

Abstract
Consumption of green tea polyphenols (GTPs) in drinking water prevents photocarcinogenesis in mice; however, the molecular mechanisms underlying this effect have not been fully elucidated. Using interleukin (IL)-12p40 knockout (IL-12-KO) mice and their wild-type counterparts and an established photocarcinogenesis protocol, we found that although administration of GTPs (0.2%, w/v) in drinking water significantly reduced UVB-induced tumor development in wild-type mice, this treatment had a non-significant effect in IL-12-KO mice. GTPs resulted in reduction in the levels of markers of inflammation (COX-2, PGE2, PCNA, cyclin D1) and proinflammatory cytokines (TNF-α, IL-6, IL-1β) in chronically UVB-exposed skin and skin tumors of wild-type mice but less effective in IL-12p40-KO mice. UVB-induced DNA damage (cyclobutane pyrimidine dimers) was resolved rapidly in GTPs-treated wild-type mice than untreated wild-type mice and this resolution followed the same time course as the GTPs-induced reduction in the levels of inflammatory responses. This effect of GTPs was less pronounced in IL-12-KO mice. The above results were confirmed by treatment of IL-12-KO mice with murine rIL-12 and treatment of wild-type mice with neutralizing anti-IL-12 antibody. To our knowledge this is previously unreported that prevention of photocarcinogenesis by GTPs is mediated through IL-12-dependent DNA repair and a subsequent reduction in skin inflammation.
Keywords: Green tea, DNA repair, cyclobutane pyrimidine dimer, interleukin-12, ultraviolet radiation
INTRODUCTION
Exposure of the skin to solar ultraviolet (UV) radiation induces inflammatory responses, oxidative stress, immunosuppression, DNA damage and gene mutations, all of which have been implicated in a variety of skin diseases including the development of skin cancers (Katiyar, 2006; Katiyar et al., 2000; Katiyar et al., 2007). UV-induced inflammatory responses, which are characterized by increased blood flow and vascular permeability, result in the development of edema, erythema, hyperplastic responses, and increases in the levels of cyclooxygenase-2 (COX-2) and prostaglandin (PG) metabolites (Black et al., 1978; Rivas and Ullrich, 1994; Katiyar and Meeran, 2007; Mukhtar and Elmets, 1996). UV-induced inflammation is considered as an early event in tumor promotion and/or tumor development. Chronic inflammation plays a crucial role in all three stages of tumor development, i.e., initiation, promotion and progression (Mukhtar and Elmets, 1996).
Interleukin (IL)-12, an immunoregulatory cytokine, is composed of two disulfide-bonded protein chains p35 and p40 (Trinchieri, 1994), and has been shown to have antitumor activity in a variety of tumor models (Brunda, 1994; Brunda et al., 1993; Zou et al., 1995; Robertson and Ritz, 1996). We and others have shown that mice deficient in IL-12 are at higher risk of UV radiation-induced skin tumors than their wild-type counterparts (Meeran et al., 2006; Maeda et al., 2006). We observed that the development of UV-induced tumors in IL-12 knockout (IL-12 KO) mice occurred earlier, was more rapid, and was associated with a significantly higher tumor multiplicity than the development of UV-induced tumors in their wild-type counterparts (Meeran et al., 2006). The demonstration of significant antitumor activity of IL-12 in preclinical animal tumor models has stimulated interest in the therapeutic use of IL-12 (Chen et al., 1997; Siders et al., 1998; Nastala et al., 1994).
Polyphenols isolated from the leaves of green tea (Camellia sinensis) have a number of beneficial health effects including anti-carcinogenic activity, which has been demonstrated in various tumor models (Katiyar and Mukhtar, 1996; Yang et al., 2002). In previous studies, we and others have shown that oral administration of an aqueous extract of green tea or green tea polyphenols (GTPs; a mixture of polyphenols) in drinking water inhibits UV radiation-induced skin carcinogenesis in mice in terms of tumor incidence, tumor multiplicity and tumor growth/size (Wang et al., 1992; Mantena et al., 2005). UV-induced DNA damage, predominantly the formation of cyclobutane pyrimidine dimers (CPDs), has been recognized as an important molecular trigger for the initiation of UVB-induced carcinogenesis in the skin (Applegate et al., 1989; Kripke et al., 1992; Yarosh et al., 1992). Reduction of CPDs through application of DNA repair enzymes considerably reduces the risk of UV-induced skin cancer in mice and in humans (Yarosh et al., 1992; Yarosh et al., 2001). UV-induced inflammation and its mediators also have been implicated in the development of skin tumors. As UV-induced inflammatory responses, such as the production of pro-inflammatory cytokines and prostaglandins, and UV-induced tumorigenesis are both causally related to UVB-induced DNA damage, we sought to determine whether the chemopreventive effects of drinking GTPs on photocarcinogenesis are mediated, at least in part, through enhancement of DNA repair and subsequent inhibition of inflammatory responses in mouse skin. As green tea is commonly used as a beverage world-wide, we assessed the mechanism of photoprotective effect of its active ingredients (polyphenols) after mixing it in drinking water and using in vivo mouse model. Our hypothesis is based on the fact that IL-12 plays a role in removal or repair of UVB-induced DNA damage, and we have found that GTPs enhance the levels of IL-12 in UV-exposed mice. We also hypothesized that, if this is the case, treatment with GTPs in drinking water would be unable to prevent UVB-induced skin carcinogenesis in IL-12-deficient mice, and unable to inhibit inflammation and inflammatory mediators, or inhibit UVB-induced DNA repair.
