

Abstract

Neighboring group participation in glycopyranosylation reactions is probed for esters at the 3-O-axial and equatorial, 4-O-axial and equatorial, and 6-O-sites of a range of donors through the use tert-butoxycarbonyl esters. The anticipated intermediate cyclic dioxanyl cation is interrupted for the axial 3-O-derivative leading to the formation of a 1,3-O-cyclic carbonate ester, with loss of a tert-butyl cation, providing convincing evidence of participation by esters at that position. However, no evidence was found for such a fragmentation of carbonate esters at the 3-O-equatorial, 4-O axial and equatorial and 6-O positions indicating that neighboring group participation from those sites does not occur under typical glycosylation conditions. Further probes employing a 4-O-(2-carboxy)benzoate ester and a 4-O-(4-methoxybenzoate) ester, the latter in conjunction with an 18O quench designed to detect bridging intermediates, also failed to provide evidence for participation by 4-O-esters in galactopyranosylation.
Introduction
Neighboring group participation, or anchimeric assistance, 1 by 2-O-carboxylate esters is of fundamental importance to carbohydrate chemistry and is responsible for the facile, reliable, highly stereoselective synthesis of the 1,2-trans class of glycosidic bonds (β-glucosides and α-mannosides and related glycosidic bonds).2 The intervention of 2-O-carboxylate esters in this manner is supported by the isolation of crystalline dioxalenium ions in some cases,3 by the spectroscopic observation of the same species as intermediates in other cases,4 by computational work,5 by rate acceleration in the case of weakly activated donors,3,6 and is implied by the highly 1,2-trans-selective nature of these reactions. Participation by other groups, such as the 2-O-(2-pyridyl)methyl ethers and several 2-O-(2-thio)ethyl ethers has been demonstrated spectroscopically recently,7 and the similar involvement of other groups such as the 2-O-phosphate esters,8,9 the 2-deoxy-2-dibenzylamino systems,10 and other bulky benzyl type ethers11 has been suggested based on stereochemical evidence.
In this paper we address the possibility of neighboring group participation by carboxylate esters located on more remote positions, specifically on O3, O4, and O6 of glycopyranosyl donors. In 2000, we observed the thioglycoside 1, with its 3-O-benzoate ester, to be highly α-selective12 in contrast to the excellent β-selectivity seen with the corresponding 3-O-benzyl ether 2. 13 The phenomenon was subsequently extended to other esters at the 3-position of mannose and was successfully exploited in the synthesis of a complex oligosaccharide.14 Our results are related to an earlier observation by van Boeckel and coworkers in which it was found that a 3-O-acetyl-protected mannopyranosyl bromide was less β-selective than the corresponding 3-O-benzyl derivative under insoluble silver salt conditions,15,16 but differ in that the changes are considerably greater in our work. At the time we speculated that the α-selectivity observed with 1 might be derived from neighboring group participation by the ester and invoked intermediates related to 3 which readily accommodate the trans-fused benzylidene acetal.12 Subsequent work from our laboratory, however, has highlighted the high sensitivity of these 4,6-O-benzylidene protected β-mannosylations to a range of substituents at the 3-position, causing us to doubt our original interpretation.17
Neighboring group participation by esters at the 3-position, both axial and equatorial, has previously been discussed by other groups,15,18 and most recently in the equatorial series by Nifantiev and coworkers for the donors 4,19 but the evidence rarely extends beyond stereochemical arguments. A similar situation pertains with respect to esters in the 4-O-position with numerous claims of neighboring group participation leading to improved synthesis of either α-galacto and fucosides, 20 or β-gluco and mannosides.21 van Boeckel argued strongly that, at least for the systems studied in his laboratory using the insoluble silver salt method for the activation of glycosyl bromides, it is not necessary to invoke neighboring group participation by esters at the 3- and/or 4-positions of glucose and mannose in order to explain the selectivities observed.15 In discussing the increased β-selectivities observed in rhamnopyranosylation reactions with donor 5, as compared to 6, under homogeneous activation conditions, we agreed with van Boeckel and inclined toward the effect of the ester being one of an electron-withdrawing group influencing the tightness of the ion pairs on activation of the donor.9 In line with this hypothesis, Takahashi and coworkers observed excellent β-selectivity in a series of glycosylations conducted with a set of donors carrying electron-withdrawing but non-participating 4-O-sulfonate esters.22 Demchenko and coworkers, on the other hand, recently came down strongly on the side of neighboring group participation from O4 with donors 6 and 7, even though the best β-selectivity recorded was only 7:1.21
With respect to the effect of esters and related functions on O6 in glycosylation reactions, early work by the Schuerch group with glucosyl sulfonates as donors indicated that 6-O-(N-phenylcarbamates) afforded high selectivity for α-glucosides,23 but the effect did not extend to the galactopyranosides.24 However, rather than postulating neighboring group participation in the glucose series, the authors inclined toward the functionality at O6 influencing stereoselectivity by modulation of the tightness of the ion pair obtained on ionization of the glycosyl donor.23 In another study by the Schuerch laboratory, investigation of a series of glucosyl donors carrying substituted 6-O-benzoates revealed that greater β-selectivity was observed with the more electron-rich esters, which was discussed in terms of stabilization of the glycosyl oxocarbenium ion by the ester group, without the formation of a cyclic intermediate such as would be required by classical neighboring group participation.25,26 In spite of this background, neighboring group participation by esters at O6, through seven-membered cyclic transition states and leading to the formation of α-glycosides continues to be invoked frequently in the literature.27
The rational development of stereocontrolled oligosaccharide synthesis requires the resolution of these fundamental issues and it is with this in mind that we advance here a series of probes capable of unambiguously establishing the presence of neighboring group participation in glycosylation reactions.
Results and Discussion
Concept
We conceived that participation by a carboxylate ester might be reliably established if the ester could be modified in such a way as to trap the intermediate dioxocarbenium ion by fragmentation. We considered that a tert-butoxycarbonyl (Boc) group would be a suitable system for use in this manner with loss of a tert-butyl-cation from the cyclic intermediate leading to the formation of a cyclic carbonate ester.
The capture of vicinal electrophilic centers by carbonate esters, especially tert-butoxycarbonates, leading to the formation of five membered cyclic carbonates with the loss of an alkyl cation or equivalent is classical in organic synthesis, 28 and the comparable use of carbamates has found application in carbohydrate chemistry.29 Although unusual, such chemistry has also been extended to the formation of a seven-membered cyclic carbonate through cyclization of a carbamate derived from a 4-pentenol with N-bromosuccinimide in a favorably conformationally constrained system. 30 More generally, the possibility of neighboring group participation in simple acyclic systems through 6- and 7-membered cyclic dioxocarbenium ions has found some support in elegant labeling experiments conducted by Wilen and coworkers.31 We note that the Hammett parameter, σp, for the CO2Me group is +0.45 which is closely related to the +0.50 of the acetyl group,32 from which we deduce that carbonate esters should be somewhat akin to acetate esters in their ability to take part in neighboring group participation. Likewise, the field effect parameter F of the CO2Me and COMe groups are very similar (0.34 and 0.33, respectively) indicating that both groups stabilize positive charge by field effects to a similar extent.32
Proof of Concept
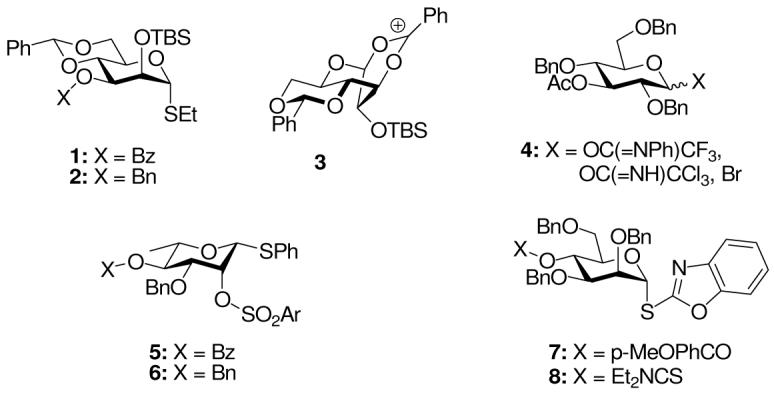
Activation of the 2-O-Boc protected thioglucoside 9, readily available through reaction of ethyl 3,4,6-tri-O-benzyl-β-D-thioglucopyranoside with Boc2O, with N-iodosuccinimide and silver triflate in dichloromethane at -10 °C afforded the cyclic carbonate 10 in 75% isolated yield (Scheme 1).
Scheme 1.
Cyclic Carbonate Formation from a 2-O-Boc Group: Glucose
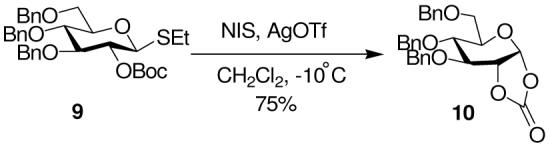
The 2-O-mannosyl carbonate 11, available from phenyl 3,4,6-tri-O-benzyl-α-D-thiomannopyranoside and Boc2O, gave the cyclic carbonate 12 in 74% yield on treatment with 1-benzenesulfinyl piperidine (BSP)33 and trifluoromethanesulfonic anhydride in dichloromethane at -60 °C (Scheme 2).
Scheme 2.
Cyclic Carbonate Formation from a 2-O-Boc Group: Mannose
These two experiments serve as proof of concept of cyclic carbonate formation from Boc esters in systems for which classical neighboring group participation by carboxylates is well established.
Axial 3-O-Esters
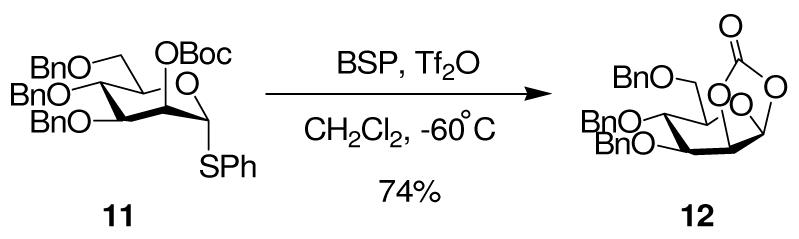
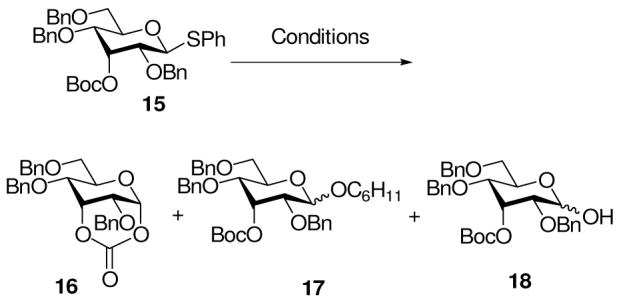
The possibility of participation by axial esters at O3 in the pyranose series was investigated with the allose donor 15, which was obtained in a straightforward manner via Dess Martin oxidation of the 2,4,6-tri-O-benzyl-β-D-thioglucoside 13, reduction with L-selectride, and condensation with Boc2O (Scheme 3).
Scheme 3.
Preparation of a 3-O-Boc-Allopyranoside Donor
The activation of 15 was investigated under three sets of conditions. Treatment with BSP and trifluoromethanesulfonic anhydride at -60 °C in dichloromethane in the absence of an external nucleophile led to the isolation of the cyclic carbonate 16 in 70% yield (Table 1, entry 1). However, inclusion of the mild, non-nucleophilic base tri-tert-butylpyrimidine (TTBP)34 in the reaction mixture and addition of cyclohexanol following activation resulted in the formation of the cyclohexyl glycoside 17 in 61% yield as an 11:1 β:α mixture, together with 7% of the expected cyclic carbonate 16 (Table 1, entry 2). Finally, activation by N-iodosuccinimide and trifluoromethanesulfonic anhydride in the absence of nucleophile gave the hydrolysis product 18 as the only isolable product (Table 1, entry 3) from a complex mixture in which the tert-butyl group was mostly retained as judged from the 1H-NMR spectrum of the crude reaction mixture.
Table 1.
The Axial 3-O-Boc Group
 | |||
|---|---|---|---|
| Entry | Conditions |
Products Yield (%) |
Anomeric Ratio (α:β) |
| 1 | BSP, Tf2O, CH2Cl2, -60°C |
16, 70 | - |
| 2 | TTBP, BSP, Tf2O, cyclohexanol, CH2Cl2, -60°C |
17, 61 16, 7 |
1:11.2 |
| 3 | NIS, Tf2O, CH2Cl2, -60°C |
18, 55 | - |
The isolation of cyclic carbonate 16 on activation with BSP and Tf2O (Table 1, entry 1) clearly demonstrates the possibility of participation by axial esters at the 3-O-position. The formation of the cyclohexyl glycosides 17 on inclusion of the nucleophile, however, strongly suggests that such neighboring group participation is not necessary for the selective formation of β-glycosides in this system and suggests that developing 1,3-diaxial interactions in transition state for formation of the α-glycoside are sufficient to explain the preferential formation of β-glycosides in this type of system. The isolation of the hydrolysis product 18 on activation with NIS/Tf2O presumably is due to trapping of a cyclic intermediate 19, before loss of the tert-butyl group, by iodide or by succinimide (vide infra) to give an unstable orthocarbonate-type species 20 which undergoes hydrolysis on work up (Scheme 4). We prefer this mechanism over the trapping of the glycosyl oxocarbenium ion by iodide or succinimide, followed by hydrolysis on work up, as both glycosyl iodides 35 and succinimides 36 have been demonstrated on numerous occasions to be isolable substances that are readily handled under standard conditions.
Scheme 4.
