SCFβ−TrCP in the ERK-induced Degradation of Xe-Cdc25A
Interestingly, the kinetics of phosphorylation and degradation of Xe-Cdc25A after MEK-CA expression (or ERK activation) in activated eggs is very similar to that after Chk1 activation in the same egg system (Shimuta et al., 2002; Kanemori et al., 2005). However, Chk1 itself was apparently not involved in the MEK-CA–induced phosphorylation or degradation of Xe-Cdc25A, because Chk1 was not activated at all in MEK-CA–expressing eggs (data not shown), just as in normal eggs (Shimuta et al., 2002). Nevertheless, given the similar phosphorylation and degradation of Xe-Cdc25A, MEK-CA-induced Xe-Cdc25A degradation might be mediated by the SCFβ-TrCP ubiquitin ligase, which is involved in Chk1-induced Xe-Cdc25A degradation (Kanemori et al., 2005). To test this possibility, we overexpressed a dominant-negative mutant of β-TrCP (β-TrCPΔF) in activated eggs (Kanemori et al., 2005). This treatment prevented the degradation (but not phosphorylation) of endogenous Xe-Cdc25A after MEK-CA expression (Figure 1C). Furthermore, when ectopically expressed in eggs, a Myc-tagged, Asp-231→Ala mutant of Xe-Cdc25A (D231A), i.e., the DDG motif mutant that cannot bind β-TrCP upon Chk1 activation (Kanemori et al., 2005), was extremely stable even after expression of MEK-CA (Figure 1D). In these experiments, even Xe-Cdc25A WT was very stable (and apparently not phosphorylated) if MKP3, an ERK (but not MEK)-inactivating phosphatase (Muda et al., 1996), was coexpressed with MEK-CA (Figure 1D). These results, together with the above-mentioned results (Figure 1A), strongly suggest that the MEK/ERK pathway, only downstream of MEK, phosphorylates Xe-Cdc25A and targets it for SCFβ-TrCP-dependent degradation.
Phosphorylation of Xe-Cdc25A by p90rsk and Its Contribution to ERK-induced Xe-Cdc25A Degradation
We attempted to identify the phosphorylation site(s) required for Xe-Cdc25A degradation after ERK activation. Previously, we showed that phosphorylation by Chk1 of four Ser residues (S120, S137, S190, and S295) in the Arg-X-X-Ser (RXXS) motifs is required for the Chk1-induced degradation of Xe-Cdc25A (Uto et al., 2004). We therefore asked whether these Ser residues would be required for ERK-induced Xe-Cdc25A degradation. When expressed in activated eggs, a Xe-Cdc25A mutant in which all of the four Ser residues were mutated to Ala (RXXS:4A) was significantly more stable than the wild type (WT), although not as stable as the completely stable D231A mutant, after MEK-CA expression (Figure 2A). Furthermore, the RXXS:4A mutant was somewhat less phosphorylated than the WT, as evidenced by its smaller size shifts, after MEK-CA expression (Figure 2A). Thus, intriguingly, the ERK pathway seems to phosphorylate at least some Chk1 phosphorylation sites for Xe-Cdc25A degradation.
Figure 2.

p90rsk phosphorylation, and its requirement for the ERK pathway-induced degradation, of Xe-Cdc25A. (A) Activated eggs were injected with 2 ng of mRNA encoding the indicated Myc-Xe-Cdc25A constructs, reinjected 2.5 h later with 9 ng of MEK-CA mRNA, and then analyzed for Myc-Xe-Cdc25A constructs by immunoblotting. (B) GST-fused Xe-Cdc25A peptides (GST-S120, GST-A120, etc., each named after the relevant Ser or substituted Ala residue numbers) were incubated with [γ-32P]ATP and p90rsk protein, subjected to SDS-polyacrylamide gel electrophoresis, stained with Coomassie Brilliant Blue (CBB), and then autoradiographed (32P). (C) Activated eggs were injected with 2 ng of mRNA encoding the indicated Myc-Xe-Cdc25A constructs, reinjected 2.5 h later with either 9 ng of MEK-CA mRNA or 36 ng of p90rsk-CA mRNA, and analyzed for Myc-Xe-Cdc25A constructs and phospho-ERK by immunoblotting. Four, three, and four independent experiments were performed for A, B, and C, respectively, and, for each, a typical result is shown.
We noted that, like Chk1, p90rsk, the kinase downstream of ERK (Sturgill et al., 1988), belongs to the Ca2+/calmodulin-regulated kinase subfamily of protein kinases and can phosphorylate RXXS motifs in its substrates (Frodin and Gammeltoft, 1999; Inoue et al., 2007). We therefore first tested whether p90rsk could phosphorylate any of the four Chk1 phosphorylation sites in Xe-Cdc25A. When analyzed by in vitro kinase assays using [γ-32P]ATP and GST-fused Xe-Cdc25A peptides (with or without Ala substitution), three (S120, S137, and S295) of the four Ser residues were significantly phosphorylated by p90rsk (Figure 2B). We then addressed whether ectopic expression of a constitutively active form of p90rsk (p90rsk-CA) can induce degradation of Xe-Cdc25A (WT or RXXS:4A) in activated eggs. Xe-Cdc25A WT was degraded after p90rsk-CA expression, albeit significantly less efficiently than after MEK-CA expression; notably, however, the RXXS:4A mutant was completely stable even after p90rsk-CA expression, although it was somewhat degraded after MEK-CA expression (Figure 2C). Together, these results suggest that p90rsk can phosphorylate Xe-Cdc25A on at least three Ser residues in the RXXS motifs and that this phosphorylation is required, in part, for ERK-induced Xe-Cdc25A degradation.