RESULTS
Stability of green tea polyphenols in drinking water
The chemical composition of GTPs was not significantly changed in drinking water for three days. As we have changed GTPs-containing water after each three days, we analyzed the polyphenolic composition of GTPs after three days using HPLC and data were compared with the original composition of GTPs used in this study. As shown in Table 1, a significant change in terms of percentage of polyphenolic constituents in GTPs was not observed which indicates the stability of GTPs in water at least for 3 days.
Table 1.
Chemical composition of green tea polyphenols (GTPs) used in this study. Analysis of GTPs was performed immediately and 3 days after dissolving it in normal drinking water.
| Constituents | Concentration (%)
|
|
|---|---|---|
| Immediate | 3 days later | |
| Epigallocatechin-3-gallate (EGCG) | 64.0 | 58.1 ±2.2 |
| Epicatechin gallate (ECG) | 5.4 | 7.0 ±0.5 |
| Epigallocatechin (EGC) | 5.3 | 4.6 ±0.4 |
| Epicatechin (EC) | 8.8 | 11.0 ±1.0 |
| Catechin | 0.9 | 1.2 ±0.2 |
| Gallocatechin gallate(GCG) | 3.1 | 2.9 ±0.4 |
| Total catechins | 87.5 | 84.8 |
Analytical conditions: Shimadzu HPLC System: LC-10ADvp Binary Pump, DGU-14A Degassing Module, SIL-HTC Auto sampler
Mobile Phase: A = 0.1% HCOOH, B = Me CN + 0.1% HCOOH
Column: Develosil C30 (Phenomenex), 2.0 × 30 mm Guard Column, 2.0 × 150 mm Analytical Column.
MDS Sciex Mass Spectrometer: API-4000 Triple Quadrupole, TurboIonspray, Negative Mode, All gases = N2
Source Temp = 350° C, Flow Rate: 0.35 ml/min
Prevention of photocarcinogenesis in mice by oral administration of GTPs is IL-12-dependent
Previously, we have shown that oral administration of GTPs (0.2%, w/v) inhibits UVB-induced tumor development in the skin of SKH-1 hairless mice (Mantena et al., 2005). As UVB-induced immunosuppression has been implicated as a risk factor for photocarcinogenesis, we investigated the effect of provision of GTPs in the drinking water on UVB-induced suppression of the contact hypersensitivity response to a contact sensitizer, dinitrofluorobenzene, and found that GTPs inhibit this reaction (Katiyar et al., unpublished data). UVB-induced suppression of contact hypersensitivity has been associated with an increase in the immunosuppressive cytokine IL-10 and suppression of the immunostimulatory cytokine IL-12 (Katiyar, 2007). We have found that GTPs affect the levels of IL-10 and IL-12 in the skin and draining lymph nodes of UVB-exposed mice and counteracts the UVB-induced reversal of the ratio of IL-10 and IL-12 (Katiyar et al., unpublished data). To test the hypothesis that drinking GTPs prevents photocarcinogenesis through the enhancement of IL-12 levels, we examined the effect of GTPs on photocarcinogenesis in IL-12p40 KO mice and their wild-type counterparts. We first confirmed that when WT mice were exposed to a standard photocarcinogenesis protocol, administration of GTPs in the drinking water resulted in a reduction in UVB-induced skin tumorigenesis in terms of tumor incidence and tumor multiplicity compared to non-GTPs-treated WT mice (Fig. 1, left panels). At the termination of the experiment at 40 weeks, the tumor incidence (% of mice with tumors) was 25% (p<0.05) lower in the wild-type mice that were administered GTPs in the drinking water as compared to non-GTPs-treated mice. Only 40% of the GTPs-treated mice developed tumors, compared to 65% of the non-GTPs-treated mice. GTPs also increased the latency period of the tumors by 2 weeks in WT mice under the experimental conditions used in these studies. A total of 33 tumors (2.5±1.0 tumors/tumor bearing animal) were recorded in the group of 20 wild-type mice that were irradiated with UVB but did not receive GTPs (non-GTPs) whereas only 15 tumors (1.87±0.5 tumors/tumor bearing mouse) were recorded in the group of 20 irradiated wild-type mice treated with GTPs (Fig. 1, left panel). The tumor multiplicity (55%, p<0.005) and tumor growth/size was significantly lower (50%, p<0.01) in the group of UVB-irradiated wild-type mice that were provided GTPs in the drinking water than in the control group of wild-type mice that were UVB-irradiated but not treated with GTPs. In addition, both the rate of appearance of the UVB-induced tumors and their development in the GTPs-treated wild-type animals was significantly lower (p<0.05, Fisher-Irwin exact test) than in control animals. Tumor formation was not observed in the skin of mice that were given either normal drinking water or GTPs in drinking water for 40 weeks but were not UV-irradiated.
