

Abstract
DNA breaks play an essential role in germinal centre B cells as intermediates to immunoglobulin class switching, a recombination process initiated by activation-induced cytidine deaminase (AID). Immunoglobulin gene hypermutation is likewise catalysed by AID but is believed to occur via single-strand DNA breaks. When improperly repaired, AID-mediated lesions can promote chromosomal translocations (CTs) that juxtapose the immunoglobulin loci to heterologous genomic sites, including oncogenes. Two of the most studied translocations are the t(8;14) and T(12;15), which deregulate cMyc in human Burkitt’s lymphomas and mouse plasmacytomas, respectively. While a complete understanding of the aetiology of such translocations is lacking, recent studies using diverse mouse models have shed light on two important issues: (1) the extent to which non-specific or AID-mediated DNA lesions promote CTs, and (2) the safeguard mechanisms that B cells employ to prevent AID tumorigenic activity. Here we review these advances and discuss the usage of pristane-induced mouse plasmacytomas as a tool to investigate the origin of Igh–cMyc translocations and B-cell tumorigenesis.
Keywords: activation-induced cytidine deaminase, chromosomal translocations, class switch recombination, somatic hypermutation, tumorigenesis
Activation-induced cytidine deaminase, a multitask enzyme
Upon antigen encounter, B lymphocytes migrate to primary follicles within peripheral lymphoid organs where they proliferate extensively and establish the germinal centre reaction. Within this specialized microenvironment, activated B cells undergo two unique genetic processes: somatic hypermutation (SHM) and class switch recombination (CSR). Somatic hypermutation is defined as the random introduction of nucleotide substitutions at the variable domains of heavy and light chain genes at a rate of 10−3–10−4 per base pair per generation.1 In conjunction with antigen-mediated selection, SHM promotes affinity maturation of antibody receptors during the humoral immune response. Class switching, on the other hand, replaces the heavy chain constant domain Cμ for one of a set of downstream CH exons (Cγ, Cε or Cα).2 The different C domains or isotypes impart antibodies with distinct physiological effector functions.
Despite expected mechanistic differences between SHM and CSR, both processes are initiated by activation-induced cytidine deiminase (AID). This B-cell-specific enzyme was first identified by the Durandy and Honjo laboratories as being essential for SHM and CSR both in humans and mice.3,4 In hens as well, AID promotes immunoglobulin gene conversion,5,6 and an AID homologue has also been recently implicated in antigen receptor diversification in hagfish.7
Precisely how AID mediates such an array of immunoglobulin gene diversification reactions is only beginning to be unravelled, but a large body of evidence clearly demonstrates that upon recruitment to the immunoglobulin loci AID deaminates cytidines into uracils.8–13 This targeted deamination occurs only in the context of single-strand (ss) DNA exposed by the RNA polII holoenzyme during transcription and stabilized by replication protein A (RPA).14 However, even within ssDNA, not all cytidines are good deamination substrates, as AID preferentially targets WRCY motifs (where W = A or T, R = A or G, and Y = C or T),12,15 an intrinsic hotspot sequence that is also evolutionarily conserved from bony fishes to humans. Based on protein sequence conservation between AID and the RNA editing enzyme APOBEC1, AID has also been proposed to deaminate RNA,16 although a direct messenger RNA (mRNA) deamination target has not yet been identified.
Somatic hypermutation
Because uracils in genomic DNA can be mutagenic, eukaryotic cells have, not surprisingly, developed several base excision repair (BER) deglycosylases to remove them. During hypermutation, uracils are primarily recognized and removed by uracil-N-glycosylase 1 (UNG), resulting in DNA abasic sites at heavy and light chain V genes.17 Like spontaneous dC deamination, a significant fraction of AID-mediated deamination events is faithfully reverted to cytidines by BER (Fig. 1). The extent of such error-free repair activity becomes evident in mouse and human B cells that are defective in BER, which accumulate large numbers of C to T (or G to A) transition mutations at immunoglobulin genes as a result of DNA replication of unrepaired uracils.18,19 In the presence of UNG, however, abasic sites in cycling cells stall the DNA replication fork and engage the activity of REV1 (REV1 in higher organisms is a deoxycytidyl transferase homologue of the Saccharomyces cerevisiae REV1 gene) and possibly other translesion synthesis (TLS) DNA polymerases.20,21 Insertion of dNTPs opposite the non-instructional abasic site results both in transition and transversion mutations at the immunoglobulin loci. Consequently, AID-mediated deamination is either faithfully repaired in G1 or becomes mutagenic if UNG recognizes the uracil at the replication fork in the S phase (see refs 22, 23 and Fig. 1). Although this model remains to be fully proven, it predicts that the extent of SHM would be directly proportional to the rate of B-cell division, i.e. how frequently the replication fork encounters unrepaired uracils at the immunoglobulin loci. Considering the extraordinary turnover of germinal centre B lymphocytes, the model would explain how mutations are favoured over error-free repair during the immune response.
Figure 1.

Current model of somatic hypermutation (based on ref. 123). Activation-induced cytidine deiminase (AID)-mediated deamination of cytidines at immunoglobulin genes results in U : G mispairs. DNA replication over uracils will promote transition mutations (C/G to T/A). Uracil can also be repaired in an error-free fashion by either base excision repair (BER) or mismatch repair (MMR). If uracils are recognized by uracil-N-glycosylase 1 (UNG) in the context of DNA replication however, abasic sites would be repaired by REV1 and other translesion polymerases leading to both transition and transversions at C : Gs. Following replication, U : G mismatches or abasic sites would engage the activity of polymerase η (pol η) resulting in mutations primarily at A : T pairs.
Uracils are not only foreign to DNA, but also create U : G mismatches, which are conventionally recognized and removed by the mismatch repair (MMR) pathway. Intriguingly, gene targeting of the MMR factors Msh2, Msh6 or Exo1 results in significant alterations in the mutation spectrum of immunoglobulin genes, indicating that MMR proteins are also directly involved in SHM.18,24,25 The MMR acts primarily in a post-replicative fashion, correcting errors of nucleotide misincorporation downstream of DNA polymerase α, β or ε.26 Like BER, MMR is a high-fidelity repair process and so it might appear counterintuitive that it potentiates DNA hypermutation. This conundrum was explained by studies demonstrating that MMR proteins recruit the error-prone translesion DNA polymerase η to the hypermutating immunoglobulin loci.27,28 This low-fidelity polymerase introduces a significant number of misincorporation errors during patch DNA synthesis, particularly at A/T pairs.29 Why DNA mismatches other than those induced by AID fail to recruit polymerase η is unclear. In short, SHM can be viewed as the result of DNA replication of unrepaired uracils, REV1/TLS polymerase mutagenic activity at C/G pairs, and nucleotide misincorporation at A/T pairs by polymerase η before or after UNG activity (Fig. 1).
Class switch recombination
Class switch recombination is targeted to specific S recombination regions by 5′ (I) promoters that mediate germline or sterile S-CH transcription.30 The 5′ and 3′ heavy chain enhancers appear to promote S-S synapses,31 which are critical to target recombination from Sμ to the appropriate downstream S domains and to impart accessibility to AID (Fig. 2). In contrast to SHM, which occurs via ssDNA lesions, CSR relies on DNA double-strand break (DSB) intermediates, which presupposes AID activity on both template and non-template DNA strands. At least two unique features of S domains may explain how DNA breaks are generated during CSR. First, S regions are characterized by a G-rich non-template strand, containing large numbers of repetitive, palindromic RGWY motifs.32 During transcription, these palindromes are displaced as R-DNA loops by the nascent germline transcripts that stably hybridize to the template strand.33–35In vitro experiments suggest that the AID/RPA complex can directly act on the displaced ssDNA.11,12,36 Antisense or bidirectional transcription of immunoglobulin genes might explain, on the other hand, how the template strand becomes accessible to deamination.37,38 The current CSR model proposes that in RGYW-packed S domains, DNA nicks on both DNA strands result in staggered DNA breaks intermediate to recombination (Fig. 2).
Figure 2.

Schematics representing class switch recombination from Sμ to Sα. Recombining switch domains are rendered accessible to activation-induced cytidine deiminase (AID) and replication protein A (RPA) by the concerted action of I-promoters and the μ (5′) and α (3′) enhancers (E). Removal of uracils by uracil-N-glycosylase 1 (UNG) and nicking of the DNA backbone by Ape nucleases results in the formation of staggered DNA breaks, which are either repaired in an error-free manner by base excision repair (BER) or serve as substrates for double-strand break (DSB) repair and/or recombination. Although not depicted, mismatch repair activity induces class switch recombination via a similar mechanism.