Phosphorylation of Xe-Cdc25A by ERK
Our observations that even the RXXS:4A mutant is somewhat degraded after MEK-CA expression (Figure 2, A and C) and that Xe-Cdc25A WT is degraded more efficiently after MEK-CA expression than after p90rsk-CA expression (Figure 2C), suggest that some other kinase(s), in addition to p90rsk, is involved in the MEK-CA-induced phosphorylation and degradation of Xe-Cdc25A. If so, the other kinase could be ERK itself, because MEK-CA expression induced a stronger phosphorylation (as well as degradation) of Xe-Cdc25A than p90rsk-CA expression (Figure 2C) and this phosphorylation (as well as degradation) was completely inhibited by ERK inactivation by MKP3 (Figure 1D). Interestingly, Xe-Cdc25A has four Ser residues (S36, S85, S129, and S198) that lie in the ERK consensus phosphorylation motif (Ser/Thr-Pro) and are mostly conserved in human Cdc25A (Figure 3A). Therefore, we first examined whether these four Ser residues could be phosphorylated by ERK in vitro, using [γ-32P]ATP and GST-fused Xe-Cdc25A peptides, essentially as performed earlier for p90rsk (Figure 2B). As shown in Figure 3B, ERK was able to phosphorylate all of the four Ser residues, particularly S85, in vitro.
Figure 3.

Phosphorylation of Xe-Cdc25A by ERK. (A) Conservation of consensus ERK phosphorylation motifs (Ser-Pro) in Xenopus and human Cdc25A proteins. (B) The indicated GST-fused Xe-Cdc25A peptides were incubated with [γ-32P]ATP and ERK protein and analyzed as described in Figure 2B. (C) GST-tagged full-length Xe-Cdc25A proteins (WT or S85A) were synthesized in wheat germ extracts, purified by GST-pull-down, incubated with either buffer (Cont.) or ERK protein, and analyzed by immunoblotting with anti-GST and anti-phospho-S85 antibodies. (D) Activated eggs were injected with 2 ng of mRNA encoding Myc-Xe-Cdc25A (WT or S85A), reinjected or not 2.5 h later with 9 ng of MEK-CA mRNA, and cultured for 50 min. Egg extracts were treated or not with λ-phosphatase and analyzed for Myc-Xe-Cdc25A and phospho-S85 by immunoblotting. (E) Activated eggs were injected with either buffer (Control), 18 ng of p21Cip1 mRNA, or 9 ng of MKP3 mRNA, reinjected 40 min later with 2 ng of Myc-Xe-Cdc25A mRNA, further injected 2.5 h later with 9 ng of MEK-CA mRNA, collected at the indicated times, and analyzed by immunoblotting as described in D. Four, three, four, and five independent experiments were performed for B, C, D, and E, respectively, and, for each, a typical result is shown.
Because S85 and its equivalent site S88 are required for the Chk1-induced degradations of Xe-Cdc25A and human Cdc25A, respectively (Busino et al., 2003; Kanemori et al., 2005), we investigated S85 phosphorylation by ERK in more detail. When phosphorylated by ERK in vitro, GST-fused full-length Xe-Cdc25A WT, but not its S85A mutant, was efficiently recognized by anti-phospho-S85 antibody (Figure 3C), confirming that S85 of Xe-Cdc25A can be phosphorylated by ERK in vitro. We then analyzed S85 phosphorylation in vivo. When coexpressed with MEK-CA in activated eggs, Xe-Cdc25A WT, but not the S85A mutant, was recognized by the anti-phospho-S85 antibody in a manner sensitive to λ-phosphatase treatment (Figure 3D). Surprisingly, however, S85 phosphorylation was detected even in the absence of MEK-CA expression (Figure 3D; see also time 0 in Figure 3E, Cont.) and after MEK-CA expression in the presence of pre-expressed MKP3 (Figure 3E, MKP3). Interestingly, this unexpected phosphorylation was strongly inhibited by prior expression of the Cdk inhibitor p21Cip1 (see times 0 and 20 min in Figure 3E, p21Cip1), indicating that it was mediated by Cdk, another proline-directed kinase. Importantly, however, despite the presence of p21Cip1, S85 phosphorylation did occur concurrently with ERK activation (and during Xe-Cdc25A degradation) after MEK-CA expression (see times 40–80 min in Figure 3E, p21Cip1); this result was even clearer when the stable D231A mutant, instead of the WT, was used for the analysis (Supplemental Figure S1A). Thus, these results show that ERK can directly phosphorylate Xe-Cdc25A on S85 both in vitro and in vivo and also on S36, S129, and S198 at least in vitro.
Contribution of ERK-catalyzed Phosphorylation to Xe-Cdc25A Degradation
To examine the requirement of ERK-catalyzed phosphorylations for Xe-Cdc25A degradation, we constructed several Xe-Cdc25A mutants, i.e., four single-Ala mutants (S36A, S85A, S129A, and S198A) and one quadruple-Ala mutant (S36/85/129/198A, termed SP:4A), and expressed them in activated eggs. After MEK-CA expression, the S129A mutant was as unstable as the WT, but both the S36A and S198A mutants were slightly more stable than the WT, and both the S85A and SP:4A mutants were significantly more stable than the WT (Figure 4A). In addition, reversion of Ala-129 to Ser (in SP:4A) had no appreciable effect on the stability of the SP:4A mutant, but reversion of Ala-36, Ala-85, or Ala-198 to Ser, particularly that of Ala-85 to Ser, significantly destabilized the SP:4A mutant (Figure 4B). Thus, these results suggest that most of the ERK-catalyzed phosphorylations, particularly phosphorylation at S85, contribute to the MEK-CA–induced degradation of Xe-Cdc25A.