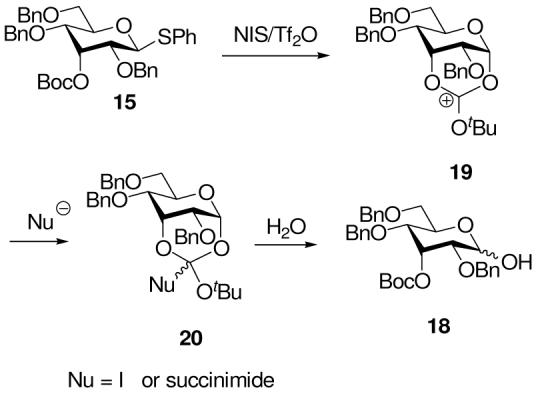
Possible Mechanisms for the Formation of 18
Equatorial O3 Esters
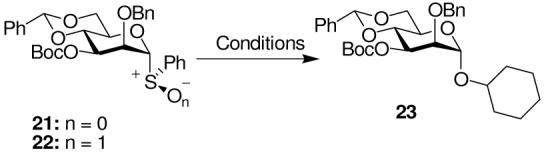
A 3-O-Boc ester 21 was readily prepared from phenyl 2-O-benzyl-4,6-O-benzylidene-α-D-thiomannoside, and a portion was converted to the sulfoxide 22, which was formed as a single diastereomer, presumed to have the (R)S configuration on the basis of previous work.37 When sulfoxide 22 was activated with Tf2O at low temperature in dichloromethane followed by aqueous work up, a complex mixture was obtained that nevertheless retained the 3-O-Boc ester as determined from the 1HNMR spectrum of the crude reaction mixture (Table 2, entry 1). On activation of the sulfoxide in the presence of TTBP followed by addition of cyclohexanol, the α-glycoside 23 was formed as a single α-anomer in 75% isolated yield (Table 2, entry 2). Activation of the thioglycoside 21 with BSP and Tf2O followed by trapping with cyclohexanol gave the same glycoside with the same exquisite α-selectivity (Table 2, entry 3). In none of the experiments conducted with donors 21 and 22 were we able to find any evidence supporting the formation of a cyclic carbonate spanning positions 1 and 3 of the pyranose ring, and it is apparent that the α-selectivity observed by ourselves with donors carrying equatorial carboxylate esters on the 3-position of glycosyl donors does not arise from classical neighboring group participation.
Table 2.
The Equatorial 3-O-Boc Group
 | |||
|---|---|---|---|
| Entry | Conditions |
Products Yield (%) |
Anomeric Ratio (α:β) |
| 1 |
22, Tf2O, CH2Cl2, -60°C |
complex mixture |
- |
| 2 |
22, TTBP, Tf2O, C6H12O, CH2Cl2, -60 °C |
23, 75 | α only |
| 3 |
21, TTBP, BSP, Tf2O, C6H12O, CH2Cl2, -60°C |
23, 69 | α only |
Axial O4 Esters
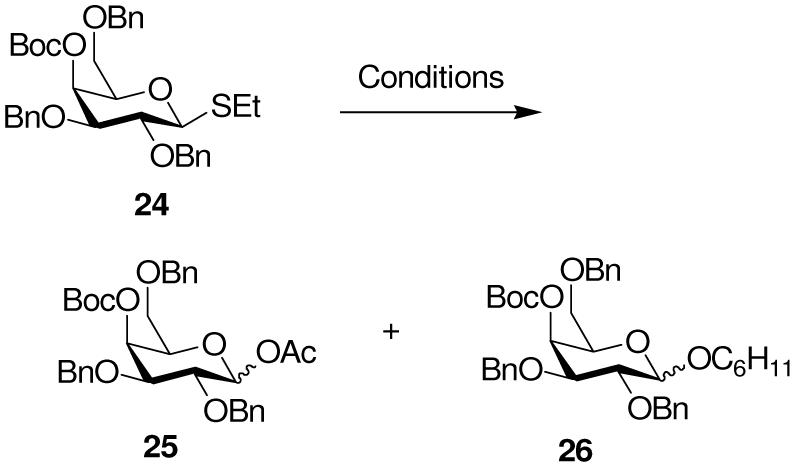
A 4-O-Boc galactosyl donor 24 was readily obtained from ethyl 2,3,6-tri-O-benzyl-β-D-thiogalactopyranoside and activated with BSP and Tf2O in ether at -60 °C. Following work up, acetylation of the crude reaction mixture enabled isolation of the glycosyl acetates 25 as an anomeric mixture (Table 3, entry 1). When cyclohexanol was added as acceptor, the cyclohexyl glycosides 26 were obtained in high yield, with the anomeric ratio being dependent on the solvent, but favoring the β-anomer in both ether and dichloromethane (Table 3, entries 2 and 3). These results effectively rule out neighboring group participation from an axial ester at O-4.
Table 3.
The Axial 4-O-Boc Group
 | |||
|---|---|---|---|
| Entry | Conditions |
Products Yield (%) |
Anomeric Ratio (α:β) |
| 1 | i) BSP, Tf2O, Et2O, -60 °C; ii) Ac2O, py |
25, 92 | 1:7.1 |
| 2 | TTBP, BSP, Tf2O, cyclohexanol, CH2Cl2, -60 °C |
26, 88 | 1:3.9 |
| 3 | TTBP, BSP, Tf2O, cyclohexanol, Et2O , -60 °C |
26, 62 | 1:1.3 |
Equatorial O4 Esters
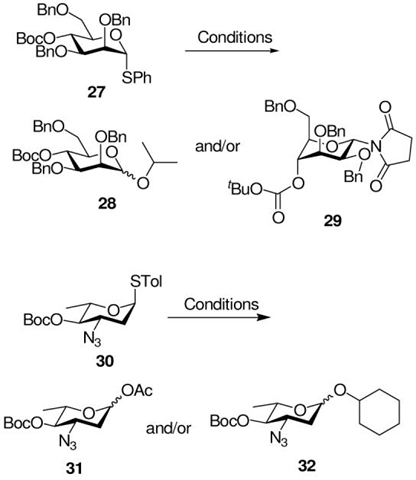
Two 4-O-Boc donors 27 and 30 were prepared in the usual manner from the corresponding alcohols and subjected to a series of activation and coupling reactions as set out in Table 4.
Table 4.
The Equatorial 4-O-Boc Group
 | ||||
|---|---|---|---|---|
| Entry | Donor | Conditions |
Products Yield (%) |
Anomeric Ratio (α:β) |
| 1 | 27 | 1.2 equiv NIS, 0.3 equiv AgOTf, 3 equiv iPrOH, CH2Cl2, -10 °C |
28, 72 | 0.9:1 |
| 2 | 27 | 1.2 equiv BSP, 1.5 equiv TTBP, 3 equiv iPrOH, CH2Cl2, -40 °C |
28, 76 | 0.8:1 |
| 3 | 27 | 1.2 equiv BSP, 1.5 equiv TTBP, 3 equiv iPrOH,, Et2O, -60 °C |
28, 65 | 0.7:1 |
| 4 | 27 | 1.2 equiv NIS, 0.3 equiv AgOTf, CH2Cl2, -10 °C |
29, 59 | α-only |
| 5 | 30 | i) BSP, Tf2O, CH2Cl2, -60 °C, ii) Ac2O, py |
31, 73% | 1:1.7 |
| 6 | 30 | TTBP, BSP, Tf2O, cyclohexanol, CH2Cl2, -60 °C |
32, 46 | 1:3.7 |
With the mannosyl donor 27, activations were conducted by the NIS and BSP/Tf2O methods with isopropanol as acceptor (Table 4, entries 1-3) with no indication of the formation of a cyclic carbonate. In each case, the isopropyl glycoside 28 was isolated as an anomeric mixture slightly favoring the β-anomer, consistent with the general results of Demchenko with donors 7 and 8. When the acceptor was omitted from the NIS-type activation, the ring-inverted α-glycosyl succinimide 29 was isolated in good yield (Table 4, entry 4) consistent with trapping of the glycosyl oxocarbenium ion being more rapid than any participation by the Boc group. With the 3-azido-2,3,6-trideoxy system 30, anticipated to undergo more facile ring inversion than 27 and so to have a greater predisposition toward neighboring group participation from the 4-position,38 no cyclic carbonate was found (Table 4, entries 5 and 6). When the activation was conducted without acceptor, only the hydrolysis product was isolated, whereas quenching with cyclohexanol allowed isolation of the glycoside as an anomeric mixture favoring the β-isomer. Overall, the results obtained argue strongly against neighboring group participation by equatorial esters at O-4 as playing a significant role in glycosylation reactions.
6-O-Esters
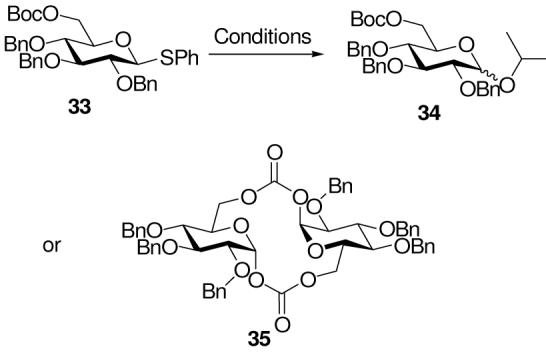
With the 6-O-Boc glucosyl donor 33 and isopropanol as acceptor, the glycoside 34 was obtained in high yield as an anomeric mixture irrespective of whether the reaction was conducted in dichloromethane or ether (Table 5, entries 1 and 2). In the absence of acceptor, a crude reaction mixture containing predominantly one product, with an anomeric doublet at δ 6.21 (J = 3.0 Hz) was obtained. Chromatographic purification was accompanied by substantial decomposition, nevertheless, the same product was isolated in 16% yield and characterized on the basis of its NMR and mass spectrometric data as the macrocyclic bis-carbonate 35. The symmetric α,α-nature of this macrolide strongly mitigates against its formation by dimerization of any monomeric cyclic carbonate as this would necessarily require inversion of configuration at the anomeric center. Neighboring group participation by esters at O-6 is deemed unlikely on the basis of these results.
Table 5.
The 6-O-Boc Group
 | |||
|---|---|---|---|
| Entry | Conditions |
Products Yield (%) |
Anomeric Ratio (α:β) |
| 1 | 1.2 equiv NIS, 0.3 equiv TfOH, 3 equiv iPrOH, CH2Cl2, 0 °C |
34, 80 | 1:1 |
| 2 | 1.2 equiv NIS, 0.3 equiv TfOH, 3 equiv iPrOH, Et2O, 0 °C |
34, 74 | 1.5:1 |
| 3 | 1.2 equiv NIS, 0.3 equiv TfOH, CH2Cl2, 0 °C |
35, 16 | |
Axial Esters at O4 Revisited
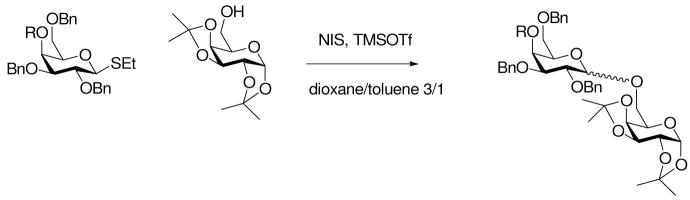
One of the more supportive papers in the literature for neighboring group participation by remote groups is that of Boons and coworkers on a series of 4-O-acyl-galactosyl donors.20c This paper appears to provide evidence for a shift in mechanism between alkanoate and arylcarboxylate esters as the data, some of which is reproduced in Table 6, clearly shows a jump in anomeric ratio between the acetate and the substituted benzoates. The situation, however, is complicated by the high α-selectivity obtained with the pivalate ester, particularly when it is realized that the Hammett σp value for a pivaloyl group is smaller than that of an acetyl group (σp pivaloyl = +0.32; σp acetyl = +0.50), and that the field effect parameter for pivaloyl is also smaller than that of an acetyl group (F pivaloyl = 0.26; F acetyl 0.33).32 Apparently, steric effects are also at play here, as is the possibility of the intermediate glycosyl oxocarbenium ions assuming different conformations depending on the size of the group at O4.
Table 6.
Literature Data on 4-O-Acyl Galactosyl Donors
 | ||
|---|---|---|
| Entry R | Yield (%) |
Anomeric Ratio (α:β) |
| 1 | acetyl | 7.2:1 |
| 2 | 4-nitrobenzoyl | 14:1 |
| 3 | benzoyl | 17:1 |
| 4 | 4-methylbenzoyl | 18:1 |
| 5 | 4-methoxybenzoyl | 14:1 |
| 6 | pivaloyl | 16:1 |
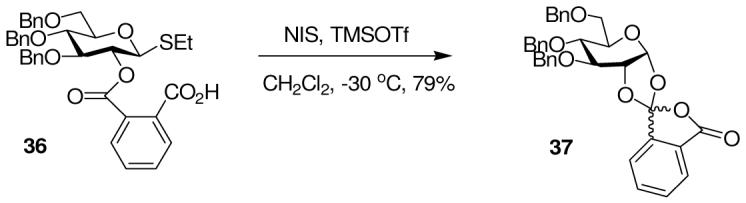
For this reason, we believed it important to develop an independent probe specifically for neighboring group participation by benzoate-type esters. We took inspiration from a seminal paper of Lemieux and Hindsgaul 39 in which it was demonstrated that 3,4,6-tri-O-acetyl-2-O-(carboxybenzoyl)-α-D-glucopyranosyl bromide underwent cyclization to an ortho-spiro ester (3,4,6-tri-O-acetyl-1,2-O-phthalidylidene-α-D-glucopyranose) on treatment with tetrabutylammonium bromide and 2,6-lutidine in 60% yield and, importantly, that one of these orthospiroesters could be isolated and fully characterized. Applying this concept, we prepared the hemiphthalate 36, by reaction of the corresponding alcohol with sodium hydride and phthalic anhydride, and treated it with NIS and TMSOTf in dichloromethane at room temperature leading to the isolation of orthoester 37 in 79% yield as an inseparable mixture of two diastereomers (Scheme 5). In addition to the 1H-NMR spectrum with a characteristic anomeric hydrogen resonance at δ 6.10, compound 37 featured a strong IR carbonyl absorption at v 1783 cm-1.
Scheme 5.
Proof of Concept for the Hemiphthalate Probe
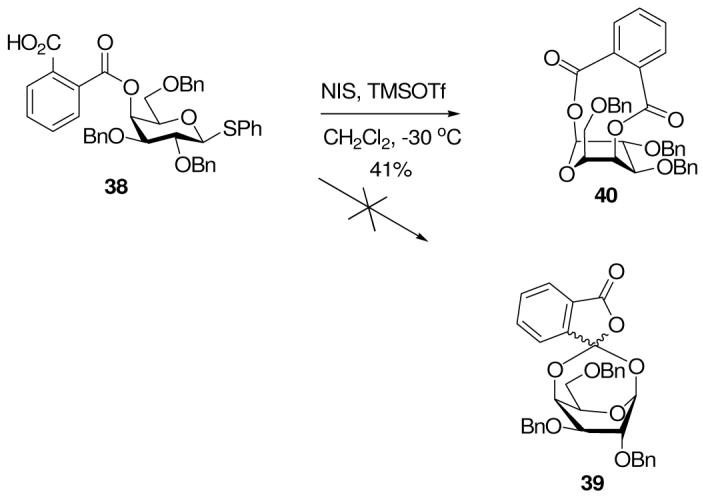
With proof of concept established, we proceeded to prepare the galactosyl hemiphthalate 38 from phenyl 2,3,6-tri-O-benzyl-β-D-thiogalactopyranoside by treatment with sodium hydride and phthalic anhydride in DMF. On activation with N-iodosuccinimide and TMSOTf at -30 °C in dichloromethane, no evidence could be found for the formation of a bridged phthalide such as 39, either by NMR or IR spectroscopy of the crude reaction mixture. Otherwise identical reactions run in CD2Cl2 with direct NMR examination also failed to provide evidence for the formation of 39. However, it was possible to isolate from these reaction mixtures a compound to which we assign the B3,O diolide structure 40 (Scheme 6).40,41 This compound might presumably arise by direct trapping of the glycosyl oxocarbenium ion by the acid group in 38, but it is also conceivably the product of the rapid rearrangement of the putative intermediate 39. This experiment was therefore deemed inconclusive and a further probe was sought.