Like other DNA DSBs occurring during pre-replicative phases of the cell cycle, AID-mediated breaks engage the non-homologous end-joining repair pathway (NHEJ). The first molecules recognizing DNA DSBs within this pathway are members of the Mre11, Rad50 and Nbs1 (MRN) complex.39 These molecules recruit and activate the serine/threonine kinase ATM, which in turn phosphorylates numerous repair factors such as MDC1, 53BP1, Nbs1 and H2AX, leading to arrest in cell cycle progression and repair.40 The importance of this pathway in mediating efficient CSR is evident in ATM−/−, 53BP1−/− or H2AX−/− B cells, where AID-induced breaks at S domains persist unrepaired throughout the cell cycle.41–43
In addition to ATM, another member of phosphatidylinositol 3-kinase family of proteins, DNA-PKcs, is involved in CSR. The DNA-PKcs acts as the catalytic subunit of the DNA–PK complex, composed of the Ku70/Ku80 heterodimer, Artemis, XRCC4, Ligase IV and Cernunnos. In the context of switching, Ku proteins appear to be absolutely required for repair of CSR DNA ends,44,45 although these experiments were complicated by the proliferation defects of Ku-deficient cells. Yet, DNA-PKcs−/− B cells or cells homozygous for a severe combined immunodeficiency (scid) mutation are severely impaired in CSR, with the exception of switching to immunoglobulin G1 (IgG1), which occurs at low but reproducible levels.46 This residual activity could be ascribed to functional redundancy between DNA-PKcs and other phosphatidylinositol 3-kinases like ATM. Alternatively or in addition, a different repair pathway other than NHEJ could mediate CSR, or at least function as a backup system when NHEJ is crippled. In keeping with this notion, XRCC4 and Ligase IV conditional knock-out B cells retain the capacity to undergo switching to nearly 50% of wild-type levels.47,48 This underlying backup repair pathway appears to rely on microhomology judging from analyses of the recombination sites at S domains of XRCC4−/− and Ligase IV−/− B cells.47–49 In conclusion, a very complex set of repair mechanisms promote and resolve DNA DSBs downstream of AID. It should be pointed out, however, that some of the pathways driving somatic hypermutation also target switch domains during CSR (see for instance ref. 50). How this plethora of enzymatic activities is regulated to ensure mostly hypermutation at V genes and recombination at C genes is under intense investigation.
Chromosomal translocations
In spite of the mechanisms outlined above, DNA lesions intermediate to CSR are occasionally misrepaired, leading to chromosomal single-stranded breaks, DSBs, translocations, deletions and aneuploidy. A well-studied case involves the t(8;14)(q24;q32) chromosomal translocations (CTs) that juxtapose immunoglobulin heavy chain (Igh) to the proto-oncogene cMyc in human Burkitt’s lymphomas.51,52 Reciprocal translocations between these two loci are also pathogenic in mouse plasmacytomas [T(12;15)] and rat immunocytomas [t(6;7)] (reviewed in ref. 53). In all cases, cell transformation is achieved by cMyc transcriptional deregulation at the hands of Eμ and/or Eα immunoglobulin enhancers. That tumours in different mammalian species deregulate cMyc in a similar fashion suggests a common underlying mechanism at the origin of Igh–cMyc translocations.
Peritoneal plasmacytomas (PCTs) in mice can be induced by injection of non-metabolizable materials such as pristane oil. This agent promotes the formation of chronic inflammatory oil granulomas (OGs) rich in interleukin-6 (IL-6) and other cytokines essential for plasma cell differentiation and transformation.54–56 Plasmacytomagenesis is highly dependent not only on the OG microenvironment but also on the genetic makeup of BALB/cAnPt (B/c) mice. Following three injections of 0·5 ml pristane given at days 0, 60 and 120, ∼ 40–60% of B/c mice develop PCTs by day 300.53 In contrast, most other inbred strains, such as DBA/2, C57BL/6 or 129, are resistant to PCT induction. Development of PCT also requires a conventional environment as tumour incidence in B/c mice raised under specific pathogen-free conditions is in the order of 5%.57
In addition to the pristane-treated B/c model, PCTs carrying T(12;15)s spontaneously develop in H-2LD-IL-6 transgenic mice.58 Overexpression of IL-6 in these animals initially results in massive hyperplasia of terminally differentiated, non-neoplastic plasma cells involving almost all lymph nodes.58 This stage is followed by the appearance of atypical plasmablast-like cells in the medullary cords of mesenteric lymph nodes. By 300 days of age, ∼60% of IL-6 transgenic mice ultimately develop PCTs.
Both pristane-induced and IL-6-induced tumour incidence can be increased and their latency can be reduced by overexpression of Bcl-2 or Bcl-xL (refs 59–61 and M.P. unpublished observations). These anti-apoptotic molecules are believed to salvage B cells carrying cMyc translocations from programmed cell death. Consequently, between 70% and 95% of Bcl-2 or Bcl-xL transgenic mice develop a PCT within 100 days. These models lend themselves to establishing chronological and anatomical relationships of CT formation and PCT development. An additional advantage of Bcl-2 and Bcl-xL transgenes is that PCT induction can be carried out in some genetically resistant strains and hybrids.
In conclusion, whether plasmacytomas arise spontaneously in IL-6 transgenic mice or are induced by pristane injections in the B/c background, they critically rely on the IL-6 cytokine and are dramatically accelerated by anti-apoptotic signals.
Detection and classification of Igh–cMyc chromosomal translocations
Highly sensitive polymerase chain reaction (PCR) methodologies have been developed for detecting CTs in different tumours and tissues. In early publications, Igh–cMyc junctions were amplified by a combination of degenerate primers and two rounds of nested PCR.62,63 More recently, a long-distance PCR protocol that uses highly specific primers has been the system of choice.61,64–66 In general, long-distance PCR generates products of considerably great fidelity but is not as sensitive as the consensus or degenerate method. On the other hand, when pushed to the limits of sensitivity, the degenerate method has the potential of yielding artefacts.67 These probably result from accidental polymerase ‘jumping’ between templates,68 or switch region insertion at cMyc.69 There are several important considerations that must be addressed when using nested PCR methodologies, such as the stringency of the annealing process (65°); appropriate, relatively low amounts of Taq polymerase (1·25 U/50 μl reaction volume); confirmatory re-isolation of the same fragment from the starting DNA; and hybridization of derivative 12 translocations with IgC region probes.
Illegitimate rearrangements between the immunoglobulin loci and cellular oncogenes were first isolated from a few human B-cell malignancies and mouse plasmacytomas.70–74 Early molecular characterization of translocation breakpoints consistently revealed the involvement of immunoglobulin switch regions, giving credence to the notion that such chromosomal abnormalities were the result of aberrant switch recombination.75–83 While mostly correct, this hypothesis has been further refined by characterization and classification of a large number of CT breakpoints isolated from mouse plasmacytomas over several years. Based on a study of 103 IL-6 transgenic and 86 pristane-induced PCTs, we classify CTs into three categories (Fig. 3). First, a significant proportion of plasmacytomas (∼10–15%) deregulate cMyc by means of variant translocations,84 which juxtapose the Igk locus and the Pvt1 gene positioned downstream of cMyc in mouse chromosome 15. Pvt1 mRNA, which does not appear to encode a protein but a cluster of microRNAs,85 is also translocated to light chain genes in Burkitt’s lymphomas.86 As most variant translocation breakpoints fall within or near Vκ-Jκ genes,84 aberrant repair of hypermutation lesions is expected to play an aetiological role to t(6;15). On the other hand, it is unclear whether Pvt1 breaks, which fall mostly near exon 5, are AID-mediated. It is interesting to note that in T-cell lymphomas, Pvt1 exon 5 is a preferred provirus integration target,87 implying that this genomic site is highly accessible in lymphocytes.
Figure 3.

Classification of chromosomal translocations isolated from mouse plasmacytomas. Upper schematic exemplifies variant translocations [t(6;15)] that juxtapose VJk domains to Pvt1 exon 5. Middle schematic shows activation-induced cytidine deiminase (AID)-mediated deamination during class switch recombination results in the formation of DNA double-strand breaks at S domains and less frequently at cMyc intron 1. Aberrant repair of these lesions can lead to the juxtaposition of cMyc to the immunoglobulin loci. Lower schematic: although at a significantly lesser extent, random, AID-independent DNA breaks can also translocate the cMyc gene to the immunoglobulin loci leading to T(12;15). Translocation breakpoints in these non-canonical translocations often map several kilobases away from S domains or the cMyc gene itself. Right table indicates the percentage of CTs involving diverse S domains in AID+/+ interleukin-6 transgenic mice, and pristane-treated AID+/+ and AID−/− B/c mice.