Figure 4.
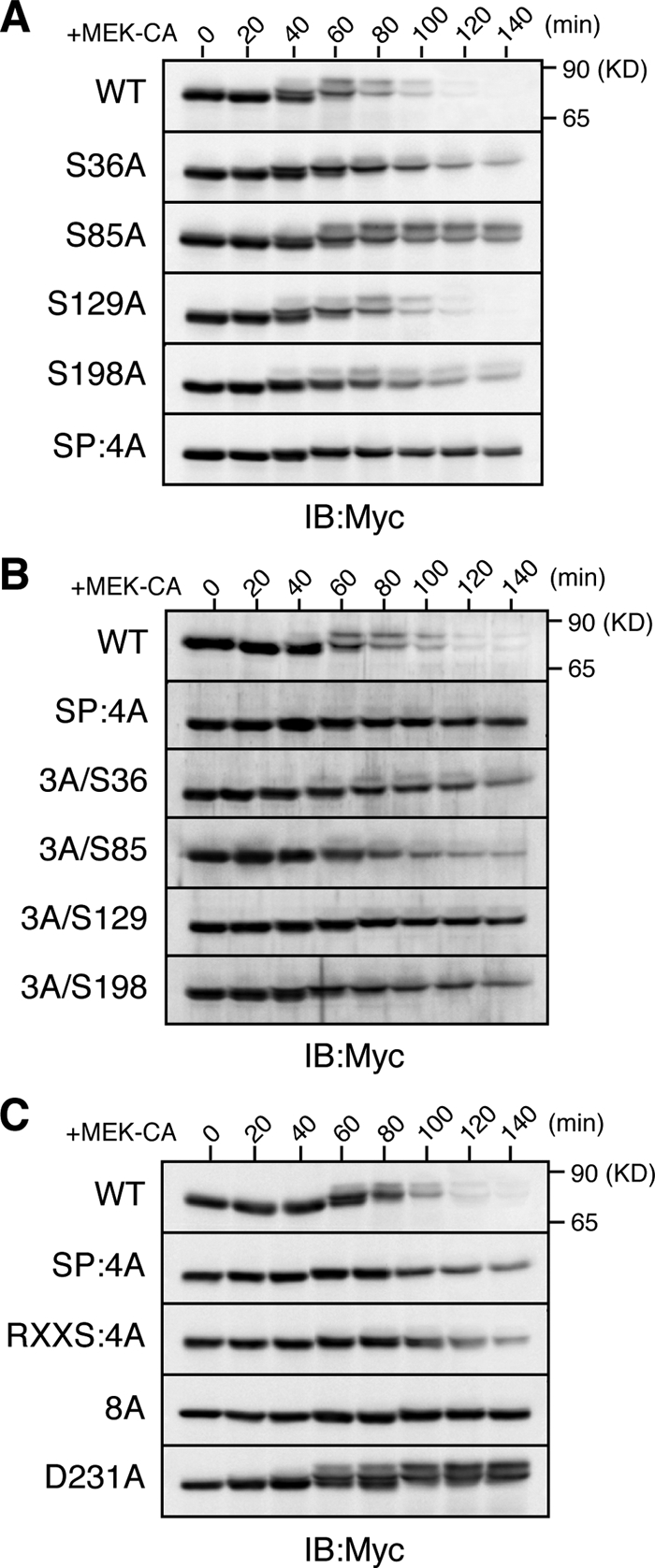
Requirement of ERK phosphorylation for Xe-Cdc25A degradation. (A–C) Activated eggs were injected with 2 ng of mRNA encoding the indicated Myc-Xe-Cdc25A constructs, reinjected 2.5 h later with 9 ng of MEK-CA mRNA, and analyzed for Myc-Xe-Cdc25A constructs by immunoblotting. In B, 3A/S36, etc., are reversion mutants of SP:4A (see text). Four independent experiments were performed for A–C, and, for each, a typical result is shown.
As for S85 phosphorylation, this phosphorylation was probably mediated in part by Cdk (Figure 3E, p21Cip1; Supplemental Figure S1A). However, even without Cdk activity (or in the presence of p21Cip1), ERK activation was able to efficiently induce the degradation of Xe-Cdc25A WT (Figure 3E, p21Cip1) but not of the S85A mutant (Supplemental Figure S1B). Thus, although Cdk also phosphorylates S85, ERK-catalyzed S85 phosphorylation alone is sufficient for the S85 phosphorylation-dependent degradation of Xe-Cdc25A.
We also compared stabilities of the SP:4A mutant, the p90rsk phosphorylation site mutant (RXXS:4A), the RXXS:4A/SP:4A double mutant (8A), and the β-TrCP binding site mutant (D231A) after MEK-CA expression. The SP:4A mutant was somewhat less stable than the completely stable D231A mutant (Figure 4C). The RXXS:4A mutant was also somewhat less stable than the D231A mutant (Figure 4C), consistent with earlier results (Figure 2A). However, the double 8A mutant was very stable, comparable with the D231A mutant (Figure 4C). Thus, these results strongly suggest that ERK phosphorylation and p90rsk phosphorylation contribute, roughly equally and additively, to the MEK-CA–induced degradation of Xe-Cdc25A. Significantly, by similar analyses, we also obtained evidence that ERK and p90rsk phosphorylations can target human Cdc25A for SCFβ-TrCP-dependent degradation in activated Xenopus eggs (Supplemental Figure S2).