Scheme 6.
Application of an Axial 4-O-Hemiphthalate Probe
To probe further the possibility of neighboring group participation by axial O4 esters, we conceived that the existence of bridging intermediates might be investigated by quenching with 18O-enriched water and determination of the site of incorporation of the label (Scheme 7). The success of this experiment is predicated upon the kinetic mode of attack on the bridging cation taking place at the cationic carbon. This has been clearly demonstrated to be the case in our earlier work when, working in the presence of a non-nucleophilic base, we were able to monitor by 13C-NMR spectroscopy the formation of glycosyl orthoesters from preformed 2-phenyl-1,3-dioxalenium ions, rather than the glycosides observed on rearrangement.4 Thus, we anticipated that low temperature activation of 41 in the absence of acceptors would lead to the glycosyl cation 42, in equilibrium with the corresponding glycosyl triflate (not shown), and that this cation would be in equilibrium with the bridged cation 44 in the event that neighboring group participation occurs. Quenching of the glycosyl cation with 18O-labelled water would lead to incorporation of the label solely at the anomeric center 43, whereas quenching of the bridged cation 44 would ultimately lead to location of the label at the carbonyl carbon, as in 46. .
Scheme 7.

An Isotopic Labeling Probe
This experiment was conducted with the 4-methoxybenzoate 41, with 95% 18O-enriched water, leading to isolation of the hydrolysis product 43/46 in 82% yield, principally as the α-anomer. Unfortunately, this sample was not amenable to mass spectrometric analysis and therefore it was converted to the acetate 47, isolated in 90% yield as a 4.1:1 α:β anomeric mixture. Mass spectrometry of 47 revealed the incorporation of one atom of 18O to the extent of approximately 50%. Inspection by 13C NMR spectroscopy revealed the presence of an isotopically shifted anomeric carbon and of a similarly shifted acetyl carbonyl group for both anomers. However, no isotopically shifted signals were apparent for either C-4 or the benzoyl carbonyl group, strongly suggesting incorporation of the label only at the anomeric site. Further confirmation of the location of the label was sought by treatment of the anomeric acetates 47 with thiophenol and BF3.OEt2 in acetonitrile resulting in the isolation of thioglycoside 41 in 97% yield as a 2.2:1 α:β-mixture. Interrogation of this mixture of anomeric thioglycosides by mass spectrometry revealed the absence of the 18O label leading to the conclusion that, in the quenching reaction, quenching only took place at the anomeric center.
Conclusion
Our results unambiguously establish that neighboring group participation by axial esters at O3 is possible in the course of glycopyranosylation reactions, in general agreement with the work of Weisner and others.18 On the other hand no evidence was found in support of neighboring group participation by an equatorial O3 ester, by axial or equatorial O4 esters, or by esters at O6. In the case of axial O4 esters, for which the strongest case had been made in the literature, an isotopic labeling probe also failed to find evidence in support of a bridging intermediate. We conclude that participation by esters at these positions is unlikely and that alternative explanations must be found for any stereochemical effects arising from the presence of esters at these positions.42
The elimination of neighboring group participation from consideration, particularly for the O3 equatorial, O4 axial and O6 esters ultimately requires the formulation of alternative hypotheses for the interesting stereochemical effects sometimes observed in the presence of these groups.43,44 Remote protecting groups and/or substituents have long been known to influence the outcome of a variety of different reaction types,45 as exemplified in glycopyranosylation by the influence of 4,6-O-alkylidene and silylene acetals and have been variously attributed to conformational, inductive, stereoelectronic and other effects13a,46 It is unlikely that one single effect will be found to be responsible for the effects of all remote esters in glycosylation reactions, but this remains to be determined by further investigation.
Experimental Section
General procedure for preparation of Boc protected alcohols
To a solution of substrate (2.0 mmol) in CH2Cl2 (30 mL), were added Boc2O (1.75 g, 8.0 mmol), Et3N (362 μL, 2.6 mmol) and DMAP (24.4 mg, 0.2 mmol) followed by stirring at room temperature overnight. The mixture was washed with saturated NaHCO3 and brine, dried, and concentrated. Chromatographic purification (Eluent: EtOAc/Hexanes = 1/5) afforded the Boc protected donors.
General procedure for thioglycoside activation by the BSP method
The donor (0.1 mmol), BSP (0.12 mmol), TTBP (0.15 mmol) and activated 4Å powdered molecular sieves (100 mg) were dissolved in dry CH2Cl2 (0.1 M) and stirred at -60 °C under N2 atmosphere for 10 minutes, then freshly distilled Tf2O (0.15 mmol) was added. The reaction mixture was stirred at -60 °C for 1 h before quenching with saturated aq. NaHCO3. After extraction with CH2Cl2 (3 × 5 mL), the combined organic phase was washed with brine, dried, and concentrated. The crude reaction mixture was purified by chromatography on silica gel.
General procedure for thioglycoside activation by the NIS method
The donor (0.1 mmol), NIS (0.15 mmol) and activated 4Å powdered molecular sieves (100 mg) were dissolved in dry CH2Cl2 (0.1 M) at room temperature, then stirred at -40 °C for 10 mins before TMSOTf (0.01 mmol) was added dropwise at -40 °C. The reaction mixture was stirred at -40 °C for 1 h before quenching with Na2S2O3 (20%) and extraction with CH2Cl2 (3 × 5 mL). The combined organic phase were washed with brine, dried, and concentrated. The crude reaction mixture was purified by chromatography on silica gel.
General procedure for glycosylation reactions by the BSP/TTBP method
The donor (0.1 mmol), BSP (0.12 mmol), TTBP (0.15 mmol) and activated 4Å powdered molecular sieves (100 mg) were dissolved in dry CH2Cl2 (0.1 M) and stirred at -60 °C under N2 atmosphere for 10 mins, before redistilled Tf2O (0.15 mmol) was added dropwise. After 10 mins, the acceptor (0.15 mmol) was added and stirring maintained at -60 °C for 2 h. The reaction mixture was then allowed to warm to room temperature before it was quenched with saturated aq. NaHCO3, extracted with CH2Cl2 (3 × 5 mL), and the combined organic phase washed with brine, dried, and concentrated. The crude reaction mixture was purified by chromatography on silica gel.
General procedure for glycosylation reactions by the NIS/AgOTf method
A solution of donor (0.05 M), acceptor (1.5 equiv) and 3 Å molecular sieves in dry CH2Cl2 was stirred at room temperature for 0.5 h under argon before it was cooled to 0 °C. NIS (1 equiv) was then added followed by AgOTf or TMSOTf (0.3 equiv), and the resulting mixture was stirred for 30 min before it was filtered. The filtrate was washed by 20% Na2S2O3 solution and brine. The aqueous layer was extracted with CH2Cl2 and the combined organic layer was dried, and concentrated under vacuum. The residue was purified by flash column chromatography on silica gel (Hex:EtOAc/5-10: 1) to afford the glycosylation products.
Ethyl 3,4,6-tri-O-benzyl-2-O-tert-butyloxycarbonyl-1-thio-β-D-glucopyranoside (9)
Compound (9) was formed from 3,4,6-tri-O-benzyl-β-D-thioglucopyranoside and Boc2O by the general procedure in 78% yield as a colorless oil. [α]D -0.8° (c, 1.2); 1H NMR δ 1.27 (t, J = 7.5 Hz, 3H), 1.49 (s, 9H), 2.72-2.76 (m, 2H), 3.48-3.51 (m, 1H), 3.67 (t, J = 9.0 Hz, 1H), 3.69-3.73 (m, 2H), 3.75 (dd, J = 2.0, 11.0 Hz, 1H), 4.40 (d, J = 10.0 Hz, 1H), 4.55 (d, J = 12.5 Hz, 1H), 4.56 (d, J = 11.0 Hz, 1H), 4.60 (d, J = 12.0Hz, 1H), 4.78-4.82 (m, 4H), 7.16-7.29 (m, 15H); δ: 14.9, 23.9, 27.8 (3C), 68.9, 73.5, 75.0, 75.2, 75.4, 77.7, 79.5, 82.8, 83.5, 84.4, 127.6-128.5 (15C), 137.9, 138.2 (2C), 152.4; ESI HRMS Calcd for C34H42O7SNa [M+Na]+: 617.2544. Found: 617.2542.
3,4,6-Tri-O-benzyl-1,2-O-carbonyl-α-D-glucopyranose (10)
Compound (10) was formed from compound (9) and NIS by the general procedure in 75% yield as a white solid. Mp, 60.1-61.2 °C; [α]D +4.8° (c, 0.5) (Lit.47 Mp, 60-61 °C; [α]23D +4.9° (c, 4.8, CHCl3)); 1H NMR δ: 3.65-3.72 (m, 2H), 3.80-3.86 (m, 2H), 3.94 (t, J = 4.0 Hz, 1H), 4.43 (d, J = 11.5 Hz, 1H), 4.49 (d, J = 12.0 Hz, 1H), 4.57 (d, J = 12.0 Hz, 1H), 4.59-4.71 (m, 4H), 6.07 (d, J = 6.5 Hz, 1H), 7.17-7.30 (m, 15H); 13C NMR δ: 68.4, 71.8, 72.7, 73.2, 73.3, 73.5, 75.9, 77.3, 97.4, 127.9-128.7 (15C), 136.9, 137.4, 137.6, 152.5; IR: 1812 cm-1.
Phenyl 2-O-tert-butoxycarbonyl-3,4,6-tri-O-benzyl-1-thio-α-D-mannopyranoside (11)
Compound (11) was formed from 3,4,6-tri-O-benzyl-α-D-thiomannopyranoside and Boc2O by the general procedure in 75% yield as a colorless oil. [α]D = 98.0° (c, 0.3); 1H NMR δ 1.47 (s, 9H), 3.74 (dd, J = 10.5, 2.0 Hz, 1H), 3.84 (dd, J = 10.5, 5.0 Hz, 1H), 3.92-3.97 (m, 2H), 4.34-4.37 (m, 1H), 4.47 (d, J = 12.0 Hz, 1H), 4.52 (d, J = 10.5 Hz, 1H), 4.61 (d, J = 11.0 Hz, 1H), 4.65 (d, J = 11.5 Hz, 1H), 4.76 (d, J = 11.0 Hz, 1H), 4.90 (d, J = 11.0 Hz, 1H), 5.37 (t, J = 2.5 Hz, 1H), 5.61 (d, J = 1.5 Hz, 1H), 7.19-7.38 (m, 18H), 7.49-7.51 (m, 2H); 13C NMR δ 27.8, 69.0, 71.9, 72.5, 73.0, 73.4, 74.7, 75.3, 78.6, 82.7, 86.1, 127.5, 127.6, 127.7, 127.8, 127.9, 128.0, 128.1, 128.3, 128.37, 128.40, 129.0, 131.9, 133.8, 137.8, 138.3, 138.4, 153.1; IR: 1097, 1277, 1740 cm-1; ESIHRMS Calcd for C38H42O7SNa [M+Na]+: 665.2544. Found: 665.2548.
3,4,6-Tri-O-benzyl-1,2-O-carbonyl-α-D-mannopyranose (12)
Compound (12) was formed from 11 and BSP by the general procedure in 74% yield as a colorless oil. [α]D = -1.5° (c, 0.7); 1H NMR δ 3.56-3.57 (m, 2H), 3.82-3.85 (m, 2H), 3.91 (t, J = 7.5 Hz, 1H), 4.50-4.53 (m, 3H), 4.73-4.79 (m, 4H), 5.79 (d, J = 5.0 Hz, 1H), 7,19-7.21 (m, 2H), 7.26-7.38 (m, 13H); 13C NMR δ 69.4, 73.0 (2C), 73.6, 74.7, 75.3, 75.9, 76.8, 96.4, 127.8, 127.9, 128.0, 128.1, 128.2, 128.46, 128.50, 128.7, 137.2, 137.6, 137.7, 153.0; IR: 1027, 1111, 1453, 1823 cm-1; ESIHRMS Calcd for C28H28O7Na [M+Na]+: 499.1728. Found: 499.1724.
Phenyl 2,4,6-Tri-O-benzyl-1-thio-β-D-allopyranoside (14)
To a stirred solution of 1348 (0.3 g, 0.55 mmol) in CH2Cl2 (5 mL), was added Dess-Martin periodinane (0.28 g, 0.66 mmol) followed by stirring at room temperature for 2 h. The reaction mixture was then diluted with CH2Cl2 (10 mL) and washed with saturated NaHCO3 and brine. The organic layer was separated and concentrated and the residue was dissolved in THF (10 mL) before L-selectride (1.1 mL, 1.1 mmol) was added at -78 °C. The reaction mixture was stirred at -78 °C for 20 mins, and then quenched with water, diluted with CH2Cl2 (10 mL) and washed with saturated NaHCO3 and brine. The organic layer was dried and concentrated and purified by column chromatography (Eluent: EtOAc/Hexanes = 1/4) to give 14 (0.14 g, 47%) as a colorless oil. [α]D = 5.0° (c, 0.2); 1H NMR δ 2.47 (s, 1H), 3.33 (dd, J = 12.2, 3.6 Hz, 1H), 3.53 (dd, J = 12.2, 3.4 Hz, 1H), 3.68-3.72 (m, 1H), 3.79 (dd, J = 13.5, 2.1 Hz, 1H), 3.94-3.98 (m, 1H), 4.33 (t, J = 3.5 Hz, 1H), 4.48-4.54 (m, 2H), 4.58-4.72 (m, 4H), 5.08 (d, J = 12.2 Hz, 1H), 7.22-7.37 (m, 18H), 7.56-7.58 (m, 2H); 13C NMR δ 66.3, 69.2, 71.4, 72.3, 73.4, 74.0, 74.5, 76.4, 83.4, 127.3, 127.5, 127.7, 128.0, 128.1, 128.2, 128.3, 128.48, 128.53, 128.8, 132.0, 133.8, 137.4, 137.6, 138.4; IR: 1066, 1283, 1454 cm-1; ESIHRMS Calcd for C33H34O5SNa [M+Na]+: 565.2020. Found: 565.2009.