A second group of CTs in mouse plasmacytomas juxtapose the Igh allele to cMyc (∼80%, Fig. 3). Based on breakpoint location, these CTs can be further subclassified into: (1) canonical, when translocations precisely join S domains and cMyc intron 1, and (2) non-canonical, when CT breakpoints are randomly distributed on both alleles (Fig. 3). Canonical translocations clearly result from AID deamination activity both at cMyc and S domains.88 In IL-6 transgenic or B/c mice treated with pristane, canonical translocations can be isolated from nearly 90% of all plasmacytomas.61 The preferred cMyc translocation partner in both tumour models is Sα (Fig. 3), suggesting that tumour precursors probably originate in gut lymphoid tissues, where IgA switching is prevalent. Some canonical translocations are complex or atypical in that in addition to S regions, they display unusual inversions or insertions of DNA from other chromosomes into cMyc (see ref. 70 for examples).
Non-canonical translocations, which randomly recombine the Igh and cMyc alleles at sites other than S domains or cMyc intron 1 comprise 10–15% of all T(12;15)s. A role for AID in the origin of these translocations was excluded based on recent studies.61,65,89 Using AID−/− IL-6 transgenic mice raised under specific pathogen-free conditions, Ramiro et al. failed to amplify T(12;15)s from hyperplastic lymphoid tissues.65 In B/c pristane-treated mice on the other hand, Igh–cMyc junctions were detected at early time-points using degenerate primers and low stringency annealing temperatures (60°), but were absent from Peyer’s patches and gut.89 A third study investigated both the development of CTs and plasma cell tumours in conventionally raised AID−/−BclxL B/c mice.61 Despite the loss of knock-out mice during the experiment to a wasting syndrome (of possible viral origin) and the slow progression of PCTs, T(12;15)s were nonetheless detected by fluorescence in situ hybridization in the absence of AID. Of the nine examples isolated, there were six T(12;15), one t(6;15) and one chr12 inversion that resulted in the juxtaposition of Igh–nMyc.61 Interestingly, none of the T(12;15) were mapped to S domains or cMyc intron 1, revealing their non-canonical origin. Non-canonical CTs have also been recently isolated from IL-6 transgenic mice (M.C. Nussenzweig, personal communication).
In summary, these studies demonstrate that whereas AID is the major catalyst for generating Igh–cMyc translocations, other cellular mechanisms can promote illegitimate recombination of these alleles in activated, cycling B lymphocytes.
Regulating AID physiological and pathological activity
Mounting evidence demonstrates that B lymphocytes minimize the potential tumour-inducing ability of AID through a variety of mechanisms. First, expression of the AID gene is largely restricted to germinal centre or T-cell-independent activated B lymphocytes.90,91 This spatiotemporal regulation is fine-tuned by the counterbalanced activities of transcriptional activators and repressors such as nuclear factor-κB, Pax5, E47, Blimp1, Id2, Id3, Irf8 and HoxC4,92–97 which are recruited to the AID gene (Aicda) basal promoter or intronic enhancer. In B cells undergoing CSR, chromatin at these two genomic sites is extensively remodelled by acetylation and methylation (Fig. 4 and ref. 91). Chromatin remodelling activity is further recruited to conserved-non-coding-sequence X (CNSX, Fig. 4). This phylogenetically conserved element is required for AID-gene expression under physiological conditions,91 but does not appear to function as a conventional enhancer based on its inability to induce luciferase transcription in reporter assays (our unpublished observations). There is also evidence of a fourth conserved element 5′ of AID (CNSVIII), which is H3K4 methylated, H4 acetylated, but surprisingly lacks H3 acetylation (Fig. 4 and ref. 91). Deciphering how all these elements confine AID transcription to germinal centre B cells awaits further biochemical and genetic characterization.
Figure 4.

Partial epigenetic map of the mouse Aicda locus (35, 400 bps) in lipopolysaccharide + interleukin-4 activated B cells depicting polymerase II binding, messenger RNA (mRNA) production, histone H3 and H4 acetylation, and histone H3 lysine 4 mono-, di-, or tri-methylation (H3K4me1-2-3). Phylogenetically conserved elements CNSVIII, activation-induced cytidine deiminase (AID) basal promoter (BP), AID intronic enhancer (AIDE), and CNSX are also highlighted. Mapping (200 base pair resolution) was produced by Solexa DNA microsequencing of immunoprecipitated chromatin (PolII, H3Ac, H4Ac, H3K4me1-2-3) or polyA+ isolated mRNA (Yamane and Casellas, manuscript in preparation).
A link between AID transcription levels and the extent of AID pathological activity was recently uncovered by studies using AID heterozygous mice, which display reduced AID mRNA and protein compared with wild-type littermates.66,98,99 In the context of isotype switching, AID+/− lymphocytes show impaired inter- and intra-switch recombination and a substantial decrease in the frequency of S mutations and chromosomal breaks. Similarly, during the humoral immune response AID+/− mice display increased numbers of germinal centre cells,99 but fewer B-cell clones with high-affinity antibodies.66 On the autoimmune MRL/lpr background, these defects result in lower levels of specific anti-dsDNA antibodies in the serum, with a concomitant delay in kidney damage relative to controls.98 Haploinsufficiency for AID is also evident by the reduced ability of AID+/− B cells to undergo intrachromosomal Igh–cMyc translocations and ultimately B-cell tumour development.66 It is important to note that the extent of off-target translocations depends on AID expression levels to a much larger extent than do on-target hypermutation or CSR. This feature, which holds true both under conditions of AID overexpression43,88 and underexpression,66 suggests that AID enzymatic activity is constrained to varying degrees depending on the genomic location100 and/or cell type.101
AID transcript amounts, and consequently AID-mediated deamination, are also indirectly regulated by at least two microRNAs. First, the lymphocyte-specific miR-155 decreases the half-life of AID mRNA by associating to its 3′ untranslated region.102,103 This regulation is physiologically relevant as demonstrated in gene targeted (Aicda155) or BAC AIDGFP transgenic mice lacking an AID miR-155 binding site, which displays enhanced CSR and SHM relative to controls.102,103 Likewise, Igh–cMyc translocations are significantly upregulated in miR-155−/− or Aicda155 B cells.102 The microRNA miR-181b has also been shown to associate with the AID 3′ untranslated region.104 Accordingly, overexpression of miR-181b in lipopolysaccharide + IL-4-stimulated B cells suppresses AID and CSR levels.104 Conditional inactivation of miR-181b in the B-cell compartment might help to determine whether it also influences the incidence of Igh–cMyc translocations and lymphomagenesis.
One puzzling feature of AID is that although it directly deaminates immunoglobulin DNA, its protein is mostly confined to the cytoplasm (refs 105,106 and Fig. 5). AID nuclear exclusion appears to be achieved by a combination of selective nuclear degradation and a CRM1-dependent export mechanism.101,107–109 Consistent with results obtained with cells overexpressing AID, increasing AID nuclear concentration enhances off-target hypermutation but has little effect on the overall mutation rate of immunoglobulin genes.101 Based on this observation it is tempting to speculate that the counterintuitive AID compartmentalization might have evolved primarily to minimize AID pathological activity during the immune response.
Figure 5.