ERK Pathway-dependent Degradation of Xe-Cdc25A during Oocyte Maturation
Given our results, Cdc25A might be degraded in an SCFβ-TrCP- dependent manner under physiological conditions in which the endogenous ERK pathway is strongly activated. During oocyte maturation in many species, the endogenous ERK pathway becomes fully activated under the control of Mos (which activates MEK) and plays an important role for meiotic division of the oocyte (Sagata, 1997; Kishimoto, 2003). Interestingly, a recent study in mice showed that Cdc25A present in immature oocytes is degraded during oocyte maturation, although the degradation mechanism is not known (Solc et al., 2008). We therefore asked whether Xe-Cdc25A could be degraded in an SCFβ-TrCP- and ERK pathway-dependent manner during Xenopus oocyte maturation. Somewhat surprisingly, unlike in mice (Solc et al., 2008), endogenous Xe-Cdc25A was not present in immature Xenopus oocytes, although it was present after fertilization, as reported previously (Kim et al., 1999) (Figure 5A; also see Figure 1, A–C). Interestingly, however, ectopically expressed Xe-Cdc25A was readily detected in immature oocytes and was degraded during progesterone-induced oocyte maturation, coincidently with germinal vesicle breakdown (a hallmark for entry into meiosis I) (Figure 5B, Control) and ERK activation (Figure 5C, Control). Importantly, this degradation of Xe-Cdc25A was dependent on both SCFβ-TrCP and ERK activity, because it was prevented either by overexpression of a dominant-negative mutant of β-TrCP (Figure 5B, β-TrCPΔF) or by treatment of the oocytes with the MEK-specific inhibitor U0126 (Figure 5C). Furthermore, and notably, both the RXXS:4A and SP:4A mutants were significantly more stable than Xe-Cdc25A WT, whereas both the double mutant (8A) and the β-TrCP-nonbinding mutant D231A were nearly completely stable, during oocyte maturation (Figure 5D). Additionally, the relative stabilities of these mutants during oocyte maturation were essentially the same as those observed in activated eggs expressing MEK-CA (compare Figures 4C and 5D). Thus, most probably, both ERK and p90rsk in the Mos–MEK–ERK–p90rsk pathway, as well as SCFβ-TrCP, are involved in the degradation of (ectopic) Xe-Cdc25A during oocyte maturation. These results suggest that, even under physiological conditions, a strongly activated ERK pathway can target coexisting Cdc25A for SCFβ-TrCP-dependent degradation.
Figure 5.

ERK pathway-dependent degradation of Xe-Cdc25A during oocyte maturation. (A) Immature oocytes (IMO) were treated with progesterone (PG) to induce maturation, whereas ovulated eggs were fertilized in vitro. Maturing oocytes and fertilized eggs were collected at the indicated times, and analyzed for endogenous Xe-Cdc25A and phospho-ERK by immunoblotting. GVBD denotes germinal vesicle breakdown. (B) Immature oocytes were coinjected with 2 ng of Myc-Xe-Cdc25A mRNA and 20 ng of either GST mRNA (Control) or β-TrCPΔF mRNA, cultured overnight, treated with progesterone, and analyzed for Myc-Xe-Cdc25A by immunoblotting. (C) Immature oocytes were injected with 2 ng of Myc-Xe-Cdc25A mRNA, cultured overnight, pretreated with dimethyl sulfoxide (Control) or 100 μM U0126 for 1 h, treated with progesterone, and analyzed for Myc-Xe-Cdc25A and phospho-ERK by immunoblotting. (D) Immature oocytes were injected with 2 ng of mRNA encoding the indicated Myc-Xe-Cdc25A constructs, cultured overnight, treated with progesterone, and analyzed for Myc-Xe-Cdc25A constructs by immunoblotting. In B–D, all the Myc-Xe-Cdc25A constructs were, in fact, phosphatase-dead C428S forms to avoid premature maturation of the oocytes. Four, three, four, and five independent experiments were performed for A, B, C, and D, respectively, and, for each, a typical result is shown.
Cell Cycle Arrest by ERK-induced Xe-Cdc25A Degradation
Cdc25A dephosphorylates Cdk1/Cdk2 on Thr-14/Tyr-15 residues and thereby promotes cell cycle progression in mammalian cells (Hoffmann et al., 1994; Molinari et al., 2000). Xe-Cdc25A begins to be synthesized after fertilization or egg activation (Figure 5A; Kim et al., 1999) and is involved in the rapid embryonic cell cycles (Kim et al., 1999). To determine whether ERK-induced Xe-Cdc25A degradation can elicit cell cycle arrest at interphase as does Chk1-induced Cdc25A degradation (Mailand et al., 2000; Shimuta et al., 2002; Busino et al., 2003), we ectopically expressed MEK-CA in one-cell embryos, and then monitored the inhibitory T14/Y15 phosphorylation of Cdk1 as well as the external morphology of embryos (Shimuta et al., 2002). In this experiment, however, we used embryos depleted of endogenous Erp1 (a maternal meiotic inhibitor) by antisense morpholino oligos (Figure 6A), because this maternal factor, if present and phosphorylated by p90rsk, induces metaphase arrest of the embryo owing to its cytostatic factor activity (Inoue et al., 2007; Nishiyama et al., 2007). Expression of MEK-CA in Erp1-depleted embryos induced both the degradation of endogenous Xe-Cdc25A and the T14/Y15 phosphorylation of Cdk1 (Figure 6B); remarkably, it also induced cleavage arrest of the embryos at the two/four-cell stages (Figure 6C). Thus, ERK activation can induce cell cycle arrest at interphase in early embryos.
Figure 6.