Phenyl 3-O-tert-butoxycarbonyl-2,4,6-tri-O-benzyl-1-thio-β-D-allopyranoside (15)
Compound 15 was formed from 14 and Boc2O by the general procedure in 91% yield as a colorless oil. [α]D = 12.3° (c, 0.13); 1H NMR δ 1.42 (s, 9H), 3.38 (dd, J = 10.0, 3.0 Hz, 1H), 3.61 (dd, J = 10.0, 3.0 Hz, 1H), 3.71 (dd, J = 11.0, 4.0 Hz, 1H), 3.78 (dd, J = 10.5, 2.0 Hz, 1H), 3.93-3.96 (m, 1H), 4.40 (d, J = 10.5 Hz, 1H), 4.49-4.52 (m, 2H), 4.58-4.61 (m, 1H), 4.67-4.71 (m, 2H), 5.06 (d, J = 6.5 Hz, 1H), 5.75 (t, J = 3.0 Hz, 1H), 7.21-7.39 (m, 18H), 7.56-7.58 (m, 2H); 13C NMR δ 27.7, 68.9, 69.0, 71.3, 71.8, 72.8, 73.4, 75.0, 75.4, 82.2, 83.6, 127.4, 127.5, 127.6, 127.9, 128.1, 128.2, 128.3, 128.7, 132.3, 133.3, 137.3, 137.5, 138.5, 153.5; IR: 1091, 1280, 1369, 1454, 1740 cm-1; ESIHRMS Calcd for C38H42O7SNa [M+Na]+: 665.2544. Found: 665.2539.
2,4,6-Tri-O-benzyl-1,3-O-carbonyl-α-D-allose (16)
Compound 16 was formed from 15 and BSP by the general procedure in 70% yield as a colorless oil. [α]D = 80.0° (c, 0.01); 1H NMR δ 3.53 (t, J = 2.0 Hz, 1H), 3.69-3.78 (m, 4H), 3.82-3.85 (m, 1H), 4.45 (dd, J = 12.0, 6.0 Hz, 1H), 4.56-4.67 (m, 4H), 4.86 (d, J = 2.0 Hz, 1H), 5.57 (t, J = 2.5 Hz, 1H), 7.22-7.34 (m, 15H); 13C NMR δ 67.1, 67.0, 69.5, 71.0, 71.6, 71.9, 73.6, 74.7, 96.2, 127.4, 127.8, 128.00, 128.02, 128.3, 128.51, 128.56, 128.63, 128.8, 129.3, 129.5, 136.1, 136.8, 137.5, 146.6; IR: 1124, 1180, 1454, 1766 cm-1; ESIHRMS Calcd for C28H28O7Na [M+Na]+: 499.1728. Found: 499.1711.
Cyclohexyl 3-O-tert-butoxycarbonyl-2,4,6-tri-O-benzyl-α-D-allopyranoside (17α) and Cyclohexyl 3-O-tert-butoxycarbonyl-2,4,6-tri-O-benzyl-β-D-allopyranoside (17β)
These glycosides were prepared by general procedure with a combined yield of 61% (α:β = 1:11.2). Chromatographic separation on silica gel eluting with EtOAc/ Hexanes (1/6) enabled pure samples of the two anomers to be obtained. 17α Colorless oil; Yield: 5%; [α]D = 22.9° (c, 0.2); 1H NMR δ 1.22-1.25 (m, 4H), 1.38-1.51 (m, 2H), 1.41 (s, 9H), 1.76-1.79 (m, 2H), 1.83-1.86 (m, 2H), 3.47-3.48 (m, 1H), 3.58-3.65 (m, 3H), 3.76-3.78 (m, 1H), 4.16 (d, J = 10.0 Hz, 1H), 4.34 (d, J = 12.0 Hz, 1H), 4.45 (d, J = 12.0 Hz, 1H), 4.55-4.61 (m, 2H), 4.68-4.73 (m, 2H), 4.97 (d, J = 4.0 Hz, 1H), 5.67 (s, 1H), 7.20-7.34 (m, 15H); 13C NMR δ 23.8, 24.1, 25.8, 27.8, 31.3, 33.3, 65.7, 68.6 (2C), 70.5, 71.4, 72.6, 72.7, 73.6, 75.6, 81.7, 94.8, 127.66, 127.73, 127.9, 128.1, 128.2, 128.3, 128.4, 137.7, 138.0, 138.1, 154.2; IR: 1100, 1284, 1367, 1454, 1735 cm-1; ESIHRMS Calcd for C38H48O8Na [M+Na]+: 655.3242. Found: 655.3237; 17β Colorless oil; Yield: 56%; [α]D = 5.1° (c, 0.2); 1H NMR δ 1.21-1.33 (m, 4H), 1.44 (s, 9H), 1.48-1.55 (m, 2H), 1.74-1.76 (m, 2H), 1.92-1.94 (m, 1H), 2.00-2.02 (m, 1H), 3.31 (dd, J = 8.0, 3.0 Hz, 1H), 3.52 (dd, J = 10.0, 3.0 Hz, 1H), 3.61-3.64 (m, 1H), 3.67-3.77 (m, 2H), 3.91 (dd, J = 10.0, 3.0 Hz, 1H), 4.35 (d, J = 11.0 Hz, 1H), 4.52-4.62 (m, 2H), 4.66-4.73 (m, 2H), 4.79 (d, J = 12.0 Hz, 1H), 4.86 (d, J = 7.5 Hz, 1H), 5.63 (t, J = 2.5 Hz, 1H), 7.22-7.38 (m, 15H); 13C NMR δ 24.2, 24.5, 25.6, 27.8, 31.6, 33.4, 67.7, 68.8, 71.2, 72.2, 73.3, 73.7, 75.8, 75.9, 76.4, 82.1, 95.6, 127.4, 127.5, 127.7, 127.8, 127.9, 128.1, 128.3, 128.4, 138.0, 138.5, 138.8, 153.4; IR: 1097, 1280, 1741 cm-1; ESIHRMS Calcd for C38H48O8Na [M+Na]+: 655.3242. Found: 655.3237.
3-O-tert-Butoxycarbonyl-2,4,6-tri-O-benzyl-D-allose (18)
The donor 15 (17.8 mg, 0.028 mmol), NIS (7.5 mg, 0.033 mmol) and activated 4Å powdered molecular sieves (80 mg) were dissolved in dried CH2Cl2 (0.1 M) at room temperature, then stirred at -60 °C for 10 mins. Tf2O (6.9 μL, 0.042 mmol) was then added dropwise at -40 °C. The reaction mixture was stirred at -40 °C for 1 h before it was quenched with Na2S2O3 (20%). The reaction mixture was extracted with CH2Cl2 (3 × 5 mL), and the combined organic phase was washed with brine, dried, and concentrated. Chromatographic purification (Eluent: EtOAc/Hexanes = 1/4 then 1/2) then gave 18 (8.4 mg, 55%) as a colorless oil. [α]D = 21.8° (c, 0.1); 1H NMR δ major isomer 1.45 (s, 9H), 3.10 (d, J = 5.5 Hz, 1H), 3.28 (dd, J = 8.0, 3.5 Hz, 1H), 3.59 (dd, J = 10.0, 3.0 Hz, 1H), 3.65 (dd, J = 10.5, 4.5 Hz, 1H), 3.71-3.73 (m, 1H), 3.96-3.99 (m, 1H), 4.36 (d, J = 10.5 Hz, 1H), 4.50 (d, J = 12.0 Hz, 1H), 4.60 (d, J = 12.0 Hz, 1H), 4.67 (d, J = 13.5 Hz, 1H), 4.76 (d, J = 12.0 Hz, 1H), 5.07 (dd, J = 7.5, 5.5 Hz, 1H), 5.71 (t, J = 3.0 Hz, 1H), 7.20-7.24 (m, 2H), 7.25-7.38 (m, 13H); minor isomer 1.43 (s, 9H), 3.53 (t, J = 2.0 Hz, 1H), 3.55-3.56 (m, 1H), 3.68-3.70 (m, 1H), 3.75-3.76 (m, 2H), 3.78-3.80 (m, 1H), 3.81-3.85 (m, 1H), 4.44-4.48 (m, 2H), 4.85-4.87 (m, 1H), 5.23 (dd, J = 9.5, 3.0 Hz, 1H), 5.58 (t, J = 2.0 Hz, 1H), 7.20-7.24 (m, 2H), 7.25-7.38 (m, 13H); 13C NMR δ major isomer 27.7, 65.7, 68.9, 69.3, 71.5, 71.8, 72.8, 72.9, 73.6, 82.2, 94.4, 127.69, 127.72, 127.80, 127.85, 127.88, 127.95, 128.02, 128.04, 128.2, 128.3, 128.40, 128.43, 128.53, 128.55, 128.6, 128.8, 137.4, 137.7, 138.0, 153.5; minor isomer: 27.6, 67.0, 67.1, 68.2, 69.5, 70.5, 71.0, 71.6, 82.9, 92.0, 95.5, 136.2, 136.8, 137.2, 137.5, 138.1, 146.6, 152.7; IR: 1096, 1280, 1369, 1454, 1740 cm-1; ESIHRMS Calcd for C32H38O8Na [M+Na]+: 573.2459. Found: 573.2448.
Phenyl 3-O-tert-butoxycarbonyl-2-O-benzyl-4,6-O-benzylidene-1-thio-α-D-manno-pyranoside (21)
This compound was formed by the general procedure in 98% yield as white foam. [α]D = 102.3° (c, 0.13); 1H NMR δ 1.48 (s, 9H), 3.88 (t, J = 10.0 Hz, 1H), 4.23 (dd, J = 10.0, 5.0 Hz, 1H), 4.28-4.33 (m, 2H), 4.38-4.42 (m, 1H), 4.67 (s, 2H), 5.10 (dd, J = 10.0, 3.5 Hz, 1H), 5.52 (s, 1H), 5.59 (s, 1H), 7.28-7.37 (m, 11H), 7.41-7.43 (m, 2H), 7.50-7.51 (m, 2H); 13C NMR δ 27.8, 65.3, 68.5, 73.3, 76.3, 77.6, 82.9, 86.8, 101.8, 126.3, 127.7, 128.0, 128.2, 128.5, 129.0, 129.2, 131.7, 133.7, 137.3, 137.4, 152.7; IR: 1099, 1253, 1282, 1742 cm-1; ESIHRMS Calcd for C31H34O7SNa [M+Na]+: 573.1918. Found: 573.1914.
Phenyl 3-O-tert-butoxycarbonyl-2-O-benzyl-4,6-O-benzylidene-1-thio-α-D-mannopyranoside S-Oxide (22)
To a solution of 21 (137.9 mg, 0.25 mmol) in CH2Cl2 (5 mL), was added m-CPBA (77%, 56.1 mg) at -78 °C after which the reaction mixture was warmed with stirring to -30 °C over 40 mins before it was quenched with saturated NaHCO3, washed with brine and dried. After concentration the crude reaction mixture was purified by column chromatography (Eluent: EtOAc/Hexanes = 1/4) to give the sulfoxide (22) (127.8 mg, 90%) as white foam. [α]D = -61.5° (c, 0.2); 1H NMR δ 1.48 (s, 9H), 3.73 (t, J = 9.5 Hz, 1H), 4.15-4.23 (m, 2H), 4.30 (t, J = 9.5 Hz, 1H), 4.50 (d, J = 10.5 Hz, 2H), 4.55 (d, J = 11.5 Hz, 1H), 4.64 (d, J = 3.0 Hz, 1H), 5.43 (dd, J = 10.0, 3.5 Hz, 1H), 5.57 (s, 1H), 7.24-7.31 (m, 5H), 7.35-7.49 (m, 3H), 7.47-7.49 (m, 2H), 7.55-7.56 (m, 3H), 7.63-7.64 (m, 2H); 13C NMR δ 27.7, 68.1, 68.9, 72.7, 73.0, 73.8, 75.5, 82.9, 97.6, 101.8, 124.6, 126.2, 128.1, 128.2, 128.4, 129.1, 129.5, 131.8, 136.9, 137.0, 141.4, 152.4; IR: 1115, 1280, 1743 cm-1; ESIHRMS Calcd for C31H34O8SNa [M+Na]+: 589.1867. Found: 589.1866.
Cyclohexyl 3-O-tert-butoxycarbonyl-2-O-benzyl-4,6-O-benzylidene-α-D-mannopyranoside (23)
Compound (23) was formed from 21 and BSP or from sulfoxide 22 and Tf2O in 69% (from 21) or 75% (from 22) yield as a colorless oil. [α]D = 35.3° (c, 1.0); 1H NMR δ 1.20-1.27 (m, 5H), 1.36-1.41 (m, 1H), 1.48 (s, 9H), 1.66-1.72 (m, 3H), 1.81-1.83 (m, 1H), 3.52-3.53 (m, 1H), 3.84 (t, J = 10.0 Hz, 1H), 3.94-3.98 (m, 2H), 4.19 (t, J = 10.0 Hz, 1H), 4.23-4.25 (m, 1H), 4.60 (d, J = 12.0 Hz, 1H), 4.76 (d, J = 12.0 Hz, 1H), 4.89 (s, 1H), 5.14 ( dd, J = 10.5, 3.5 Hz, 1H), 5.57 (s, 1H), 7.28-7.42 (m, 7H), 7.46-7.50 (m, 2H), 7.56-7.59 (m, 1H); 13C NMR δ 23.8, 24.0, 25.6, 27.8, 31.1, 33.2, 64.0, 68.8, 73.5, 73.9, 75.3, 82.5, 97.1, 101.6, 126.2, 127.6, 127.9, 128.1, 128.5, 128.8, 128.9, 129.4, 131.4, 133.6, 136.6, 137.4, 137.8, 152.9; IR v 1070, 1274, 1451, 1720 cm-1; ESIHRMS Calcd for C31H40O8Na [M+Na]+: 563.2616. Found: 563.2611.