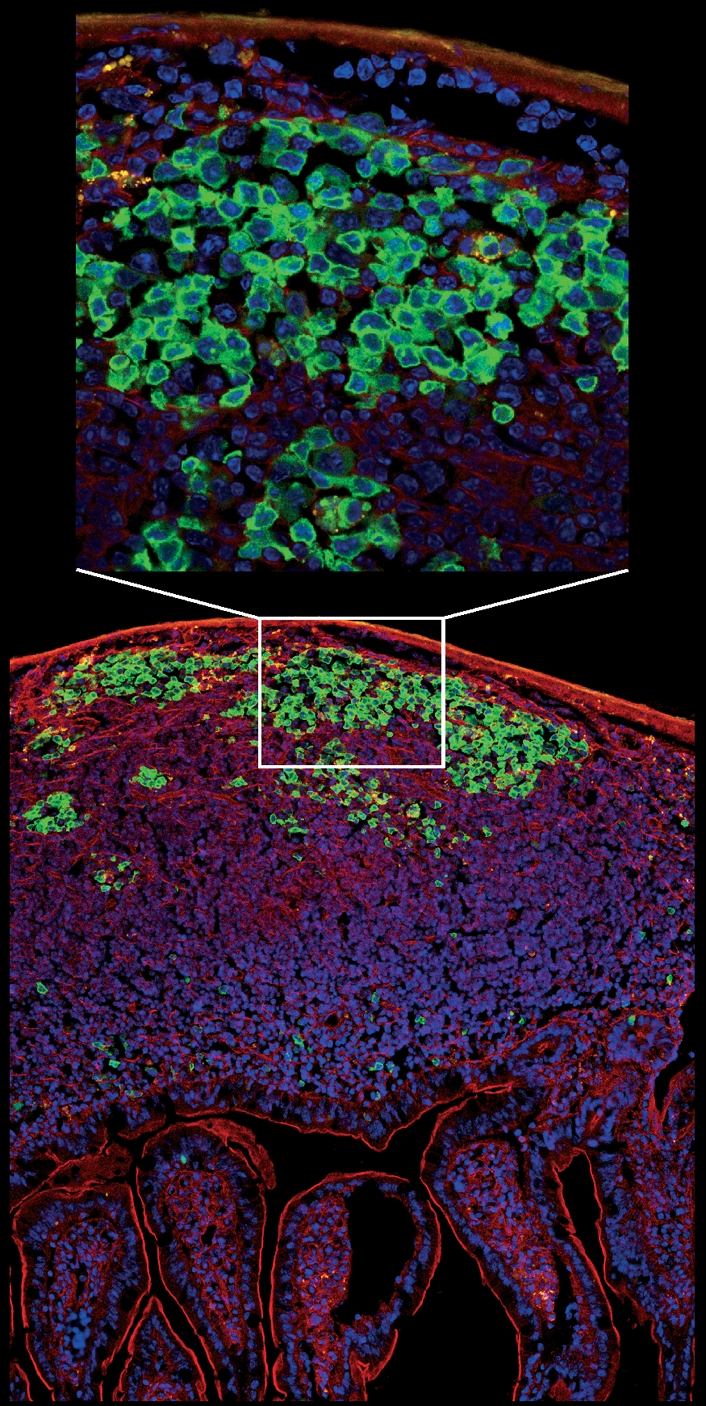
Micrograph showing a longitudinal section of intestinal villi and Peyer’s patches from the jejunum of activation-induced cytidine deiminase-green fluorescent protein (AID-GFP) mice. A close-up view of the germinal centre reveals AID localization is mostly cytoplasmic. Cells were stained with DAPI (blue) and F-actin was labelled with phalloidin (red). The image was prepared by Susan Lim and collected by Kristien Zaal using a Leica SP5 confocal microscope.
Post-translational phosphorylation constitutes another level of AID regulation. Mass spectrometry and biochemical analyses have identified at least two phosphorylation target sites in AID: serine 38 and threonine 140.99,110–113 Phosphorylation of S38, carried out either by protein kinase A and/or protein kinase C,99,111 is thought to facilitate AID interaction with RPA, which imparts accessibility to transcribed DNA.14 Mice carrying an S38A mutation show 20–35% of wild-type CSR and SHM levels.99 The origin of T140 phosphorylation is less clear, but T140A mutant mice show preferential defects in hypermutation levels.99 Considering the effect of AID expression levels on AID physiological and pathological activity, it is reasonable to expect that AID hypo- or hyperphosphorylation would have a more dramatic impact in the incidence of Igh breaks and chromosomal translocations. In this regard, it has been recently shown that SHM species lacking CSR (teleost) harbour a ‘constitutively phosphorylated’ form of AID, while those capable of inducing CSR (tetrapods) require S38 phosphorylation to fully activate AID.114 This post-translational control mechanism might thus have evolved to minimize AID-mediated lesions and translocations.114
One of the most remarkable aspects of SHM and CSR is that they rely on DNA repair factors from the BER and MMR pathways that commonly correct DNA deamination. To explain this apparent contradiction it has been hypothesized that AID simply overwhelms error-free repair or subverts it into an error-prone mechanism in B lymphocytes. As discussed previously, the engagement of low-fidelity DNA polymerases during hypermutation and DSB repair enzymes during CSR supports the latter scenario. On the other hand, there is clear evidence that AID overcomes UNG/Msh2 error-free repair at non-immunoglobulin genes.100 This is also the case at the immunoglobulin loci because absence of UNG and/or Msh2 significantly increases the mutation frequency at S domains during isotype switching.18,115 Based on analyses of polymerase β−/− B cells activated under suboptimal conditions it has also been proposed that DNA lesions intermediate to CSR (and by extrapolation to chromosomal translocations) are decreased by error-free BER.116 The underlying assumption is that faithful correction of dC deamination at S domains by polymerase β, and possibly translesion synthesis polymerases normally prevent DNA break formation. At the same time, it should be remembered that staggered DNA breaks at switch regions would also result from canonical, high-fidelity repair on both DNA strands (Fig. 2). A role for high-fidelity repair in DNA break formation is clear in AID+/− mice, which display CSR and chromosomal translocations at ∼ 50% of wild-type levels despite the fact that virtually all S mutations are repaired in an error-free fashion.66 Furthermore, cMyc and other oncogenes are frequently translocated to the immunoglobulin loci in an unmutated state.88,100,117,118 These observations have profound implications for B-cell tumorigenesis because they raise the intriguing possibility that while protecting the B-cell genome from mistargeted hypermutation, DNA repair pathways recruited downstream of AID might in fact promote chromosomal translocations.
Concluding remarks
We have summarized various cellular mechanisms that help restrain AID deamination activity (Fig. 6). How efficiently these processes prevent tumorigenesis in the B-cell compartment is clear from studies with transgenic mice expressing AID ectopically.119 Surprisingly, these animals do not display significant abnormalities in B cells but instead develop spontaneous T-cell lymphomas carrying extensive hypermutation at T-cell receptor, cMyc, Pim1 and other genes. In addition, the mice often develop multiple lung microadenomas and less frequently melanomas, hepatocellular carcinomas, and sarcomas.120 It is tempting to speculate that some of the mechanisms outlined above might be B-cell-specific, and therefore unable to protect other somatic cells from tumorigenesis when AID is aberrantly expressed. In this context, there is evidence that signalling pathways, including nuclear factor-κB, can upregulate AID in non-B cells leading to neoplastic progression.121 In the B-cell compartment, transforming viruses can also sustain AID expression for long periods of time,122 a feature that could promote malignancy in susceptible individuals. It will be interesting to determine whether tumour susceptibility is in fact related to failure in AID regulatory mechanisms.
Figure 6.

Summary of activation-induced cytidine deiminase (AID) regulatory mechanisms: 1- Spatiotemporal targeting of AID gene transcription to activated and germinal centre B cells. This is achieved by the concerted action of transcription factors and the Aicda epigenetic landscape. 2- microRNA regulation of AID messenger RNA half-life. 3- Protein kinase A (PKA) and/or protein kinase C (PKC) -mediated AID phosphorylation. 4- AID nuclear export mediated by CRM1. 5- Nuclear proteasomal degradation. 5- AID interaction with single-stranded (ss) DNA binding protein replication protein A (RPA), which allows AID to deaminate transcribed ssDNA. 7- Preferential recruitment of the AID–RPA complex to immunoglobulin genes. 8- Mistargeted hypermutation to non-immunoglobulin genes like cMyc counteracted by error-free base excision repair (BER) and mismatch repair. 9- Immunoglobulin-oncogene translocations eliminated by apoptosis via P53 activation. This safeguard mechanism might also get rid of B lymphocytes carrying extensive DNA damage.
Acknowledgments
The authors thank Susan Lim and Kristien Zaal for providing the AID-GFP micrograph, Konrad Huppi for sharing unpublished data, and Michel Nussenzweig, Javier DiNoia and Siegfried Janz for critically reading the manuscript. The writing of this review was supported (in part) by the Intramural Research Program of NIAMS and NCI of the National Institutes of Health.