Cell cycle arrest in early embryos by ERK-induced Xe-Cdc25A degradation. (A) One-cell embryos 25 min after fertilization were injected with either water (Control) or 200 ng of Erp1 antisense morpholino oligos (Erp1-MO) and cultured for 1.5 h; embryo extracts were treated with λ-phosphatase and analyzed for endogenous Erp1 by immunoblotting. (B) One-cell embryos injected with Erp1 morpholino oligos as above were cultured for 35 min, injected with either water (Control), 5 ng of MEK-CA mRNA, or both 5 ng of MEK-CA mRNA and 200 pg of Myc-Xe-Cdc25A (WT or 8A) mRNA, cultured for the indicated times, and analyzed for Xe-Cdc25A (endogenous plus exogenous), phospho-ERK, and phospho-T14/Y15 by immunoblotting. (C) The embryos treated as in B were photographed 3 h after injection of MEK-CA mRNA (together with or without Myc-Xe-Cdc25A mRNA). Three and four independent experiments were performed for A and B, respectively, and, for each, a typical result is shown.
To test whether the cell cycle arrest by ERK activation was a result of ERK-induced Xe-Cdc25A degradation, we coexpressed MEK-CA and either Xe-Cdc25A WT or its stable 8A mutant in Erp1-depleted embryos. Under the present experimental conditions (see Figure 6B legend), coexpression of Xe-Cdc25A WT did not appreciably affect Cdk1 T14/Y15 phosphorylation or cleavage arrest induced by MEK-CA expression. However, coexpression of the stable 8A mutant greatly reduced the levels of Cdk1 T14/Y15 phosphorylation (Figure 6B) and largely restored embryonic cell divisions (Figure 6C). Thus, clearly, the stable 8A mutant, but not the WT, can overcome ERK-induced cell cycle arrest, indicating that Xe-Cdc25A degradation is responsible for the ERK-induced cell cycle arrest. These results suggest that strong ERK activation can induce cell cycle arrest at interphase by targeting Cdc25A for degradation.
DISCUSSION
Chk1 phosphorylates and targets Cdc25A for SCFβ-TrCP-dependent degradation both in mammalian cells and Xenopus embryos (Busino et al., 2003; Jin et al., 2003; Kanemori et al., 2005). In this study, we have found that the ERK pathway can also phosphorylate and target Xe-Cdc25A for SCFβ-TrCP-dependent degradation in Xenopus eggs. Interestingly, the mechanism of Xe-Cdc25A degradation induced by the ERK pathway is very similar to that induced by Chk1, as summarized in Figure 7.
Figure 7.

Model for the mechanism of Xe-Cdc25A degradation and cell cycle arrest induced by the ERK pathway. On strong activation of the ERK pathway, ERK phosphorylates Xe-Cdc25A on S85 (which normally is phosphorylated by Cdk) and other Ser residues (omitted), whereas the downstream kinase p90rsk phosphorylates Xe-Cdc25A on other multiple Ser residues (which overlap with Chk1 phosphorylation sites). These phosphorylations facilitate ubiquitination of Xe-Cdc25A by SCFβ-TrCP, thereby targeting the phosphatase for degradation and causing cell cycle arrest at interphase. For details, see text. RD, regulatory domain; DDG, DDG motif; CD, catalytic domain.
Our results show that, when strongly activated in Xenopus eggs, the ERK–MAPK pathway, but not the JNK or p38 MAPK pathways, can induce prominent phosphorylation and degradation of Xe-Cdc25A (Figure 1, A and B). This degradation involves the SCFβ-TrCP ubiquitin ligase (Figure 1, C and D), similar to Chk1-induced degradation (Kanemori et al., 2005). Furthermore, p90rsk, the kinase downstream of ERK, can phosphorylate Xe-Cdc25A on three of the four Chk1 phosphorylation sites (in the RXXS motifs) (Figure 2B) and thereby can target the phosphatase for degradation, albeit less efficiently than MEK (Figure 2C). Thus, it seems that the ERK pathway can induce Xe-Cdc25A degradation partly because p90rsk phosphorylates Xe-Cdc25A on sites overlapping with Chk1 phosphorylation sites (Figure 7). In this context, it is somewhat surprising that the p38 pathway, which has the Chk1-like kinase MK2 downstream of p38 (Manke et al., 2005) and perhaps thereby targets mammalian Cdc25A for degradation under certain conditions (Khaled et al., 2005; Reinhardt et al., 2007), cannot efficiently induce Xe-Cdc25A degradation in eggs (Figure 1A). This is likely due, at least in part, however, to a relatively low abundance of MK2 protein in Xenopus eggs, because overexpression of MK2 (together with the p38 activator MKK6) can induce much more efficient degradation of Xe-Cdc25A in eggs (Supplemental Figure S3). Given our results and the structural analogy between the p38 and the ERK pathways (Raman et al., 2007), MK2 may phosphorylate, at least in part, p90rsk phosphorylation sites, whereas p38 itself may phosphorylate ERK phosphorylation sites (see below), for Xe-Cdc25A degradation.