Ethyl 4-O-tert-butoxycarbonyl-2,3,6-tri-O-benzyl-1-thio-β-D-galactopyranoside (24)
Compound (24) was formed from 2,3,6-tri-O-benzyl-β-D-thiogalactopyranoside and Boc2O by the general procedure in 90% yield as a colorless oil. [α]D = 16.8° (c, 1.7); 1H NMR δ 1.31 (t, J = 7.4 Hz, 3H), 1.49 (s, 9H), 2.70-2.80 (m, 2H), 5.57-3.72 (m, 5H), 4.46 (d, J = 9.3 Hz, 1H), 4.53 (s, 2H), 4.55 (d, J = 8.3 Hz, 1H), 4.79-4.84 (m, 3H), 5.43 (dd, J = 3.1, 0.7 Hz, 1H), 7.26-7.38 (m, 15H); 13C NMR δ 15.1, 24.9, 27.7, 68.5, 69.7, 71.9, 73.9, 75.8, 75.9, 76.8, 81.2, 82.0, 85.3, 127.6, 127.7, 127.8, 127.9, 128.0, 128.2, 128.3, 128.4, 137.8, 138.0, 138.3, 153.5; IR: 1104, 1280, 1741 cm-1; ESIHRMS Calcd for C34H42O7SNa [M+Na]+: 617.2544. Found: 617.2532.
4-O-tert-Butoxycarbonyl-2,3,6-O-tribenzyl-α-D-galactopyranosyl acetate (25α) and 4-O-tertbutoxycarbonyl-2,3,6-O-benzyl-β-D-galactopyranosyl acetate (25β)
To a solution of compound 24 (32.4 mg, 0.054 mmol), BSP (13.7 mg, 0.065 mmol) and 4Å powdered molecular sieves (100 mg) in Et2O (1.0 mL), was added Tf2O (11.0 μL, 0.065 mmol) at -60 °C under N2. The reaction mixture was stirred at -60 °C for 30 minutes, and then quenched with saturated NaHCO3, extracted with CH2Cl2 (3 × 5 mL), and the combined organic phase washed with brine, dried, and concentrated. The residue was dissolved in Ac2O (1.0 mL), pyridine (1.0 mL) was added and the mixture was stirred at room temperature overnight. After removal of the solvent the residue was dissolved in CH2Cl2 (5 mL), washed with saturated NaHCO3, and the organic phase was washed with brine and dried. After concentration the crude reaction mixture was purified by column chromatography (eluent: EtOAc/Hexanes = 1/4) to give 25 (29.7 mg, 92%, α:β = 1:7.1) as a colorless oil. 25α Yield: 10%; [α]D = 14.7° (c, 0.2); 1H NMR δ 1.47 (s, 9H), 2.10 (s, 3H), 3.51-3.59 (m, 2H), 3.90-3.97 (m, 2H), 4.16 (t, J = 7.5 Hz, 1H), 4.50 (s, 2H), 4.59 (d, J = 11.0 Hz, 1H), 4.66-4.73 (m, 2H), 4.81 (d, J = 10.5 Hz, 1H), 5.49 (s, 1H), 6.35 (d, J = 3.0 Hz, 1H),7.20-7.38 (m, 15H); 13C NMR δ 21.0, 27.7, 68.2, 70.2, 70.3, 72.1, 73.6, 73.9, 74.7, 76.0, 82.3, 90.6, 127.5, 127.6, 127.8, 127.9, 128.1, 128.3, 128.4, 137.7, 138.1, 138.2, 153.2, 169.3; IR: 1108, 1279, 1369, 1454, 1744 cm-1; ESIHRMS Calcd for C34H40O9Na [M+Na]+: 615.2565. Found: 615.2559; 25β Yield: 71%; [α]D = 11.2° (c, 0.2); 1H NMR δ 1.48 (s, 9H), 2.04 (s, 3H), 3.56 (t, J = 8.5 Hz, 1H), 3.65 (t, J = 10.0 Hz, 2H), 3.79 (t, J = 8.5 Hz, 1H), 3.85 (t, J = 5.0 Hz, 1H), 4.51 (s, 2H), 4.55 (d, J = 11.5 Hz, 1H), 4.69 (d, J = 11.0 Hz, 1H), 4.82-4.86 (m, 2H), 5.44 (s, 1H), 5.59 (d, J = 8.0 Hz, 1H), 7.26-7.35 (m, 15H); 13C NMR δ 21.0, 27.8, 67.7, 69.1, 72.1, 72.8, 73.9, 75.5, 77.6, 79.8, 82.3, 94.0, 127.69, 127.72, 127.8, 127.9, 128.0, 128.1, 128.3, 128.4, 128.5, 137.6, 137.8, 138.4, 153.3, 169.3; IR: 1105, 1280, 1368, 1454, 1743 cm-1; ESIHRMS Calcd for C34H40O9Na [M+Na]+: 615.2565. Found: 615.2560.
Cyclohexyl 4-O-tert-butoxycarbonyl-2,3,6-tri-O-benzyl-α-D-galactopyranoside (26α) and Cyclohexyl 4-O-tert-butoxycarbonyl-2,3,6-tri-O-benzyl-β-D-galactopyranoside (26β)
These glycosides were obtained by the general procedure with a combined yield of 88% (α:β = 1:3.9). When the reaction was conducted in diethyl ether as solvent the yield was 62% and the α:β ratio 1:1.3. 26α Colorless oil; Yield: 18%;[α]D = 88.6° (c, 0.2); 1H NMR δ 1.15-1.26 (m, 3H), 1.31-1.38 (m, 1H), 1.44-1.53 (m, 11H), 1.71-1.78 (m, 2H), 1.86-1.91 (m, 2H), 3.54-3.59 (m, 3H), 3.82 (dd, J = 10.0, 3.5 Hz, 1H), 3.98 (dd, J = 9.5, 3.0 Hz, 1H), 4.21 (t, J = 6.0 Hz, 1H), 4.49-4.54 (m, 2H), 4.59-4.65 (m, 2H), 4.78-4.82 (m, 2H), 4.99 (d, J = 3.5 Hz, 1H), 5.42 (s, 1H), 7.20-7.38 (m, 15H); 13C NMR δ 24.2, 24.5, 25.6, 27.8, 31.6, 33.4, 67.7, 68.8, 71.2, 72.2, 73.3, 73.7, 75.8, 75.9, 76.4, 82.1, 95.7, 127.4, 127.5, 127.7, 127.8, 127.9, 128.1, 128.3, 128.4, 128.0, 138.6, 138.8, 153.4; IR: 1102, 1279, 1453, 1741 cm-1; ESIHRMS Calcd for C38H48O8Na [M+Na]+: 655.3242. Found: 655.3245; 26β Colorless oil; Yield: 70%; [α]D = 17.4° (c, 0.4); 1H NMR δ 1.22-1.30 (m, 4H), 1.40-1.53 (m, 11H), 1.74-1.76 (m, 2H), 1.90-1.97 (m, 2H), 3.55 (dd, J = 9.5, 3.5 Hz, 1H), 3.60-3.69 (m, 5H), 4.48 (d, J = 8.0 Hz, 1H), 4.54 (d, J = 2.5 Hz, 2H), 4.57 (d, J = 11.0 Hz, 1H), 4.72 (d, J = 11.0 Hz, 1H), 4.79 (d, J = 11.5 Hz, 1H), 4.92 (d, J = 10.5 Hz, 1H), 5.34 (d, J = 3.5 Hz, 1H), 7.26-7.39 (m, 15H); 13C NMR δ 24.0, 24.1, 25.7, 27.8, 31.9, 33.7, 68.7, 69.8, 72.2, 73.9, 75.3, 76.8, 77.7, 79.0, 79.6, 81.9, 102.0, 127.5, 127.8, 127.9, 128.0, 128.1, 128.2, 128.3, 128.5, 138.0, 138.2, 138.9, 153.6; IR: 1080, 1280, 1454, 1741 cm-1; ESIHRMS Calcd for C38H48O8Na [M+Na]+: 655.3242. Found: 655.3231.
Phenyl 2,3,6-tri-O-benzyl-4-O-tert-butyloxycarbonyl-1-thio-α-D-mannopyranoside (27)
Compound (27) was formed from 2,3,6-tri-O-benzyl-α-D-thiomannopyranoside and Boc2O by the general procedure in 76% yield as a colorless oil. [α]D +49.0° (c, 1.45); 1H NMR δ: 1.47 (s, 9H), 3.68 (dd, J = 3.0, 11.0 Hz, 1H), 3.73 (dd, J = 6.0, 11.0 Hz, 1H), 3.85 (dd, J = 3.0, 9.5 Hz, 1H), 4.00 (d, J = 2.0, 2.5 Hz, 1H), 4.43-4.47 (m, 1H), 4.53 (d, J = 10.0 Hz, 1H), 4.54 (s, 2H), 4.57 (d, J = 10.0 Hz, 1H), 4.64 (d, J = 12.0 Hz, 1H), 4.70 (d, J = 12.0 Hz, 1H), 5.24 (t, J = 10.0 Hz, 1H), 5.57 (d, J = 1.5 Hz, 1H), 7.19-7.39 (m, 13H), 7.49 (d, J = 7.5 Hz, 2H); 13C NMR δ: 27.8 (3C), 69.6, 71.2, 71.8, 71.9, 72.1, 73.4, 75.8, 77.5, 82.5, 85.8, 127.4-128.5 (18C), 129.0, 131.9, 134.0, 137.8, 138.0, 138.3, 152.7; ESI HRMS Calcd for C38H42O7 SNa [M+Na]+: 665.2544. Found 665.2540.
Isopropyl 2,3,6-tri-O-benzyl-4-O-tert-butyloxycarbonyl-α-D-mannopyranoside (28α) and isopropyl 2,3,6-tri-O-benzyl-4-O-tert-butyloxycarbonyl-β-D-mannopyranoside (28β)
28α. [α]D +21.6° (c, 0.3); 1H NMR δ: 1.06 (d, J = 6.0 Hz, 3H), 1.16 (d, J = 6.0 Hz, 3H), 1.42 (s, 9H), 3.60-3.68 (m, 2H), 3.73 (t, J = 2.5 Hz, 1H), 3.88-3.98 (m, 3H), 4.54 (dd, J = 4.5, 12.0 Hz, 2H), 4.60 (dd, J = 1.5, 12.0 Hz, 2H), 4.67 (d, J = 12.0 Hz, 1H), 4.78 (d, J = 12.5 Hz, 1H), 4.93 (d, J = 1.5 Hz, 1H), 5.16 (t, J = 10.0 Hz, 1H), 7.20-7.40 (m, 15H); 13C NMR δ: 21.2, 23.2, 27.7, 69.0, 69.8, 70.2, 72.0, 72.2, 72.8, 73.4, 74.6, 77.7, 82.2, 95.9, 127.3-128.3 (15C), 138.4 (3C), 152.7; IR: 1746 cm-1; ESI HRMS Calcd for C35H44O8Na [M+Na]+: 615.2929. Found 615.2930; 28β [α]D -54.3° (c, 0.3); 1H NMR δ: 1.16 (d, J = 6.0 Hz, 3H), 1.30 (d, J = 6.0 Hz, 3H), 1.46 (s, 9H), 3.46 (dd, J = 3.0, 9.5 Hz, 1H), 3.56-3.59 (m, 1H), 3.67-3.74 (m, 2H), 3.84 (d, J = 3.0 Hz, 1H), 4.01 (m, 1H), 4.35 (d, J = 12.0 Hz, 1H), 4.44 (d, J = 12.0 Hz, 1H), 4.47 (br. s, 1H), 4.54 (d, J = 12.0, 1H), 4.61 (d, J = 12.0 Hz, 1H), 4.88 (d, J = 12.5 Hz, 1H), 4.96 (d, J = 12.5 Hz, 1H), 5.04 (t, J = 10.0 Hz, 1H), 7.20-7.49 (m, 15H); 13C NMR δ: 21.8, 23.6, 27.7 (3C), 70.4, 71.3, 71.5, 72.2, 73.6, 73.7, 74.3, 76.4, 78.1, 79.8, 82.4, 99.4, 126.7-128.9 (15C), 138.1, 138.3, 138.6, 152.7; ESI HRMS Calcd for C35H44O8Na [M+Na]+: 615.2929. Found 615.2925.
N(2′,3′,6′-Tri-O-benzyl-4′-O-tert-butyloxycarbonyl-α-D-mannopyranosyl)-succinimide (29)
[α]23D +62.0° (c, 0.5); 1H NMR δ: 1.49 (s, 9H), 2.48-2.56 (m, 2H), 3.67 (dd, J = 5.0, 11.0 Hz, 1H), 3.78 (dd, J = 6.5, 11.0 Hz, 1H), 3.96 (t, J = 2.5 Hz, 1H), 4.36 (d, J = 12.0 Hz, 1H), 4.43 (t, J = 5.0 Hz, 1H), 4.48 (d, J = 12.0 Hz, 1H), 4.54 (s, 2H), 4.71 (d, J = 12.0 Hz, 1H), 4.75 (d, J = 12.0 Hz, 1H), 4.78 (dd, J = 3.0, 8.0 Hz, 1H), 4.94 (dd, J = 3.0, 5.0 Hz, 1H), 5.77 (d, J = 9.0 Hz, 1H), 7.18-7.40 (m, 17H); 13C NMR δ: 27.8, 28.0, 68.2, 71.6, 71.8, 72.1, 72.3, 73.4, 74.4, 75.3, 83.0, 127.6-128.4 (15C), 137.8, 138.1 (2C), 152.6, 176.7 (2C); IR: 1711, 1720, 1739 cm-1; ESI HRMS Calcd for C36H41O9NNa [M+Na]+: 654.2674. Found 654.2669.
p-Tolyl 4-O-tert-butoxycarbonyl-3-azido-2,3,6-trideoxy-1-thio-α-L-lyxopyranoside (30)
Compound 30 was formed by general procedure in 98% yield as a colorless oil. [α]D = -287.1° (c, 0.24); 1H NMR δ 1.21 (d, J = 6.0 Hz, 3H), 1.53 (s, 9H), 2.12 (td, J = 13.5, 5.5 Hz, 1H), 2.33 (s, 3H), 2.36 (dd, J = 13.5, 5.5 Hz, 1H), 3.89-3.95 (m, 1H), 4.34-4.37 (m, 1H), 4.43 (t, J = 9.5 Hz, 1H), 5.49 (d, J = 5.5 Hz, 1H), 7.11 (d, J = 8.0 Hz, 2H), 7.32 (d, J = 8.0 Hz, 2H); 13C NMR δ 17.2, 21.1, 27.7, 36.0, 58.3, 66.7, 78.7, 83.2, 83.4, 129.9, 130.3, 132.1, 137.8, 152.7; IR: 1097, 1274, 1750, 2104 cm-1; ESIHRMS Calcd for C18H25N4SNa [M+Na]+: 402.1458. Found: 402.1456.