References
- 1.Neuberger MS. Antibody diversification by somatic mutation: from Burnet onwards. Immunol Cell Biol. 2008;86:124–32. doi: 10.1038/sj.icb.7100160. [DOI] [PubMed] [Google Scholar]
- 2.Honjo T, Kinoshita K, Muramatsu M. Molecular mechanism of class switch recombination: linkage with somatic hypermutation. Annu Rev Immunol. 2002;20:165–96. doi: 10.1146/annurev.immunol.20.090501.112049. [DOI] [PubMed] [Google Scholar]
- 3.Revy P, Muto T, Levy Y, et al. Activation-induced cytidine deaminase (AID) deficiency causes the autosomal recessive form of the Hyper-IgM syndrome (HIGM2). [see comments] Cell. 2000;102:565–75. doi: 10.1016/s0092-8674(00)00079-9. [DOI] [PubMed] [Google Scholar]
- 4.Muramatsu M, Kinoshita K, Fagarasan S, Yamada S, Shinkai Y, Honjo T. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. [see comments] Cell. 2000;102:553–63. doi: 10.1016/s0092-8674(00)00078-7. [DOI] [PubMed] [Google Scholar]
- 5.Harris RS, Sale JE, Petersen-Mahrt SK, Neuberger MS. AID is essential for immunoglobulin V gene conversion in a cultured B cell line. Curr Biol. 2002;12:435–8. doi: 10.1016/s0960-9822(02)00717-0. [DOI] [PubMed] [Google Scholar]
- 6.Arakawa H, Hauschild J, Buerstedde JM. Requirement of the activation-induced deaminase (AID) gene for immunoglobulin gene conversion. Science. 2002;295:1301–6. doi: 10.1126/science.1067308. [DOI] [PubMed] [Google Scholar]
- 7.Rogozin IB, Iyer LM, Liang L, Glazko GV, Liston VG, Pavlov YI, Aravind L, Pancer Z. Evolution and diversification of lamprey antigen receptors: evidence for involvement of an AID-APOBEC family cytosine deaminase. Nat Immunol. 2007;8:647–56. doi: 10.1038/ni1463. [DOI] [PubMed] [Google Scholar]
- 8.Sohail A, Klapacz J, Samaranayake M, Ullah A, Bhagwat AS. Human activation-induced cytidine deaminase causes transcription-dependent, strand-biased C to U deaminations. Nucleic Acids Res. 2003;31:2990–4. doi: 10.1093/nar/gkg464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ramiro AR, Stavropoulos P, Jankovic M, Nussenzweig MC. Transcription enhances AID-mediated cytidine deamination by exposing single-stranded DNA on the nontemplate strand. Nat Immunol. 2003;4:452–6. doi: 10.1038/ni920. [DOI] [PubMed] [Google Scholar]
- 10.Dickerson SK, Market E, Besmer E, Papavasiliou FN. AID mediates hypermutation by deaminating single stranded DNA. J Exp Med. 2003;197:1291–6. doi: 10.1084/jem.20030481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chaudhuri J, Tian M, Khuong C, Chua K, Pinaud E, Alt FW. Transcription-targeted DNA deamination by the AID antibody diversification enzyme. Nature. 2003;422:726–30. doi: 10.1038/nature01574. [DOI] [PubMed] [Google Scholar]
- 12.Pham P, Bransteitter R, Petruska J, Goodman MF. Processive AID-catalysed cytosine deamination on single-stranded DNA simulates somatic hypermutation. Nature. 2003;424:103–7. doi: 10.1038/nature01760. [DOI] [PubMed] [Google Scholar]
- 13.Petersen-Mahrt SK, Harris RS, Neuberger MS. AID mutates E. coli suggesting a DNA deamination mechanism for antibody diversification. Nature. 2002;418:99–103. doi: 10.1038/nature00862. [DOI] [PubMed] [Google Scholar]
- 14.Chaudhuri J, Khuong C, Alt FW. Replication protein A interacts with AID to promote deamination of somatic hypermutation targets. Nature. 2004;430:992–8. doi: 10.1038/nature02821. [DOI] [PubMed] [Google Scholar]
- 15.Huang FT, Yu K, Hsieh CL, Lieber MR. Downstream boundary of chromosomal R-loops at murine switch regions: implications for the mechanism of class switch recombination. Proc Natl Acad Sci USA. 2006;103:5030–5. doi: 10.1073/pnas.0506548103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Honjo T, Muramatsu M, Fagarasan S. AID: how does it aid antibody diversity? Immunity. 2004;20:659–68. doi: 10.1016/j.immuni.2004.05.011. [DOI] [PubMed] [Google Scholar]
- 17.Di Noia J, Neuberger MS. Altering the pathway of immunoglobulin hypermutation by inhibiting uracil-DNA glycosylase. Nature. 2002;419:43–8. doi: 10.1038/nature00981. [DOI] [PubMed] [Google Scholar]
- 18.Rada C, Di Noia JM, Neuberger MS. Mismatch recognition and uracil excision provide complementary paths to both Ig switching and the A/T-focused phase of somatic mutation. Mol Cell. 2004;16:163–71. doi: 10.1016/j.molcel.2004.10.011. [DOI] [PubMed] [Google Scholar]
- 19.Kavli B, Otterlei M, Slupphaug G, Krokan HE. Uracil in DNA – general mutagen, but normal intermediate in acquired immunity. DNA Repair (Amst) 2007;6:505–16. doi: 10.1016/j.dnarep.2006.10.014. [DOI] [PubMed] [Google Scholar]
- 20.Simpson LJ, Sale JE. Rev1 is essential for DNA damage tolerance and non-templated immunoglobulin gene mutation in a vertebrate cell line. EMBO J. 2003;22:1654–64. doi: 10.1093/emboj/cdg161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jansen JG, Langerak P, Tsaalbi-Shtylik A, van den Berk P, Jacobs H, de Wind N. Strand-biased defect in C/G transversions in hypermutating immunoglobulin genes in Rev1-deficient mice. J Exp Med. 2006;203:319–23. doi: 10.1084/jem.20052227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Peled JU, Kuang FL, Iglesias-Ussel MD, Roa S, Kalis SL, Goodman MF, Scharff MD. The biochemistry of somatic hypermutation. Annu Rev Immunol. 2008;26:481–511. doi: 10.1146/annurev.immunol.26.021607.090236. [DOI] [PubMed] [Google Scholar]
- 23.Di Noia JM, Rada C, Neuberger MS. SMUG1 is able to excise uracil from immunoglobulin genes: insight into mutation versus repair. EMBO J. 2006;25:585–95. doi: 10.1038/sj.emboj.7600939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Martomo SA, Yang WW, Gearhart PJ. A role for Msh6 but not Msh3 in somatic hypermutation and class switch recombination. J Exp Med. 2004;200:61–8. doi: 10.1084/jem.20040691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wiesendanger M, Kneitz B, Edelmann W, Scharff MD. Somatic hypermutation in MutS homologue (MSH)3-, MSH6-, and MSH3/MSH6-deficient mice reveals a role for the MSH2–MSH6 heterodimer in modulating the base substitution pattern. J Exp Med. 2000;191:579–84. doi: 10.1084/jem.191.3.579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jiricny J. The multifaceted mismatch-repair system. Nat Rev Mol Cell Biol. 2006;7:335–46. doi: 10.1038/nrm1907. [DOI] [PubMed] [Google Scholar]
- 27.Zeng X, Winter DB, Kasmer C, Kraemer KH, Lehmann AR, Gearhart PJ. DNA polymerase eta is an A–T mutator in somatic hypermutation of immunoglobulin variable genes. Nat Immunol. 2001;2:537–41. doi: 10.1038/88740. [DOI] [PubMed] [Google Scholar]
- 28.Delbos F, Aoufouchi S, Faili A, Weill JC, Reynaud CA. DNA polymerase eta is the sole contributor of A/T modifications during immunoglobulin gene hypermutation in the mouse. J Exp Med. 2007;204:17–23. doi: 10.1084/jem.20062131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pavlov YI, Rogozin IB, Galkin AP, Aksenova AY, Hanaoka F, Rada C, Kunkel TA. Correlation of somatic hypermutation specificity and A–T base pair substitution errors by DNA polymerase eta during copying of a mouse immunoglobulin kappa light chain transgene. Proc Natl Acad Sci USA. 2002;99:9954–9. doi: 10.1073/pnas.152126799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stavnezer J, Guikema JE, Schrader CE. Mechanism and regulation of class switch recombination. Annu Rev Immunol. 2008;26:261–92. doi: 10.1146/annurev.immunol.26.021607.090248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wuerffel R, Wang L, Grigera F, et al. S–S synapsis during class switch recombination is promoted by distantly located transcriptional elements and activation-induced deaminase. Immunity. 2007;27:711–22. doi: 10.1016/j.immuni.2007.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tashiro J, Kinoshita K, Honjo T. Palindromic but not G-rich sequences are targets of class switch recombination. Int Immunol. 2001;13:495–505. doi: 10.1093/intimm/13.4.495. [DOI] [PubMed] [Google Scholar]