In addition to p90rsk, ERK itself also phosphorylates Xe-Cdc25A, but, in this case, on several SP motifs (Figure 3, A–D). This phosphorylation, as well as p90rsk phosphorylation (Figure 2A), is required, in part, for MEK-induced Xe-Cdc25A degradation (Figure 4, A and B). Double mutation of the ERK and p90rsk phosphorylation sites, however, renders Xe-Cdc25A (as well as human Cdc25A; Supplemental Figure S2) nearly completely resistant to MEK-induced degradation (Figure 4C). Thus, it seems that ERK phosphorylation and p90rsk phosphorylation contribute, roughly equally and additively, to the MEK-induced degradation of Xe-Cdc25A (as well as of human Cdc25A) (Figure 7). Interestingly, however, S85, the most important ERK phosphorylation site (Figure 4A), is also phosphorylated by Cdk (perhaps Cdk2; Ducruet and Lazo, 2003) in eggs (Figure 3, D and E), although this phosphorylation (by Cdk) is not required for ERK-induced Xe-Cdc25A degradation (because of its phosphorylation by ERK itself) (Figure 3E and Supplemental Figure S1A). Notably, S85 and phosphorylation of its equivalent site S88 are required, in part, for the Chk1-induced degradations of Xe-Cdc25A and human Cdc25A, respectively (Busino et al., 2003; Kanemori et al., 2005), and Cdk activity is required for Chk1-induced Xe-Cdc25A degradation (our unpublished data). Therefore, S85 of Xe-Cdc25A (as well as S88 of human Cdc25A) is probably phosphorylated by Cdk in the case of Chk1-induced degradation (Figure 7).
How might phosphorylation of the RXXS and SP motifs (by p90rsk and ERK, respectively) act to target Xe-Cdc25A for SCFβ-TrCP-dependent degradation? This question is important because both the RXXS and SP motifs are located dispersedly far from the DDG motif (to which β-TrCP binds) (Figure 7). Concerning this issue, our preliminary experiments suggested that, whereas the phosphorylation of the RXXS and SP motifs is not necessarily required for β-TrCP binding of Xe-Cdc25A upon ERK activation, it is required significantly for efficient ubiquitination of the phosphatase (data not shown). (This contrasts with the case of human Wee1, in which Cdk phosphorylation of an SP motif near the DDG-like motif promotes both β-TrCP binding and ubiquitination; Watanabe et al., 2005). Thus, it is conceivable that phosphorylation of the RXXS/SP motifs might act to facilitate ubiquitination of Xe-Cdc25A by the bound SCFβ-TrCP, perhaps by causing conformational changes of the phosphatase (Figure 7). A similar role has been suggested for Chk1-phosphorylated RXXS motifs in both human Cdc25A and Xe-Cdc25A (Busino et al., 2003; Kanemori et al., 2005).
The ERK pathway can target (ectopic) Xe-Cdc25A for SCFβ-TrCP-dependent degradation even under physiological conditions, or during oocyte maturation, in which the endogenous ERK pathway is fully activated (Figure 5). This result could explain the mechanism for degradation of endogenous Cdc25A during mouse oocyte maturation, which has recently been shown to occur and to be important for meiotic progression of the oocyte (Solc et al., 2008).
Importantly, we find that strong ERK activation can induce cell cycle arrest in early embryos by targeting Xe-Cdc25A for degradation (Figure 6; also see Figure 7). This finding can explain why activating ERK early in cycling egg extracts can cause a G2 arrest (Walter et al., 1997; Bitangcol et al., 1998; Murakami and Vande Woude, 1998). Furthermore, our finding seems to have an important implication in the effects of strongly activated ERK on somatic cells. As is well known, whereas weak or sustained ERK activation is generally mitogenic, strong ERK activation by oncogenic Ras or activated Raf leads to cell cycle arrest in many cell types (and also differentiation in some cases) (Ebisuya et al., 2005; Meloche and Pouyssegur, 2007). This cell cycle arrest may be elicited and maintained primarily by the ERK-induced expression of Cdk inhibitors, such as p21Cip1 and p16Ink4a (Pumiglia and Decker, 1997; Serrano et al., 1997; Zhu et al., 1998). Given our results and the degradation of Cdc25A in TPA-treated human cells (Supplemental Figure S4); however, such cell cycle arrest could also be contributed to by an ERK-induced degradation of Cdc25A, similar to cell cycle arrest by Chk1-induced Cdc25A degradation (Donzelli and Draetta, 2003; Bartek and Lukas, 2003). The Ras/Raf- or ERK-induced cell cycle arrest is followed by senescence, a cellular response currently considered to be a defense against neoplasmic transformation (Serrano et al., 1997; Zhu et al., 1998). Thus, degradation of Cdc25A, together with the expression of Cdk inhibitors, might act as a fail-safe mechanism to limit the well-known transforming potential of excessive ERK mitogenic signaling (Lin et al., 1998). In any case, our results suggest that the ERK pathway, when strongly activated, negatively influences cell cycle progression by targeting Cdc25A for SCFβ-TrCP-dependent degradation.
Supplementary Material
ACKNOWLEDGMENTS
We thank members of the Sagata laboratory for discussions and K. Gotoh for typing the manuscript. This work was supported by grants from the CREST Research Project of the Japan Science and Technology Agency and from the Ministry of Education, Culture, Sports, Science and Technology of Japan (to N. S.).
Abbreviations used:
- ERK
extracellular signal-regulated kinase
- MAPK
mitogen-activated protein kinase
- Mkp3
mitogen-activated protein kinase phosphatase 3
- SCF
Skp1/Cullin1/F-box protein
- Xe
Xenopus.
Footnotes
This article was published online ahead of print in MBC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E09-01-0008) on February 25, 2009.