4-O-tert-butoxycarbonyl-3-azido-2,3,6-trideoxy-1-L-lyxopyranosyl acetate (31)
Application of the BSP protocol to compound 30 gave 4-O-tert-butoxycarbonyl-3-azido-2,3,6-trideoxy-1-α-L-lyxopyranose in 30% yield as a colorless oil with 1H NMR δ 1.21 (d, J = 6.0 Hz, 3H), 1.52 (s, 9H), 1.79 (td, J = 13.0, 3.5 Hz, 1H), 2.05 (dd, J = 13.0, 5.0 Hz, 1H), 3.75-3.78 (m, 1H), 3.86-3.91 (m, 1H), 4.42 (t, J = 10.0 Hz, 1H), 5.12 (d, J = 3.0 Hz, 1H); 13C NMR δ 17.4, 27.7, 34.8, 57.2, 66.5, 76.4, 91.4, 96.1, 152.6; IR: 1132, 1255, 1749, 2103 cm-1. Acetylation of this pyranose according to the procedure used for 25 gave the title compound 31 in 73% combined yield (α:β = 1:1.7) as a colorless oil. [α]D = -21.9° (c, 0.2); 1H NMR (400 MHz) δ major isomer β 1.27 (d, J = 6.0 Hz, 3H), 1.51 (s, 9H), 1.73-1.82 (m, 1H), 2.12 (s, 3H), 2.23-2.28 (m, 1H), 3.57-3.67 (m, 2H), 4.43 (t, J = 9.6 Hz, 1H), 5.72 (dd, J = 9.6, 1.6 Hz, 1H); minor isomer α 1.22 (d, J = 6.0 Hz, 3H), 1.57 (s, 9H), 1.85-1.89 (m, 1H), 2.09 (s, 3H), 2.15-2.20 (m, 1H), 3.86-3.93 (m, 2H), 4.46 (t, J = 10.0 Hz, 1H), 6.15 (d, J = 2.4 Hz, 1H); 13C NMR (100 MHz) δ major isomer (β) 17.5, 21.2, 27.9, 35.2, 59.6, 71.9, 77.4, 83.5, 91.5, 152.8, 169.2; minor isomer (α) 17.6, 31.1, 34.3, 57.4, 68.5, 78.1, 83.7, 90.6, 152.1, 169.1; IR: 1259, 1752, 2101 cm-1; ESIHRMS Calcd for C13H21N3O6Na [M+Na]+ : 338.1328. Found: 338.1322.
Cyclohexyl 4-O-tert-butoxycarbonyl-3-azido-2,3,6-trideoxy-L-lyxopyranoside (32)
Compound 32 was formed from 30 by the general procedure in 46% yield (α:β = 1:3.7) as a colorless oil. [α]D = -3.3° (c, 0.15); 1H NMR δ major isomer 1.26 (d, J = 6.5 Hz, 3H), 1.22-1.32 (m, 4H), 1.34-1.41 (m, 2H), 1.51 (s, 9H), 1.69-1.77 (m, 3H), 1.85-1.94 (m, 2H), 2.17-2.21 (m, 1H), 3.41-3.46 (m, 1H), 3.52-3.57 (m, 1H), 3.61-3.66 (m, 1H), 4.36-4.42 (m, 1H), 4.62 (dd, J = 9.5, 2.0 Hz, 1H); minor isomer 1.19 (d, J = 6.5 Hz, 3H), 1.55 (s, 9H), 2.09-2.13 (m, 1H), 3.88-3.97 (m, 2H), 3.52-3.57 (m, 1H), 5.00 (d, J = 3.0 Hz, 1H) 13C NMR δ 17.5, 24.0, 24.2, 25.6, 27.7, 31.8, 33.5, 36.8, 59.9, 70.6, 76.4, 77.3, 78.0, 78.1, 83.2, 96.1, 97.2, 152.6; IR: 1130, 1749, 2103 cm-1; ESIHRMS Calcd for C17H29N3O5Na [M+Na]+: 378.2000. Found: 378.2005.
Phenyl 2,3,4-tri-O-benzyl-6-O-tert-butyloxycarbonyl-1-thio-β-D-glucopyranoside (33)
Compound (33) was formed from 2,3,4-tri-O-benzyl-β-D-thioglucopyranoside and Boc2O by the general procedure in 80% yield as a colorless oil. [α]D +2.7° (c, 0.8); 1H NMR δ: 1.53 (s, 9H), 3.55 (t, J = 9.0 Hz, 1H), 3.60-3.64 (m, 2H), 3.74-3.79 (m, 1H), 4.33 (dd, J = 5.0 Hz, 1H), 4.39 (dd, J = 1.5, 10.0 Hz, 1H), 4.63 (d, J = 10.5 Hz, 1H), 4.70 (d, J = 10.0 Hz, 1H), 4.77 (d, J = 10.0 Hz, 1H), 4.89 (d, J = 10.0 Hz, 1H), 4.90 (d, J = 10.0 Hz, 1H), 4.94-4.99 (m, 2H), 7.26-7.50 (m, 18H), 7.61 (d, J = 7.5, 1H); 13C NMR δ: 27.9, 65.7, 75.3, 75.6, 75.9, 77.0, 77.7, 80.9, 82.3, 86.7, 87.7, 127.6-129.0 (18C), 132.0 (2C), 133.8, 137.7, 138.0, 138.3, 153.5; ESI HRMS Calcd for C38H42O7SNa [M+Na]+: 665.2544. Found 665.2539.
Isopropyl 2,3,4-tri-O-benzyl-6-O-tert-butyloxycarbonyl-α-D-glucopyranoside (34α) and isopropyl 2,3,4-tri-O-benzyl-6-O-tert-butyloxycarbonyl-β-D-glucopyranoside (34β)
Compounds 34α and 34β were formed from 33 by the general NIS procedure in 80% yield (α:β = 1:1, dichloromethane) or 74% yield (α:β = 1.5:1, diethyl ether) as a colorless oily mixture with ESI HRMS Calcd for C35H44O8Na [M+Na]+: 615.2929. Found 615.2922. 34α 1H NMR δ: 1.18 (d, J = 6.0 Hz, 3H), 1.22 (d, J = 6.0 Hz, 3H), 1.46 (s, 9H), 3.54-3.57 (m, 1H), 3.87 (m, J = 6.0 Hz, 1H), 3.91 (m, 1H), 4.01 (t, J = 9.0 Hz, 1H), 4.19 (dd, J = 2.0, 12.0 Hz, 1H), 4.28 (m, 1H), 4.33 (dd, J = 4.5, 11.5 Hz, 1H), 4.55 (d, J = 12.0 Hz, 1H), 4.69 (d, J = 10.5 Hz, 1H), 4.76 (d, J = 12.0 Hz, 1H), 4.82 (d, J = 10.5 Hz, 1H), 4.84 (d, J = 3.0 Hz, 1H), 4.88 (d, J = 11.0 Hz, 1H), 5.02 (d, J = 11.0 Hz, 1H), 7.21-7.29 (m, 15H); δ: 21.4, 23.4, 28.0 (3 C), 65.7, 68.9, 69.5, 73.4, 75.5, 78.0, 80.1, 82.2, 82.3, 95.0, 127.8-128.7 ( 15 C), 138.2, 138.4, 139.1, 153.7; 34β. 1H NMR δ: 1.23 (d, J = 6.0 Hz, 3H), 1.28 (d, J = 6.0 Hz, 3H), 1.47 (s, 9H), 3.42 (t, J = 8.0 Hz, 1H), 3.46-3.54 (m, 3H), 3.64 (t, J = 9.0 Hz, 1H), 4.00 (m, 1H), 4.27 (m, 1H), 4.46 (d, J = 8.0 Hz), 4.56 (d, J = 10.5 Hz, 1H), 4.64 (d, J = 12.0 Hz, 1H), 4.78 (d, J = 10.5 Hz, 1H), 4.85 (d, J = 10.5 Hz, 1H), 4.93 (d, J = 12.5 Hz, 1H), 4.96 (d, J = 12.0 Hz, 1H), 7.21-7.29 (m, 15H); 13C NMR δ: 22.4, 23.9, 28.0 (3 C), 66.0, 72.6, 73.1, 75.1, 75.3, 75.9, 78.1, 82.4, 85.0, 102.3, 127.8-128.7 (15 C), 138.0, 138.7, 138.8, 153.6.
Di-(2,3,4-tri-O-benzyl-D-glucopyranoside)-(1,6),(6,1)-dicarbonate (35)
[α]D +88.7° (c, 0.3); 1H NMR (C6D6) δ 3.12 (t, J = 9.0 Hz, 2H), 3.53 (ddd, J = 1.2, 4.0, 9.0 Hz, 2H), 4.14 (t, J = 9.0 Hz, 2H), 4.22 (t, J = 9.0 Hz, 2H), 4.33 (t, J = 9.0 Hz, 2H), 4.48 (dd, J = 12.0, 20.0 Hz, 4H), 4.61 (d, J = 11.5 Hz, 2H), 4.68 (d, J = 11.0 Hz, 2H), 4.81 (d, J = 11.5 Hz, 2H), 4.93 (d, J = 11.5 Hz, 2H), 6.21 (d, J = 3.0 Hz, 2H), 7.14-7.37 (m, 30H); 13C NMR δ: 67.0, 72.5, 72.6, 75.4, 77.3, 78.7, 81.7, 93.6, 127.6-128.4 (30C), 138.0, 138.1, 139.1, 153.0; IR: 1768 cm-1; ESI HRMS Calcd for C56H56O14Na [M+Na]+: 975.3568. Found: 975.3582.
Ethyl 3,4,6-tri-O-benzyl-2-O-(2-carboxybenzoyl)-1-thio-β-D-glucopyranoside (36)
To a solution of ethyl 3,4,6-tri-O-benzyl-β-D-thioglucopyranoside (460 mg, 0.92 mmol) in dry DMF (10.0 mL), was added NaH (60%, 73.6 mg, 1.84 mmol) at room temperature. The reaction mixture was stirred for 10 mins, and then phthalic anhydride (272.5 mg, 1.84 mmol) was added and the mixture was stirred at room temperature overnight. After removal of the solvent, the residue was taken up in 5% HCl (20 mL), and extracted with CH2Cl2 (3 × 20 mL). The combined organic phase was washed with brine and dried. After concentration the residue was purified by column chromatography (eluent: EtOAc/Hexanes = 1/1) to give the compound 36 in 37% yield as a colorless oil. [α]D = 15.9° (c, 4.3); 1H NMR (400 MHz) δ 1.27 (t, J = 7.6 Hz, 3H), 2.70-2.80 (m, 2H), 3.59 (dd, J = 9.6, 2.4 Hz, 1H), 3.70-3.80 (m, 3H), 3.88 (t, J = 8.8 Hz, 1H), 4.55-4.64 (m, 4H), 4.76-4.82 (m, 3H), 5.29-5.36 (m, 1H), 7.15-7.35 (m, 15H), 7.51-7.57 (m, 2H), 7.76-7.84 (m, 2H), 9.22 (br. s, 1H); 13C NMR (100 MHz) δ 15.1, 24.0, 69.1, 73.5, 73.7, 75.3, 75.3, 78.1, 79.7, 83.4, 84.2, 127.82, 127.84, 128.0, 128.1, 128.2, 128.3, 128.4, 128.5, 128.61, 128.64, 129.5, 130.2, 130.7, 131.4, 132.0, 132.7, 138.2, 138.3, 138.4, 165.7, 172.6; IR: 1070, 1266, 1454, 1704, 1729 cm-1; ESIHRMS Calcd for C37H38O8SNa [M+Na]+: 665.2185. Found: 665.2186.
3,4,6-Tri-O-benzyl-1,2-O-phthalidylidene-α-D-glucopyranose (37) (endo and exo mixture)
Compound 37 was formed from 36 and NIS by the general procedure in 79% yield as a colorless oil. [α]D = 48.2° (c, 0.6); 1H NMR (400 MHz) δ major isomer 3.70-3.81 (m, 3H), 4.02 (t, J = 3.6 Hz, 1H), 4.10-4.14 (m, 1H), 4.40 (d, J = 11.6 Hz, 1H), 4.51-4.63 (m, 4H), 4.71-4.76 (m, 2H), 6.09-6.11 (m, 1H), 7.18-7.20 (m, 2H), 7.26-7.34 (m, 13H), 7.61-7.67 (m, 3H), 7.86-7.88 (m, 1H); minor isomer 3.85 (dd, J = 10.8, 3.2 Hz, 1H), 4.19-4.23 (m, 1H), 4.43-4.46 (m, 1H), 4.81-4.87 (m, 2H), 7.21-7.23 (m, 2H), 7.57 (d, J = 7.2 Hz, 1H), 7.75 (t, J = 7.2 Hz, 1H); 13C NMR (100 MHz) δ major isomer 69.4, 71.4, 72.2, 73.2, 73.8, 74.8, 75.8, 80.4, 82.8, 98.9, 123.6, 125.5, 127.94, 127.99, 128.03, 128.1, 128.2, 128.3, 128.4, 128.5, 128.6, 128.8, 132.4, 135.0, 137.3, 137.8, 138.2, 140.9, 165.7; minor isomer 68.4, 72.1, 73.5, 74.9, 75.0, 101.7, 123.1, 125.4, 127.89, 132.2, 135.1, 138.0, 138.4, 142.6; IR: 891, 1061, 1108, 1283, 1453, 1783 cm-1; ESIHRMS Calcd for C35H32O8Na [M+Na]+: 603.1995. Found: 603.1994.