- 33.Daniels GA, Lieber MR. Transcription targets recombination at immunoglobulin switch sequences in a strand-specific manner. Curr Top Microbiol Immunol. 1996;217:171–89. doi: 10.1007/978-3-642-50140-1_12. [DOI] [PubMed] [Google Scholar]
- 34.Reaban ME, Griffin JA. Induction of RNA-stabilized DNA conformers by transcription of an immunoglobulin switch region. Nature. 1990;348:342–4. doi: 10.1038/348342a0. [DOI] [PubMed] [Google Scholar]
- 35.Shinkura R, Tian M, Smith M, Chua K, Fujiwara Y, Alt FW. The influence of transcriptional orientation on endogenous switch region function. Nat Immunol. 2003;4:435–41. doi: 10.1038/ni918. [DOI] [PubMed] [Google Scholar]
- 36.Barreto V, Reina-San-Martin B, Ramiro AR, McBride KM, Nussenzweig MC. C-terminal deletion of AID uncouples class switch recombination from somatic hypermutation and gene conversion. Mol Cell. 2003;12:501–8. doi: 10.1016/s1097-2765(03)00309-5. [DOI] [PubMed] [Google Scholar]
- 37.Ronai D, Iglesias-Ussel MD, Fan M, Li Z, Martin A, Scharff MD. Detection of chromatin-associated single-stranded DNA in regions targeted for somatic hypermutation. J Exp Med. 2007;204:181–90. doi: 10.1084/jem.20062032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chaudhuri J, Basu U, Zarrin A, et al. Evolution of the immunoglobulin heavy chain class switch recombination mechanism. Adv Immunol. 2007;94:157–214. doi: 10.1016/S0065-2776(06)94006-1. [DOI] [PubMed] [Google Scholar]
- 39.Petrini JH. The mammalian Mre11-Rad50-nbs1 protein complex: integration of functions in the cellular DNA-damage response. Am J Hum Genet. 1999;64:1264–9. doi: 10.1086/302391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shiloh Y. ATM and related protein kinases: safeguarding genome integrity. Nat Rev. 2003;3:155–68. doi: 10.1038/nrc1011. [DOI] [PubMed] [Google Scholar]
- 41.Petersen S, Casellas R, Reina-San-Martin B, et al. AID is required to initiate Nbs1/gamma-H2AX focus formation and mutations at sites of class switching. Nature. 2001;414:660–5. doi: 10.1038/414660a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Franco S, Gostissa M, Zha S, et al. H2AX prevents DNA breaks from progressing to chromosome breaks and translocations. Mol Cell. 2006;21:201–14. doi: 10.1016/j.molcel.2006.01.005. [DOI] [PubMed] [Google Scholar]
- 43.Ramiro AR, Jankovic M, Callen E, et al. Role of genomic instability and p53 in AID-induced c-myc–Igh translocations. Nature. 2006;440:105–9. doi: 10.1038/nature04495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Manis JP, Gu Y, Lansford R, Sonoda E, Ferrini R, Davidson L, Rajewsky K, Alt FW. Ku70 is required for late B cell development and immunoglobulin heavy chain class switching. J Exp Med. 1998;187:2081–9. doi: 10.1084/jem.187.12.2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Casellas R, Nussenzweig A, Wuerffel R, et al. Ku80 is required for immunoglobulin isotype switching. EMBO J. 1998;17:2404–11. doi: 10.1093/emboj/17.8.2404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Manis JP, Dudley D, Kaylor L, Alt FW. IgH class switch recombination to IgG1 in DNA-PKcs-deficient B cells. Immunity. 2002;16:607–17. doi: 10.1016/s1074-7613(02)00306-0. [DOI] [PubMed] [Google Scholar]
- 47.Yan CT, Boboila C, Souza EK, et al. IgH class switching and translocations use a robust non-classical end-joining pathway. Nature. 2007;449:478–82. doi: 10.1038/nature06020. [DOI] [PubMed] [Google Scholar]
- 48.Soulas-Sprauel P, Le Guyader G, Rivera-Munoz P, Abramowski V, Olivier-Martin C, Goujet-Zalc C, Charneau P, de Villartay JP. Role for DNA repair factor XRCC4 in immunoglobulin class switch recombination. J Exp Med. 2007;204:1717–27. doi: 10.1084/jem.20070255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pan-Hammarstrom Q, Jones AM, Lahdesmaki A, Zhou W, Gatti RA, Hammarstrom L, Gennery AR, Ehrenstein MR. Impact of DNA ligase IV on nonhomologous end joining pathways during class switch recombination in human cells. J Exp Med. 2005;201:189–94. doi: 10.1084/jem.20040772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Faili A, Aoufouchi S, Weller S, Vuillier F, Stary A, Sarasin A, Reynaud CA, Weill JC. DNA polymerase eta is involved in hypermutation occurring during immunoglobulin class switch recombination. J Exp Med. 2004;199:265–70. doi: 10.1084/jem.20031831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kuppers R. Mechanisms of B-cell lymphoma pathogenesis. Nat Rev. 2005;5:251–62. doi: 10.1038/nrc1589. [DOI] [PubMed] [Google Scholar]
- 52.Klein U, Dalla-Favera R. Germinal centres: role in B-cell physiology and malignancy. Nat Rev Immunol. 2008;8:22–33. doi: 10.1038/nri2217. [DOI] [PubMed] [Google Scholar]
- 53.Potter M. Neoplastic development in plasma cells. Immunol Rev. 2003;194:177–95. doi: 10.1034/j.1600-065x.2003.00061.x. [DOI] [PubMed] [Google Scholar]
- 54.Nordan RP, Neckers LM, Rudikoff S, Potter M. A growth factor required by plasmacytoma cells in vitro. Curr Top Microbiol Immunol. 1986;132:114–20. doi: 10.1007/978-3-642-71562-4_15. [DOI] [PubMed] [Google Scholar]
- 55.Suematsu S, Matsusaka T, Matsuda T, Ohno S, Miyazaki J, Yamamura K, Hirano T, Kishimoto T. Generation of plasmacytomas with the chromosomal translocation t(12;15) in interleukin 6 transgenic mice. Proc Natl Acad Sci USA. 1992;89:232–5. doi: 10.1073/pnas.89.1.232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kovalchuk AL, Kim JS, Park SS, et al. IL-6 transgenic mouse model for extraosseous plasmacytoma. Proc Natl Acad Sci USA. 2002;99:1509–14. doi: 10.1073/pnas.022643999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Byrd LG, McDonald AH, Gold LG, Potter M. Specific pathogen-free BALB/cAn mice are refractory to plasmacytoma induction by pristane. J Immunol. 1991;147:3632–7. [PubMed] [Google Scholar]
- 58.Suematsu S, Matsuda T, Aozasa K, et al. IgG1 plasmacytosis in interleukin 6 transgenic mice. Proc Natl Acad Sci USA. 1989;86:7547–51. doi: 10.1073/pnas.86.19.7547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cheung WC, Kim JS, Linden M, Peng L, Van Ness B, Polakiewicz RD, Janz S. Novel targeted deregulation of c-Myc cooperates with Bcl-X(L) to cause plasma cell neoplasms in mice. J Clin Invest. 2004;113:1763–73. doi: 10.1172/JCI20369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Silva S, Kovalchuk AL, Kim JS, Klein G, Janz S. BCL2 accelerates inflammation-induced BALB/c plasmacytomas and promotes novel tumors with coexisting T(12;15) and T(6;15) translocations. Cancer Res. 2003;63:8656–63. [PubMed] [Google Scholar]
- 61.Kovalchuk AL, duBois W, Mushinski E, et al. AID-deficient Bcl-xL transgenic mice develop delayed atypical plasma cell tumors with unusual Ig/Myc chromosomal rearrangements. J Exp Med. 2007;204:2989–3001. doi: 10.1084/jem.20070882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Janz S, Muller J, Shaughnessy J, Potter M. Detection of recombinations between c-myc and immunoglobulin switch alpha in murine plasma cell tumors and preneoplastic lesions by polymerase chain reaction. Proc Natl Acad Sci USA. 1993;90:7361–5. doi: 10.1073/pnas.90.15.7361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Muller JR, Jones GM, Potter M, Janz S. Detection of immunoglobulin/c-myc recombinations in mice that are resistant to plasmacytoma induction. Cancer Res. 1996;56:419–23. [PubMed] [Google Scholar]