REFERENCES
- Alonso G., Ambrosino C., Jones M., Nebreda A. R. Differential activation of p38 mitogen-activated protein kinase isoforms depending on signal strength. J. Biol. Chem. 2000;275:40641–40648. doi: 10.1074/jbc.M007835200. [DOI] [PubMed] [Google Scholar]
- Bartek J., Lukas J. Chk1 and Chk2 kinases in checkpoint control and cancer. Cancer Cell. 2003;3:421–429. doi: 10.1016/s1535-6108(03)00110-7. [DOI] [PubMed] [Google Scholar]
- Bitangcol J. C., Chau A.S.S., Stadnick E., Lohka M. J., Dicken B., Shibuya E. K. Activation of the p42 mitogen-activated protein kinase pathway inhibits Cdc2 activation and entry into M-phase in cycling Xenopus egg extracts. Mol. Biol. Cell. 1998;9:451–467. doi: 10.1091/mbc.9.2.451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busino L., Donzelli M., Chiesa M., Guadavaccaro D., Ganoth D., Dorrello N. V., Hershko A., Pagano M., Draetta G. F. Degradation of Cdc25A by β-TrCP during S phase and in response to DNA damage. Nature. 2003;426:87–91. doi: 10.1038/nature02082. [DOI] [PubMed] [Google Scholar]
- Donzelli M., Draetta G. F. Regulating mammalian checkpoints through Cdc25 inactivation. EMBO Rep. 2003;4:671–677. doi: 10.1038/sj.embor.embor887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ducruet A. P., Lazo J. S. Regulation of Cdc25A half-life in interphase by cyclin-dependent kinase 2 activity. J. Biol. Chem. 2003;278:31838–31842. doi: 10.1074/jbc.M303604200. [DOI] [PubMed] [Google Scholar]
- Ebisuya M., Kondoh K., Nishida E. The duration, magnitude and compartmentalization of ERK MAP kinase activity: mechanisms for providing signaling specificity. J. Cell Sci. 2005;118:2997–3002. doi: 10.1242/jcs.02505. [DOI] [PubMed] [Google Scholar]
- Frescas D., Pagano M. Deregulated proteolysis by the F-box proteins SKP2 and β-TrCP: tipping the scales of cancer. Nat. Rev. Cancer. 2008;8:438–449. doi: 10.1038/nrc2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frodin M., Gammeltoft S. Role and regulation of 90 kDa ribosomal S6 kinase (RSK) in signal transduction. Mol. Cell. Endocrinol. 1999;151:65–77. doi: 10.1016/s0303-7207(99)00061-1. [DOI] [PubMed] [Google Scholar]
- Hoffmann I., Draetta G., Karsenti E. Activation of the phosphatase activity of human cdc25A by a cdk2-cyclin E dependent phosphorylation at the G1/S transition. EMBO J. 1994;13:4302–4310. doi: 10.1002/j.1460-2075.1994.tb06750.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue D., Ohe M., Kanemori Y., Nobui T., Sagata N. A direct link of the Mos-MAPK pathway to Erp1/Emi2 in meiotic arrest of Xenopus laevis eggs. Nature. 2007;446:1100–1104. doi: 10.1038/nature05688. [DOI] [PubMed] [Google Scholar]
- Jin J., Shirogane T., Xu L., Nalepa G., Qin J., Elledge S. J., Harper J. W. SCFβ-TRCP links Chk1 signaling to degradation of the Cdc25A protein phosphatase. Genes Dev. 2003;17:3062–3074. doi: 10.1101/gad.1157503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanemori Y., Uto K., Sagata N. β-TrCP recognizes a previously undescribed nonphosphorylated destruction motif in Cdc25A and Cdc25B phosphatases. Proc. Natl. Acad. Sci. USA. 2005;102:6279–6284. doi: 10.1073/pnas.0501873102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khaled A. R., Bulavin D. V., Kittipatarin C., Li W. Q., Alvarez M., Kim K., Young H. A., Fornace A. J., Durum S. K. Cytokine-driven cell cycling is mediated through Cdc25A. J. Cell Biol. 2005;169:755–763. doi: 10.1083/jcb.200409099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S.H.Li.C., Maller J. L. A maternal form of the phosphatase Cdc25A regulates early embryonic cell cycles in Xenopus laevis. Dev. Biol. 1999;212:381–391. doi: 10.1006/dbio.1999.9361. [DOI] [PubMed] [Google Scholar]
- Kishimoto T. Cell cycle control during meiotic maturation. Curr. Opin. Cell Biol. 2003;15:654–663. doi: 10.1016/j.ceb.2003.10.010. [DOI] [PubMed] [Google Scholar]
- Lei K., Nimnual A., Zong W. X., Kennedy N. J., Flavell R. A., Thompson C. B., Bar-Sagi D., Davis R. J. The Bax subfamily of Bcl2-related proteins is essential for apoptotic signal transduction by c-Jun NH(2)-terminal kinase. Mol. Cell. Biol. 2002;22:4929–4942. doi: 10.1128/MCB.22.13.4929-4942.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin A. W., Barradas M., Stone J. C., van Aelst L., Serrano M., Lowe S. W. Premature senescence involving p53 and p16 is activated in response to constitutive MEK/MAPK mitogenic signaling. Genes Dev. 1998;12:3008–3019. doi: 10.1101/gad.12.19.3008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mailand N., Falck J., Lukas C., Syljuåsen R. G., Welcker M., Bartek J., Lukas J. Rapid destruction of human Cdc25A in response to DNA damage. Science. 2000;288:1425–1429. doi: 10.1126/science.288.5470.1425. [DOI] [PubMed] [Google Scholar]
- Manke I. A., Nguyen A., Lim D., Stewart M. Q., Elia A. E., Yaffe M. B. MAPKAP kinase-2 is a cell cycle checkpoint kinase that regulates the G2/M transition and S phase progression in response to UV irradiation. Mol. Cell. 2005;17:37–48. doi: 10.1016/j.molcel.2004.11.021. [DOI] [PubMed] [Google Scholar]