Phenyl 2,3,6-tri-O-benzyl-4-O-(2-carboxybenzoyl)-1-thio-β-D-galactopyranoside (38)
Compound 38 was formed from phenyl 2,3,6-tri-O-benzyl-β-D-thiogalactopyranoside and phthalic anhydride by the same procedure as compound 36 in 48% yield as a colorless oil. [α]D = 16.2° (c, 1.1); 1H NMR (400 MHz) δ 3.67-3.77 (m, 4H), 3.84 (t, J = 6.0 Hz, 1H), 4.49-4.56 (m, 3H), 4.71-4.79 (m, 3H), 4.82 (d, J = 11.2 Hz, 1H), 5.85 (d, J = 1.6 Hz, 1H), 7.15-7.38 (m, 18H), 7.49-7.58 (m, 4H), 7.68 (d, J = 8.0 Hz, 1H), 7.77 (d, J = 8.0 Hz, 1H); 13C NMR (100 MHz) δ 68.7, 69.0, 72.3, 73.9, 76.0, 76.6, 77.0, 81.5, 88.0, 127.6, 127.98, 128.02, 128.1, 128.3, 128.5, 128.61, 128.63, 128.7, 129.1, 129.67, 129.74, 130.4, 131.3, 131.5, 131.8, 132.0, 132.1, 133.9, 137.8, 138.1, 138.4, 166.9, 171.1; IR: 1067, 1102, 1280, 1728 cm-1; ESIHRMS Calcd for C41H38O8SNa [M+Na]+: 713.2185. Found: 713.2191.
3,4,6-Tri-O-benzyl-1,4-O-phthaloyl-α-D-galactopyranose (40)
Compound 40 was formed from 38 and NIS by the general procedure in 41% yield as a colorless oil. [α]D = 80.0° (c, 0.3);1H NMR (400 MHz) δ 3.19-3.27 (m, 2H), 3.71 (dd, J = 10.0, 2.4 Hz, 1H), 3.79-3.81 (m, 1H), 4.13 (d, J = 12.0 Hz, 1H), 4.32 (dd, J = 10.0 4.4 Hz, 1H), 4.35 (d, J = 12.0 Hz, 1H), 4.68 (d, J = 12.4 Hz, 1H), 4.81 (d, J = 10.4 Hz, 1H), 4.87 (d, J = 10.4 Hz, 1H), 4.93 (d, J = 13.2 Hz, 1H), 5.85 (d, J = 2.4 Hz, 1H), 6.58 (d, J = 4.0 Hz, 1H), 6.97-6.99 (m, 3H), 7.03-7.04 (m, 2H), 7.09 (d, J = 8.0 Hz, 1H), 7.20-7.31 (m, 6H), 7.39-7.43 (m, 5H), 7.51-7.54 (m, 1H), 8.30 (d, J = 8.0 Hz, 1H); 13C NMR (100 Hz) δ 67.0, 68.6, 69.9, 70.9, 73.8, 74.9, 75.2, 75.7, 77.4, 93.1, 125.7, 127.6, 127.7, 127.8, 127.9, 128.1, 128.3, 128.47, 128.51, 128.8, 129.6, 132.0, 133.0, 135.9, 137.4, 138.3, 138.6, 164.0, 167.1; IR: 1067, 1105, 1261, 1724 cm-1; ESIHRMS Calcd for C27H28O5SNa [M-C8H4O3(phthalic anhydride)+Na]+: 455.1834. Found: 455.1855.
Phenyl 2,3,6-tri-O-benzyl-4-O-(4-methoxybenzoyl)-1-thio-β-D-galactopyranoside (41)
To a solution of 4-methoxybenzoic acid (102.1 mg, 0.76 mmol) in dry CH3CN (5 mL), was added 1,1′-carbonyl diimidazole (129.2 mg, 0.80 mmol) under N2. The reaction mixture was stirred at 60-70 °C for 2 h then cooled to room temperature before phenyl 2,3,6-tri-O-benzyl-β-D-thiogalactopyranoside (196.6 mg, 0.36 mmol) in CH3CN (4 mL) was added by dropwise followed by DBU (121.0 μL, 0.76 mmol). The reaction mixture was stirred at 60 °C overnight, cooled, poured into saturated aqueous NaHCO3, and extracted with CH2Cl2 (3 × 20 mL). The combined organic phase was washed with brine, dried, and concentrated. The residue was purified by column chromatography (eluent: EtOAc/Hexanes = 1/4) to give the 41 in 93% yield as a colorless oil. [α]D = 22.5° (c, 2.2); 1H NMR δ 3.60 (dd, J = 10.0, 6.5 Hz, 1H), 3.69-3.78 (m, 3H), 3.89 (d, J = 7.5 Hz, 1H), 3.91 (s, 3H), 4.46-4.55 (m, 3H), 4.72 (d, J = 9.5 Hz, 1H), 4.76 (s, 2H), 4.87 (d, J = 11.5 Hz, 1H), 5.88 (d, J = 3.0 Hz, 1H), 6.93-6.96 (m, 2H), 7.24-7.41 (m, 18H), 7.65-7.67 (m, 2H), 7.96-7.99 (m, 2H); 13C NMR δ 55.7, 67.3, 68.8, 72.0, 74.0, 75.9, 76.6, 76.9, 81.7, 87.4, 113.9, 122.4, 127.8, 127.9, 128.0, 128.1, 128.4, 128.5, 128.56, 128.58, 128.6, 129.1, 132.3, 133.1, 133.3, 137.90, 137.92, 138.5, 163.8, 165.6; IR: 1102, 1257, 1454, 1605, 1716 cm-1; ESIHRMS Calcd for C41H40O7SNa [M+Na]+: 699.2392. Found: 699.2397.
2,3,6-Tri-O-benzyl-4-O-(4-methoxybenzoyl)-D-[1-16/18O]-galactopyranosyl acetate (47)
To a solution of donor 41 (35.7 mg, 0.053 mmol), BSP (13.2 mg, 0.063 mmol) and activated 4Å molecular sieves (100 mg) in dichloromethane (1 mL), was added Tf2O (13.3 μL, 0.079 mmol) at -60 °C. The mixture was stirred at -60 °C for 2 h before H2182O (95% 18O, 50 μL) was added. The reaction mixture was filtered, dried and concentrated. The residue was purified by column chromatography (eluent: EtOAc/Hexanes = 1/2) to give 2,3,6-tri-O-benzyl-4-O-(4-methoxybenzoyl)-α-D-[1-16/18O]-galactopyranose 43 (38.1 mg, 82%) as a colorless oil. From the 13C NMR, 16O/18O is around 4:1. [α]D = 98.3° (c, 1.0); 1H NMR δ 3.49 (d, J = 6.5 Hz, 2H), 3.89 (s, 3H), 3.95 (dd, J = 10.0, 3.5 Hz, 1H), 4.13 (dd, J = 10.0, 3.5 Hz, 1H), 4.36 (d, J = 12.5 Hz, 1H), 4.44 (d, J = 11.5 Hz, 1H), 4.56 (d, J = 11.5 Hz, 1H), 4.60 (t, J = 6.0 Hz, 1H), 4.68-4.71 (m, 2H), 4.86 (d, J = 11.5 Hz, 1H), 5.35 (d, J = 3.5 Hz, 1H), 5.83 (d, J = 2.5 Hz, 1H), 6.90 (d, J = 8.5 Hz, 2H), 7.19-7.27 (m, 13H), 7.32 (d, J = 6.5 Hz, 2H), 7.96 (d, J = 9.0 Hz, 2H); 13C NMR δ 55.7, 68.7, 68.9, 69.2, 71.8, 73.5, 73.8, 75.0, 76.6, 94.1(94.050, 94.072), 113.8, 122.7, 127.6, 127.69, 127.73, 127.9, 128.06, 128.08, 128.4, 128.45, 128.50, 132.2, 138.1, 138.5, 138.7, 163.7, 165.7; IR: 1100, 1257, 1454, 1605, 1717 cm-1. This pyranose was dissolved in Ac2O pyridine was added and the mixture was stirred at 80 °C overnight. After removal of the solvents the residue was dissolved in CH2Cl2, washed with saturated NaHCO3, and brine and dried. After concentration the crude reaction mixture was purified by column chromatography (eluent: EtOAc/Hexanes = 1/4) to give compound 47 (90%, α:β = 4.1:1) as a colorless oil. From the 13C NMR the 16O/18O ratio is ∼1:1. Mass spectrometry also displayed a 1:1 16O/18O ratio for the sodiated molecular ion. [α]D = 40.9° (c, 0.5); 1H NMR δ major isomer 3.48-3.56 (m, 3H), 3.88 (s, 3H), 3.98-4.03 (m, 2H), 4.25 (t, J = 6.5 Hz, 1H), 4.39-4.41 (m, 1H), 4.46-4.50 (m, 1H), 4.54-4.59 (m, 1H), 4.66-4.70 (m, 3H), 4.87 (d, J = 11.5 Hz, 2H), 5.91 (s, 1H), 6.41 (d, J = 3.0 Hz, 1H), 6.92 (d, J = 9.0 Hz, 2H), 7.21-7.33 (m, 15H), 7.97 (d, J = 9.0 Hz, 2H); minor isomer 3.60-3.62 (m, 1H), 3.89 (s, 3H), 3.95 (t, J = 6.0 Hz, 1Hz), 4.81 (d, J = 12.0 Hz, 1H), 5.67 (d, J = 7.5 Hz, 1H), 5.86 (d, J = 2.5 Hz, 1H), 6.94 (d, J = 9.0 Hz, 2H), 7.21-7.36 (m, 15H), 8.05 (d, J = 9.0 Hz, 2H); 13C NMR δ major isomer 21.4, 55.7, 67.8, 68.5, 70.8, 72.0, 74.0, 74.6, 76.4, 80.1, 91.0 (high resolution: 90.991, 91.020), 113.9, 127.9, 128.0, 128.07, 128.14, 128.18, 128.23, 128.3, 128.4, 128.52, 128.55, 128.59, 132.2, 137.8, 138.2, 138.3, 163.79, 165.6, 169.7 (169.739, 169.752); minor isomer 21.2, 66.8, 67.9, 72.1, 73.5, 73.86, 73.90, 75.6, 77.8, 94.3 (94.243, 94.267), 122.4, 127.7, 132.3, 137.7, 137.9, 138.5, 163.83, 156.5, 169.4; IR ν 1100, 1257, 1605, 1717, 1750 cm-1; ESIHRMS Calcd for C37H38O9Na [M+Na]+: 649.2414. Found: 649.2404; ESIHRMS Calcd for C37H38O818ONa [M+Na]+: 651.2456. Found: 651.2465.
Phenyl 2,3,6-tri-O-benzyl-4-O-(4-methoxybenzoyl)-1-thio-α,β-D-galactopyranoside (41α and 41β)
To a solution of phenyl 2,3,6-tri-O-benzyl-4-O-(4-methoxybenzoyl)-D-[1-16/18O]- galactopyranosyl acetate (47) (13.9 mg, 0.022 mmol) in CH3CN (1 mL), was added thiophenol (34 μL, 0.033 mmol) and BF3·Et2O (9.3 μL, 0.07 mmol) under N2 at room temperature. The reaction mixture was stirred at room temperature overnight then was poured into saturated aqueous NaHCO3, and extracted with CH2Cl2 (3 × 5 mL). The combined organic phase was washed with brine and dried. Concentration and purification of the residue by column chromatography (eluent: EtOAc/Hexanes = 1/4) gave 41 (α:β = 2.2:1) in 97% yield as a colorless oil. [α]D = 58.4° (c, 0.5); 1H NMR δ major isomer 3.48-3.53 (m, 2H), 3.67-3.76 (m, 1H), 3.80 (s, 3H), 3.87 (dd, J = 10.0, 3.0 Hz, 1H), 4.15 (dd, J = 10.0, 5.05 Hz, 1H), 4.38-4.43 (m, 1H), 4.56 (d, J = 11.5 Hz, 1H), 4.66-4.80 (m, 3H), 4.78 (d, J = 12.0 Hz, 1H), 5.65 (d, J = 5.5 Hz, 1H), 5.79 (d, J = 3.0 Hz, 1H), 6.83-6.86 (m, 2H), 7.13-7.27 (m, 16H), 7.29-7.31 (m, 2H), 7.46-7.48 (m, 2H), 7.90-7.91 (m, 2H); minor isomer 3.67-3.76 (m, 1H), 3.83 (s, 3H), 4.38-4.43 (m, 2H), 4.44- 4.46 (m, 1H), 4.62 (d, J = 9.0 Hz, 1H), 4.77 (d, J = 11.0 Hz, 1H), 5.79 (d, J = 3.0 Hz, 1H), 6.83-6.86 (m, 2H), 7.13-7.27 (m, 16H), 7.29-7.31 (m, 2H), 7.56-7.58 (m, 2H), 7.87-7.89 (m, 2H); 13C NMR δ major isomer 55.70, 68.5, 69.0, 69.3, 72.2, 73.1, 73.7, 75.5, 76.8, 88.1, 113.9, 122.5, 127.5, 127.7, 127.8, 127.9, 127.95, 127.97, 128.06, 128.12, 128.2, 128.3, 128.4, 128.46, 128.52, 128.56, 128.61, 129.10, 132.2, 132.5, 133.0, 138.2, 138.3, 163.7, 165.6; minor isomer 55.73, 67.2, 68.8, 71.9, 73.9, 75.9, 76.6, 77.0, 81.7, 87.4, 122.4, 129.1, 132.3, 133.2, 134.2, 137.8, 138.0, 138.5, 163.7, 165.6; IR: 1101, 1256, 1605, 1716 cm-1; ESIHRMS Calcd for C41H40O7SNa [M+Na]+: 699.2392. Found: 699.2372. Inspection by 13C NMR spectroscopy revealed no isotopically shifted signal for the anomeric carbon. No evidence was found by mass spectrometry for the presence of 18O.
Supplementary Material
Acknowledgment
We thank the NIH (GM62160) for support of this work, Professor Todd Lowary (University of Alberta) for donation of the immediate precursor to compound 30, and an anonymous reviewer for an insightful suggestion regarding the effect of esters at O4.