- 64.Kovalchuk AL, Potter M, Janz S. DNA sequence analysis of the genetic recombination between Igh6 and Myc in an uncommon BALB/c plasmacytoma, TEPC 1194. Immunogenetics. 1996;44:151–6. doi: 10.1007/BF02660065. [DOI] [PubMed] [Google Scholar]
- 65.Ramiro AR, Jankovic M, Eisenreich T, et al. AID is required for c-myc/IgH chromosome translocations in vivo. Cell. 2004;118:431–8. doi: 10.1016/j.cell.2004.08.006. [DOI] [PubMed] [Google Scholar]
- 66.Takizawa M, Tolarova H, Li Z, et al. AID expression levels determine the extent of cMyc oncogenic translocations and the incidence of B cell tumor development. J Exp Med. 2008;205:1949–57. doi: 10.1084/jem.20081007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ramiro AR, Jankovic M, Nussenzweig MC. Amplifying Igh translocations. Nat Immunol. 2005;6:117. doi: 10.1038/ni0205-117. author reply 8. [DOI] [PubMed] [Google Scholar]
- 68.Paabo S, Irwin DM, Wilson AC. DNA damage promotes jumping between templates during enzymatic amplification. J Biol Chem. 1990;265:4718–21. [PubMed] [Google Scholar]
- 69.Kovalchuk AL, Kim JS, Janz S. E mu/S mu transposition into Myc is sometimes a precursor for T(12;15) translocation in mouse B cells. Oncogene. 2003;22:2842–50. doi: 10.1038/sj.onc.1206345. [DOI] [PubMed] [Google Scholar]
- 70.Potter M, Wiener F. Plasmacytomagenesis in mice: model of neoplastic development dependent upon chromosomal translocations. Carcinogenesis. 1992;13:1681–97. doi: 10.1093/carcin/13.10.1681. [DOI] [PubMed] [Google Scholar]
- 71.Gauwerky CE, Croce CM. Chromosomal translocations in leukaemia. Semin Cancer Biol. 1993;4:333–40. [PubMed] [Google Scholar]
- 72.Kuppers R, Dalla-Favera R. Mechanisms of chromosomal translocations in B cell lymphomas. Oncogene. 2001;20:5580–94. doi: 10.1038/sj.onc.1204640. [DOI] [PubMed] [Google Scholar]
- 73.Leder P, Battey J, Lenoir G, Moulding C, Murphy W, Potter H, Stewart T, Taub R. Translocations among antibody genes in human cancer. Science. 1983;222:765–71. doi: 10.1126/science.6356357. [DOI] [PubMed] [Google Scholar]
- 74.Rabbitts TH. Chromosomal translocations in human cancer. Nature. 1994;372:143–9. doi: 10.1038/372143a0. [DOI] [PubMed] [Google Scholar]
- 75.Taub R, Kirsch I, Morton C, Lenoir G, Swan D, Tronick S, Aaronson S, Leder P. Translocation of the c-myc gene into the immunoglobulin heavy chain locus in human Burkitt lymphoma and murine plasmacytoma cells. Proc Natl Acad Sci USA. 1982;79:7837–41. doi: 10.1073/pnas.79.24.7837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Stanton LW, Watt R, Marcu KB. Translocation, breakage and truncated transcripts of c-myc oncogene in murine plasmacytomas. Nature. 1983;303:401–6. doi: 10.1038/303401a0. [DOI] [PubMed] [Google Scholar]
- 77.Marcu KB, Harris LJ, Stanton LW, Erikson J, Watt R, Croce CM. Transcriptionally active c-myc oncogene is contained within NIARD, a DNA sequence associated with chromosome translocations in B-cell neoplasia. Proc Natl Acad Sci USA. 1983;80:519–23. doi: 10.1073/pnas.80.2.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hamlyn PH, Rabbitts TH. Translocation joins c-myc and immunoglobulin gamma 1 genes in a Burkitt lymphoma revealing a third exon in the c-myc oncogene. Nature. 1983;304:135–9. doi: 10.1038/304135a0. [DOI] [PubMed] [Google Scholar]
- 79.Erikson J, ar-Rushdi A, Drwinga HL, Nowell PC, Croce CM. Transcriptional activation of the translocated c-myc oncogene in Burkitt lymphoma. Proc Natl Acad Sci USA. 1983;80:820–4. doi: 10.1073/pnas.80.3.820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Dalla-Favera R, Martinotti S, Gallo RC, Erikson J, Croce CM. Translocation and rearrangements of the c-myc oncogene locus in human undifferentiated B-cell lymphomas. Science. 1983;219:963–7. doi: 10.1126/science.6401867. [DOI] [PubMed] [Google Scholar]
- 81.Crews S, Barth R, Hood L, Prehn J, Calame K. Mouse c-myc oncogene is located on chromosome 15 and translocated to chromosome 12 in plasmacytomas. Science. 1982;218:1319–21. doi: 10.1126/science.7146913. [DOI] [PubMed] [Google Scholar]
- 82.Adams JM, Gerondakis S, Webb E, Mitchell J, Bernard O, Cory S. Transcriptionally active DNA region that rearranges frequently in murine lymphoid tumors. Proc Natl Acad Sci USA. 1982;79:6966–70. doi: 10.1073/pnas.79.22.6966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Adams JM, Gerondakis S, Webb E, Corcoran LM, Cory S. Cellular myc oncogene is altered by chromosome translocation to an immunoglobulin locus in murine plasmacytomas and is rearranged similarly in human Burkitt lymphomas. Proc Natl Acad Sci USA. 1983;80:1982–6. doi: 10.1073/pnas.80.7.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Siwarski D, Muller U, Andersson J, Notario V, Melchers F, Rolink A, Huppi K. Structure and expression of the c-Myc/Pvt 1 megagene locus. Curr Top Microbiol Immunol. 1997;224:67–72. doi: 10.1007/978-3-642-60801-8_6. [DOI] [PubMed] [Google Scholar]
- 85.Beck-Engeser GB, Lum AM, Huppi K, Caplen NJ, Wang BB, Wabl M. Pvt1-encoded microRNAs in oncogenesis. Retrovirology. 2008;5:4. doi: 10.1186/1742-4690-5-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Graham M, Adams JM. Chromosome 8 breakpoint far 3′ of the c-myc oncogene in a Burkitt’s lymphoma 2;8 variant translocation is equivalent to the murine pvt-1 locus. EMBO J. 1986;5:2845–51. doi: 10.1002/j.1460-2075.1986.tb04578.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Tsichlis PN, Lee JS, Bear SE, et al. Activation of multiple genes by provirus integration in the Mlvi-4 locus in T-cell lymphomas induced by moloney murine leukemia virus. J Virol. 1990;64:2236–44. doi: 10.1128/jvi.64.5.2236-2244.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Robbiani DF, Bothmer A, Callen E, et al. Activation induced deaminase is required for the chromosomal translocations in c-myc that lead to c-myc/IgH translocations. Cell. 2008;135:1028–38. doi: 10.1016/j.cell.2008.09.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Unniraman S, Zhou S, Schatz DG. Identification of an AID-independent pathway for chromosomal translocations between the Igh switch region and Myc. Nat Immunol. 2004;5:1117–23. doi: 10.1038/ni1127. [DOI] [PubMed] [Google Scholar]
- 90.Muramatsu M, Sankaranand VS, Anant S, Sugai M, Kinoshita K, Davidson NO, Honjo T. Specific expression of activation-induced cytidine deaminase (AID), a novel member of the RNA-editing deaminase family in germinal center B cells. J Biol Chem. 1999;274:18470–6. doi: 10.1074/jbc.274.26.18470. [DOI] [PubMed] [Google Scholar]
- 91.Crouch EE, Li Z, Takizawa M, et al. Regulation of AID expression in the immune response. J Exp Med. 2007;204:1145–56. doi: 10.1084/jem.20061952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Gonda H, Sugai M, Nambu Y, Katakai T, Agata Y, Mori KJ, Yokota Y, Shimizu A. The balance between Pax5 and Id2 activities is the key to AID gene expression. J Exp Med. 2003;198:1427–37. doi: 10.1084/jem.20030802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Sayegh CE, Quong MW, Agata Y, Murre C. E-proteins directly regulate expression of activation-induced deaminase in mature B cells. Nat Immunol. 2003;4:586–93. doi: 10.1038/ni923. [DOI] [PubMed] [Google Scholar]
- 94.Shaffer AL, Lin KI, Kuo TC, et al. Blimp-1 orchestrates plasma cell differentiation by extinguishing the mature B cell gene expression program. Immunity. 2002;17:51–62. doi: 10.1016/s1074-7613(02)00335-7. [DOI] [PubMed] [Google Scholar]