- Mansour S. J., Matten W. T., Hermann A. S., Candia J. M., Rong S., Fukasawa K., Vande Woude G. F., Ahn N. G. Transformation of mammalian cells by constitutively active MAP kinase kinase. Science. 1994;265:966–970. doi: 10.1126/science.8052857. [DOI] [PubMed] [Google Scholar]
- Meloche S., Pouyssegur J. The ERK1/2 mitogen-activated protein kinase pathway as a master regulator of the G1- to S-phase transition. Oncogene. 2007;26:3227–3239. doi: 10.1038/sj.onc.1210414. [DOI] [PubMed] [Google Scholar]
- Molinari M., Mercurio C., Dominguez J., Goubin F., Draetta G. F. Human Cdc25A inactivation in response to S phase inhibition and its role in preventing premature mitosis. EMBO Rep., 2000;1:71–79. doi: 10.1093/embo-reports/kvd018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muda M., Theodosiou A., Rodrigues N., Boschert U., Camps M., Gillieron C., Davies K., Ashworth A., Arkinstall S. The dual specificity phosphatases M3/6 and MKP-3 are highly selective for inactivation of distinct mitogen-activated protein kinases. J. Biol. Chem. 1996;271:27205–27208. doi: 10.1074/jbc.271.44.27205. [DOI] [PubMed] [Google Scholar]
- Murakami M. S., Vande Woude G. F. Analysis of the early embryonic cell cycles of Xenopus; regulation of cell cycle length by Xe-wee1 and Mos. Development. 1998;125:237–248. doi: 10.1242/dev.125.2.237. [DOI] [PubMed] [Google Scholar]
- Nishiyama T., Ohsumi K., Kishimoto T. Phosphorylation of Erp1 by p90rsk is required for cytostatic factor arrest in Xenopus laevis eggs. Nature, 2007;446:1096–1099. doi: 10.1038/nature05696. [DOI] [PubMed] [Google Scholar]
- Okamoto K., Sagata N. Mechanism for inactivation of the mitotic inhibitory kinase Wee1 at M phase. Proc. Natl. Acad. Sci. USA. 2007;104:3753–3758. doi: 10.1073/pnas.0607357104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pumiglia K. M., Decker S. J. Cell cycle arrest mediated by the MEK/mitogen-activated protein kinase pathway. Proc. Natl. Acad. Sci. USA. 1997;94:448–452. doi: 10.1073/pnas.94.2.448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raman M., Chen W., Cobb M. H. Differential regulation and properties of MAPKs. Oncogene. 2007;26:3100–3112. doi: 10.1038/sj.onc.1210392. [DOI] [PubMed] [Google Scholar]
- Reinhardt H. C., Aslanian A. S., Lees J. A., Yaffe M. B. p53-deficient cells rely on ATM- and ATR-mediated checkpoint signaling through the p38MAPK/MK2 pathway for survival after DNA damage. Cancer Cell. 2007;11:175–189. doi: 10.1016/j.ccr.2006.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagata N. What does Mos do in oocytes and somatic cells? Bioessays. 1997;19:13–21. doi: 10.1002/bies.950190105. [DOI] [PubMed] [Google Scholar]
- Serrano M., Lin A. W., McCurrach M. E., Beach D., Lowe S. W. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 1997;88:593–602. doi: 10.1016/s0092-8674(00)81902-9. [DOI] [PubMed] [Google Scholar]
- Shimuta K., Nakajo N., Uto K., Hayano Y., Okazaki K., Sagata N. Chk1 is activated transiently and targets Cdc25A for degradation at the Xenopus midblastula transition. EMBO J. 2002;21:3694–3703. doi: 10.1093/emboj/cdf357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solc P., Saskova A., Baran V., Kubelka M., Schultz R. M., Motlik J. CDC25A phosphatase controls meiosis I progression in mouse oocytes. Dev. Biol. 2008;317:260–269. doi: 10.1016/j.ydbio.2008.02.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturgill T. M., Ray L. B., Erikson E., Maller J. L. Insulin-stimulated MAP-2 kinase phosphorylates and activates ribosomal protein S6 kinase II. Nature. 1988;334:715–718. doi: 10.1038/334715a0. [DOI] [PubMed] [Google Scholar]
- Uto K., Inoue D., Shimuta K., Nakajo N., Sagata N. Chk1, but not Chk2, inhibits Cdc25 phosphatases by a novel common mechanism. EMBO J. 2004;23:3386–3396. doi: 10.1038/sj.emboj.7600328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter S. A., Guadagno T. M., Ferrell J. E., Jr Induction of a G2-phase arrest in Xenopus egg extracts by activation of p42 mitogen-activated protein kinase. Mol. Biol. Cell. 1997;8:2157–2169. doi: 10.1091/mbc.8.11.2157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamanaka H., Moriguchi T., Masuyama N., Kusakabe M., Hanafusa H., Takeda R., Takeda S., Nishida E. JNK functions in the non-canonical Wnt pathway to regulate convergent extension movements in vertebrates. EMBO Rep. 2002;3:69–75. doi: 10.1093/embo-reports/kvf008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe N., Arai H., Iwasaki J. -I., Shiina M., Ogata K., Hunter T., Osada H. Cyclin-dependent kinase (CDK) phosphorylation destabilizes somatic Wee1 via multiple pathways. Proc. Natl. Acad. Sci. USA. 2005;102:11663–11668. doi: 10.1073/pnas.0500410102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J., Woods D., McMahon M., Bishop J. M. Senescence of human fibroblasts induced by oncogenic Raf. Genes Dev. 1998;12:2997–3007. doi: 10.1101/gad.12.19.2997. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.