References
- 1.Capon B, McManus SP. Neighboring Group Participation. Plenum; New York: 1976. [Google Scholar]
- 2 A).Capon B. Chem. Rev. 1969;69:407–498. [Google Scholar]; B) Bochkov AF, Zaikov GE. Chemistry of the O-Glycosidic Bond. Pergamon; Oxford: 1979. [Google Scholar]; C) Ernst B, Hart GW, Sinaÿ P, editors. Carbohydrates in Chemistry and Biology. Wiley-VCH; Weinheim: 2000. [Google Scholar]; D) Fraser-Reid B, Kuniaki T, Thiem J, editors. Glycoscience: Chemistry and Chemical Biology. Springer-Verlag; Berlin: 2001. [Google Scholar]; E) Demchenko AV, editor. Handbook of Chemical Glycosylation: Advances in Stereoselectivity and Therapeutic Relevance. Wiley-VCH; Weinheim: 2008. [Google Scholar]; F) Green LG, Ley SV. In: Carbohydrates in Chemistry and Biology. Ernst B, Hart GW, Sinaÿ P, editors. Vol. 1. Wiley-VCH; Weinheim: 2000. pp. 427–448. [Google Scholar]; G) Sinnott ML. Carbohydrate Chemistry and Biochemistry. RSC Publishing; Cambridge: 2007. [Google Scholar]
- 3.Paulsen H, Herold C-P. Chem. Ber. 1970;103:2450–2462. [Google Scholar]
- 4.Crich D, Dai Z, Gastaldi S. J. Org. Chem. 1999;64:5224–5229. doi: 10.1021/jo990424f. [DOI] [PubMed] [Google Scholar]
- 5.Nukada T, Berces A, Zgierski MZ, Whitfield DM. J. Am. Chem. Soc. 1998;120:13291–13295. [Google Scholar]
- 6 a).Crich D, Li M. Org. Lett. 2007;9:4115–4118. doi: 10.1021/ol701466u. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Konradsson P. In: Glycoscience: Chemistry and Chemical Biology. Fraser-Reid B, Tatsuta K, Thiem J, editors. Vol. 1. Springer; Berlin: 2001. pp. 535–549. [Google Scholar]
- 7 a).Smoot JT, Pornsuriyasak P, Demchenko AV. Angew. Chem. Int. Ed. 2005;44:7123–7126. doi: 10.1002/anie.200502694. [DOI] [PubMed] [Google Scholar]; b) Kim J-H, Yang H, Boons G-J. Angew. Chem. Int. Ed. 2005;44:947–949. doi: 10.1002/anie.200461745. [DOI] [PubMed] [Google Scholar]; c) Kim J-H, Yang H, Park J, Boons G-J. J. Am. Chem. Soc. 2005;127:12090–12097. doi: 10.1021/ja052548h. [DOI] [PubMed] [Google Scholar]
- 8.Yamada T, Takemura K, Yoshida J.-i., Yamago S. Angew. Chem. Int. Ed. 2006;45:7575–7578. doi: 10.1002/anie.200602699. [DOI] [PubMed] [Google Scholar]
- 9.For a different opinion on participation by 2-O-phosphate esters see Crich D, Hutton TK, Banerjee A, Jayalath P, Picione J. Tetrahedron: Asymmetry. 2005;16:105–119.
- 10.Jiao H, Hindsgaul O. Angew. Chem. Int. Ed. 1999;38:346–348. doi: 10.1002/(SICI)1521-3773(19990201)38:3<346::AID-ANIE346>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 11.Kulkarni SS, Liu Y-H, Hung S-C. J. Org. Chem. 2005;70:2808–2811. doi: 10.1021/jo047794a. [DOI] [PubMed] [Google Scholar]
- 12.Crich D, Cai W, Dai Z. J. Org. Chem. 2000;65:1291–1297. doi: 10.1021/jo9910482. [DOI] [PubMed] [Google Scholar]
- 13 a).Crich D, Sun S. Tetrahedron. 1998;54:8321–8348. [Google Scholar]; b) Crich D, Lim LBL. Org. React. 2004;64:115–251. [Google Scholar]
- 14 a).Crich D, Vinod AU, Picione J. J. Org. Chem. 2003;68:8453–8458. doi: 10.1021/jo035003j. [DOI] [PubMed] [Google Scholar]; b) Crich D, Yao Q. J. Am. Chem. Soc. 2004;126:8232–8236. doi: 10.1021/ja048070j. [DOI] [PubMed] [Google Scholar]
- 15.van Boeckel CAA, Beetz T, van Aelst SF. Tetrahedron. 1984:4097–4107. [Google Scholar]
- 16.Also see Paulsen H, Lebuhn R. Liebigs. 1983:1047–1072.
- 17 a).Crich D, Li L. J. Org. Chem. 2007;72:1681–1690. doi: 10.1021/jo062294y. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Crich D, Vinogradova O. J. Org. Chem. 2006;71:8473–8480. doi: 10.1021/jo061417b. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Crich D, Jayalath P, Hutton TK. J. Org. Chem. 2006;71:3064–3070. doi: 10.1021/jo0526789. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Crich D, Wu B. Org. Lett. 2006;8:4879–4882. doi: 10.1021/ol061938l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18 a).Tsai TYR, Jin H, Wiesner K. Helv. Chim. Acta. 1984;62:1403–1405. [Google Scholar]; b) Smid P, de Ruiter GA, van der Marel G, van Boom JH. J. Carbohydr. Chem. 1991;10:833–849. [Google Scholar]; c) Jin H, Tsai R, Wiesner K. Can. J. Chem. 1983;61:2442. [Google Scholar]; d) Chiba S, Kitamura M, Narasaka K. J. Am. Chem. Soc. 2006;128:6931–6937. doi: 10.1021/ja060408h. [DOI] [PubMed] [Google Scholar]
- 19.Ustyuzhanina N, Komarova B, Zlotina N, Krylov V, Gerbst A, Tsvetkov Y, Nifantiev N. Synlett. 2006:921–923. [Google Scholar]
- 20 a).Dejter-Juszynski M, Flowers HM. Carbohydr. Res. 1972;23:41–45. doi: 10.1016/s0008-6215(00)87031-7. [DOI] [PubMed] [Google Scholar]; b) Corey EJ, Carpino P. J. Am. Chem. Soc. 1989;111:5472–5473. [Google Scholar]; c) Demchenko AV, Rousson E, Boons G-J. Tetrahedron Lett. 1999;40:6523–6536. [Google Scholar]; d) Mukaiyama T, Suenaga M, Chiba H, Jona H. Chem. Lett. 2002:56–57. [Google Scholar]; e) Cheng Y-P, Chen H-T, Lin C-C. Tetrahedron Lett. 2002;43:7721–7723. [Google Scholar]
- 21.De Meo C, Kamat MN, Demchenko AV. Eur. J. Org. Chem. 2005:706–711. [Google Scholar]
- 22.Tanaka H, Yoshizawa A, Takahashi T. Angew. Chem. Int. Ed. 2007;46:2505–2507. doi: 10.1002/anie.200604031. [DOI] [PubMed] [Google Scholar]
- 23.Eby R, Schuerch C. Carbohydr. Res. 1974;34:79–90. [Google Scholar]
- 24 a).Lucas TJ, Schuerch C. Carbohydr. Res. 1975;39:39–45. doi: 10.1016/s0008-6215(00)82649-x. [DOI] [PubMed] [Google Scholar]; b) Marousek V, Lucas TJ, Wheat PE, Schuerch C. Carbohydr. Res. 1978;60:85–96. [Google Scholar]
- 25.Fréchet JM, Schuerch C. J. Am. Chem. Soc. 1972;94:604–609. [Google Scholar]
- 26 a).Note that classical neighboring group participation from an O6 ester leading to β-glycosides is stereoelectronically improbable as the intermediate would correspond to an inside, outside bicyclo[5.3.1]undecane-type system: Alder RW, East SP. Chem. Rev. 1996;96:2097–2111. doi: 10.1021/cr940246k.; Kim S, Winkler J. Chem. Soc. Rev. 1997;26:387–400. [Google Scholar]
- 27 a).Mukaiyama T, Suenaga M, Chiba H, Jona H. Chem. Lett. 2002:56–57. [Google Scholar]; b) Cheng Y-P, Chen H-T, Lin C-C. Tetrahedron Lett. 2002;43:7721–7723. [Google Scholar]
- 28.Bartlett PA, Meadows JD, Brown EG, Morimoto A, Jernstedt KK. J. Org. Chem. 1982;47:4013–4018. [Google Scholar]
- 29.Guenther W, Kunz H. Carbohydr. Res. 1992;228:217–241. doi: 10.1016/s0008-6215(00)90561-5. [DOI] [PubMed] [Google Scholar]
- 30.Kocovsky P, Stieborova I. J. Chem. Soc., Perkin Trans. 1. 1987:1969–1974. [Google Scholar]
- 31.Wilen SH, Delguzzo L, Saferstein R. Tetrahedron. 1987;43:5089–5094. [Google Scholar]
- 32.Hansch C, Leo A, Taft RW. Chem. Rev. 1991;91:165–195. [Google Scholar]
- 33.Crich D, Smith M. J. Am. Chem. Soc. 2001;123:9015–9020. doi: 10.1021/ja0111481. [DOI] [PubMed] [Google Scholar]
- 34.Crich D, Smith M, Yao Q, Picione J. Synthesis. 2001:323–326. [Google Scholar]
- 35 a).Caputo R, Kunz H, Mastroianni D, Palumbo G, Pedatella S, Solla F. Eur. J. Org. Chem. 1999:3147–3150. [Google Scholar]; b) Bickley J, Cottrell JA, Ferguson JR, Field RA, Harding JR, Hughes DL, Kartha KPR, Law JL, Scheinman F, Stachulski AV. Chem. Commun. 2003:1266–1267. doi: 10.1039/b302629a. [DOI] [PubMed] [Google Scholar]; c) E.-Badri MH, Willenbring D, Tantillo DJ, Gervay-Hague J. J. Org. Chem. 2007;72:4663–4672. doi: 10.1021/jo070229y. [DOI] [PubMed] [Google Scholar]; d) Perrie JA, Harding JR, King C, Sinnott D, Stachulski AV. Org. Lett. 2003;5:4545–4548. doi: 10.1021/ol035475k. [DOI] [PubMed] [Google Scholar]
- 36 a).Krog-Jensen C, Oscarson S. J. Org. Chem. 1996;61:1234–1238. doi: 10.1021/jo960776b. [DOI] [PubMed] [Google Scholar]; b) Bai Y, Lowary TL. J. Org. Chem. 2006;71:9672–9680. doi: 10.1021/jo061821a. [DOI] [PubMed] [Google Scholar]; c) Grayson EJ, Ward SJ, Hall AJ, Rendle PM, Gamblin DP, Batsanov AS, Davis BG. J. Org. Chem. 2005;70:9740–9754. doi: 10.1021/jo051374j. [DOI] [PubMed] [Google Scholar]
- 37 a).Crich D, Mataka J, Sun S, Lam K-C, Rheingold AR, Wink DJ. Chem. Commun. 1998:2763–2764. [Google Scholar]; b) Crich D, Mataka J, Zakharov LN, Rheingold AL, Wink DJ. J. Am. Chem. Soc. 2002;124:6028–6036. doi: 10.1021/ja0122694. [DOI] [PubMed] [Google Scholar]
- 38.Fan E, Shi W, Lowary TL. J. Org. Chem. 2007;72:2917–2928. doi: 10.1021/jo062542q. [DOI] [PubMed] [Google Scholar]
- 39.Lemieux RU, Hindsgaul O. Carbohydr. Res. 1980;82:195–206. [Google Scholar]
- 40.The structure of this compound is based on the examination of 1H-NMR data which display 3J1,2, 3J2,3, and 3J3,4 coupling constants of 4.0, 10.0, and 2.4 Hz consistent with a B3,O conformation for the pyranose ring. The mass spectrum, does not display a molecular ion, but has [M-phthalic anhydride]+ (10%).
- 41.A Chemical Abstracts search revealed 22 ten-membered diolides derived from phthalic anhydride thereby indicating the accessibility of this structural class.
- 42.Our conclusions largely parallel those of Woerpel in his recent investigation of the transannular participation of 4-thioethers in the reactions of cyclic tetrahydropyranyl oxocarbenium ions: Beaver MG, Billings SB, Woerpel KA. J. Am. Chem. Soc. 2008;130:2082–2086. doi: 10.1021/ja0767783.
- 43.We consider the effect of the O4 equatorial esters to be satisfactorily explained by their effect on the equilibrium between the covalent glycosyl donors and the various ion pairs implicated in glycosylation as discussed by van Boeckel.15
- 44.An anonymous reviewer has offered the insightful suggestion that the effect of esters at O4 may be explained by their influence on the conformation about the C5-C6 bond, thereby affecting the proximity of the O6 group to the ring oxygen.
- 45 a).Barton DHR, McCapra F, May PJ, Thudium F. J. Chem. Soc. 1960:1297–1311. [Google Scholar]; b) Magnus N, Magnus P. Tetrahedron Lett. 1997;38:3491–3494. [Google Scholar]; c) Cieplak AS. Chem. Rev. 1999;99:1265–1336. doi: 10.1021/cr980381n. [DOI] [PubMed] [Google Scholar]
- 46 a).Fraser-Reid B, Wu ZC, Andrews W, Skowronski E. J. Am. Chem. Soc. 1991;113:1434–1435. [Google Scholar]; b) Imamura A, Ando H, Korogi S, Tanabe G, Muraoka O, Ishida H, Kiso M. Tetrahedron Lett. 2003;44:6725–6728. [Google Scholar]; c) Jensen HH, Nordstrom M, Bols M. J. Am. Chem. Soc. 2004;126:9205–9213. doi: 10.1021/ja047578j. [DOI] [PubMed] [Google Scholar]; d) McDonnell C, López O, Murphy PV, Bolaños J. G. Fernández, Hazell RG, Bols M. J. Am. Chem. Soc. 2004;126:12374–12385. doi: 10.1021/ja047476t. [DOI] [PubMed] [Google Scholar]; e) Pedersen CM, Nordstrom LU, Bols M. J. Am. Chem. Soc. 2007;129:9222–9235. doi: 10.1021/ja071955l. [DOI] [PubMed] [Google Scholar]; f) Jensen HH, Pedersen CM, Bols M. Chem. Eur, J. 2007;13:7576–7581. doi: 10.1002/chem.200700947. [DOI] [PubMed] [Google Scholar]; g) Okada Y, Mukae T, Okajimia K, Taira M, Fujita M, Yamada H. Org. Lett. 2007;8:1573–1576. doi: 10.1021/ol070427b. [DOI] [PubMed] [Google Scholar]; h) Okada Y, Nagata O, Taira M, Yamada H. Org. Lett. 2007;9:2755–2758. doi: 10.1021/ol070720b. [DOI] [PubMed] [Google Scholar]; i) Jensen HH, Bols M. Acc. Chem. Res. 2006;39:259–265. doi: 10.1021/ar050189p. [DOI] [PubMed] [Google Scholar]
- 47.Chiara JL, García Á. Synlett. 2005:2607–2610. [Google Scholar]
- 48.Suzuki K, Ohtsuka I, Kanemitsu T, Ako T, Kanie O. J. Carbohydr. Chem. 2005;24:219–236. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.