- 95.Lin KI, Angelin-Duclos C, Kuo TC, Calame K. Blimp-1-dependent repression of Pax-5 is required for differentiation of B cells to immunoglobulin M-secreting plasma cells. Mol Cell Biol. 2002;22:4771–80. doi: 10.1128/MCB.22.13.4771-4780.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Sciammas R, Shaffer AL, Schatz JH, Zhao H, Staudt LM, Singh H. Graded expression of interferon regulatory factor-4 coordinates isotype switching with plasma cell differentiation. Immunity. 2006;25:225–36. doi: 10.1016/j.immuni.2006.07.009. [DOI] [PubMed] [Google Scholar]
- 97.Dedeoglu F, Horwitz B, Chaudhuri J, Alt FW, Geha RS. Induction of activation-induced cytidine deaminase gene expression by IL-4 and CD40 ligation is dependent on STAT6 and NFkappaB. Int Immunol. 2004;16:395–404. doi: 10.1093/intimm/dxh042. [DOI] [PubMed] [Google Scholar]
- 98.Jiang C, Zhao ML, Diaz M. Activation-induced deaminase heterozygous MRL/lpr mice are delayed in the production of high-affinity pathogenic antibodies and in the development of lupus nephritis. Immunology. 2008;126:102–13. doi: 10.1111/j.1365-2567.2008.02882.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.McBride KM, Gazumyan A, Woo EM, Schwickert TA, Chait BT, Nussenzweig MC. Regulation of class switch recombination and somatic mutation by AID phosphorylation. J Exp Med. 2008;205:2585–94. doi: 10.1084/jem.20081319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Liu M, Duke JL, Richter DJ, Vinuesa CG, Goodnow CC, Kleinstein SH, Schatz DG. Two levels of protection for the B cell genome during somatic hypermutation. Nature. 2008;451:841–5. doi: 10.1038/nature06547. [DOI] [PubMed] [Google Scholar]
- 101.McBride KM, Barreto V, Ramiro AR, Stavropoulos P, Nussenzweig MC. Somatic hypermutation is limited by CRM1-dependent nuclear export of activation-induced deaminase. J Exp Med. 2004;199:1235–44. doi: 10.1084/jem.20040373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Dorsett Y, McBride KM, Jankovic M, et al. MicroRNA-155 suppresses activation-induced cytidine deaminase-mediated Myc–Igh translocation. Immunity. 2008;28:1–9. doi: 10.1016/j.immuni.2008.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Teng G, Hakimpour P, Landgraf P, Rice A, Tuschl T, Casellas R, Papavasiliou FN. MicroRNA-155 is a negative regulator of activation-induced cytidine deaminase. Immunity. 2008;28:621–9. doi: 10.1016/j.immuni.2008.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.de Yebenes VG, Belver L, Pisano DG, Gonzalez S, Villasante A, Croce C, He L, Ramiro AR. miR-181b negatively regulates activation-induced cytidine deaminase in B cells. J Exp Med. 2008;205:2199–206. doi: 10.1084/jem.20080579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Rada C, Jarvis JM, Milstein C. AID-GFP chimeric protein increases hypermutation of Ig genes with no evidence of nuclear localization. Proc Natl Acad Sci USA. 2002;99:7003–8. doi: 10.1073/pnas.092160999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Cattoretti G, Buettner M, Shaknovich R, Kremmer E, Alobeid B, Niedobitek G. Nuclear and cytoplasmic AID in extrafollicular and germinal center B cells. Blood. 2006;107:3967–75. doi: 10.1182/blood-2005-10-4170. [DOI] [PubMed] [Google Scholar]
- 107.Ito S, Nagaoka H, Shinkura R, Begum N, Muramatsu M, Nakata M, Honjo T. Activation-induced cytidine deaminase shuttles between nucleus and cytoplasm like apolipoprotein B mRNA editing catalytic polypeptide 1. Proc Natl Acad Sci USA. 2004;101:1975–80. doi: 10.1073/pnas.0307335101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Brar SS, Watson M, Diaz M. Activation-induced cytosine deaminase (AID) is actively exported out of the nucleus but retained by the induction of DNA breaks. J Biol Chem. 2004;279:26395–401. doi: 10.1074/jbc.M403503200. [DOI] [PubMed] [Google Scholar]
- 109.Aoufouchi S, Faili A, Zober C, D’Orlando O, Weller S, Weill JC, Reynaud CA. Proteasomal degradation restricts the nuclear lifespan of AID. J Exp Med. 2008;205:1357–68. doi: 10.1084/jem.20070950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Pasqualucci L, Kitaura Y, Gu H, Dalla-Favera R. PKA-mediated phosphorylation regulates the function of activation-induced deaminase (AID) in B cells. Proc Natl Acad Sci USA. 2006;103:395–400. doi: 10.1073/pnas.0509969103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Basu U, Chaudhuri J, Alpert C, et al. The AID antibody diversification enzyme is regulated by protein kinase A phosphorylation. Nature. 2005;438:508–11. doi: 10.1038/nature04255. [DOI] [PubMed] [Google Scholar]
- 112.McBride KM, Gazumyan A, Woo EM, Barreto VM, Robbiani DF, Chait BT, Nussenzweig MC. Regulation of hypermutation by activation-induced cytidine deaminase phosphorylation. Proc Natl Acad Sci USA. 2006;103:8798–803. doi: 10.1073/pnas.0603272103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Pham P, Smolka MB, Calabrese P, Landolph A, Zhang K, Zhou H, Goodman MF. Impact of phosphorylation and phosphorylation-null mutants on the activity and deamination specificity of activation-induced cytidine deaminase. J Biol Chem. 2008;283:17428–39. doi: 10.1074/jbc.M802121200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Basu U, Wang Y, Alt FW. Evolution of phosphorylation-dependent regulation of activation-induced cytidine deaminase. Mol Cell. 2008;32:285–91. doi: 10.1016/j.molcel.2008.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Xue K, Rada C, Neuberger MS. The in vivo pattern of AID targeting to immunoglobulin switch regions deduced from mutation spectra in msh2−/− ung−/− mice. J Exp Med. 2006;203:2085–94. doi: 10.1084/jem.20061067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Wu X, Stavnezer J. DNA polymerase beta is able to repair breaks in switch regions and plays an inhibitory role during immunoglobulin class switch recombination. J Exp Med. 2007;204:1677–89. doi: 10.1084/jem.20070756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Pasqualucci L, Neumeister P, Goossens T, Nanjangud G, Chaganti RS, Kuppers R, Dalla-Favera R. Hypermutation of multiple proto-oncogenes in B-cell diffuse large-cell lymphomas. Nature. 2001;412:341–6. doi: 10.1038/35085588. [DOI] [PubMed] [Google Scholar]
- 118.Shen HM, Peters A, Baron B, Zhu X, Storb U. Mutation of BCL-6 gene in normal B cells by the process of somatic hypermutation of Ig genes. Science. 1998;280:1750–2. doi: 10.1126/science.280.5370.1750. [DOI] [PubMed] [Google Scholar]
- 119.Okazaki IM, Hiai H, Kakazu N, Yamada S, Muramatsu M, Kinoshita K, Honjo T. Constitutive expression of AID leads to tumorigenesis. J Exp Med. 2003;197:1173–81. doi: 10.1084/jem.20030275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Okazaki IM, Kotani A, Honjo T. Role of AID in tumorigenesis. Adv Immunol. 2007;94:245–73. doi: 10.1016/S0065-2776(06)94008-5. [DOI] [PubMed] [Google Scholar]
- 121.Matsumoto Y, Marusawa H, Kinoshita K, et al. Helicobacter pylori infection triggers aberrant expression of activation-induced cytidine deaminase in gastric epithelium. Nat Med. 2007;13:470–6. doi: 10.1038/nm1566. [DOI] [PubMed] [Google Scholar]
- 122.Gourzi P, Leonova T, Papavasiliou FN. A role for activation-induced cytidine deaminase in the host response against a transforming retrovirus. Immunity. 2006;24:779–86. doi: 10.1016/j.immuni.2006.03.021. [DOI] [PubMed] [Google Scholar]
- 123.Neuberger MS, Rada C. Somatic hypermutation: activation-induced deaminase for C/G followed by polymerase eta for A/T. J Exp Med. 2007;204:7–10. doi: 10.1084/jem.20062409. [DOI] [PMC free article] [PubMed] [Google Scholar]