Figure 1.
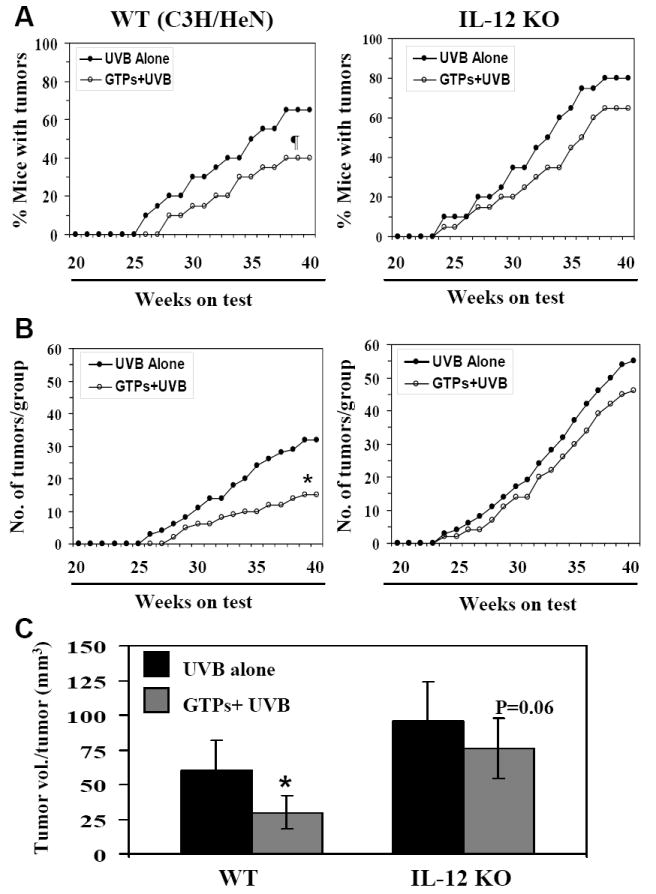
Administration of GTPs in drinking water inhibits UVB-induced skin tumor development in wild-type (WT; C3H/HeN) mice but does not inhibit it significantly in IL-12 KO mice. The percent of mice with tumors (Panel A) and the total number of tumors per group (Panel B) are plotted as a function of the number of weeks on treatment. The tumor data for WT mice are shown in the left panels while the tumor data for IL-12 KO mice are shown in the right panels. Tumor volume per tumor (mm3) was recorded at the termination of the experiment at 40th week, and tumor volume is represented as mean± SD, as shown in Panel C. Each treatment group had 20 mice. Significant inhibition versus UVB alone at the termination of the experiment, *p<0.01, ¶p<0.05.
Analysis of the effects of administration of GTPs in the drinking water of IL-12 KO mice indicated that the GTPs did not exert a significant protective effect against photocarcinogenesis in the IL-12 KO mice (Fig. 1; right panels). The IL-12 KO mice appeared to be more susceptible to UVB radiation-induced skin tumor development than their wild-type counterparts, as is evident from the data concerning tumor incidence and tumor multiplicity (Fig. 1, right panels vs left panels). Although the difference in susceptibility in terms of percent of mice with tumors was not statistically significant, 80% of the irradiated IL-12 KO mice developed tumors whereas only 65% of their wild-type counterparts did. A total of 55 tumors were recorded in the group of IL-12 KO mice compared to only 33 tumors in the group of wild-type mice (Fig. 1). The first appearance of UV-induced tumors was noted at least 2 weeks earlier in the IL-12 KO mice than in the wild-type mice (difference is not statistically significant). As shown in Fig. 1 (right panels), the administration of GTPs in drinking water of the IL-12 KO mice has less pronounced protective effects from UVB-induced skin tumor development as measured in terms of either tumor incidence (15%, p=0.13), tumor multiplicity (16%, p=0.11) or tumor volume per tumor (21%, p<0.06). This non-significant chemopreventive effect of GTPs on UVB-induced tumor development in IL-12 KO mice, however, suggests that in addition to IL-12, other protective mechanisms may have a role. It means, one protective mechanism of GTPs is IL-12-dependent and another is not. Histopathologic analyses of H&E-stained sections of UVB-induced skin tumors harvested from both wild-types and IL-12 KO mice revealed that majority of the tumors were squamous cell papillomas and were of epidermal origin.
Effect of GTPs on UVB-induced inflammation and its mediators in mice
(i) Administration of GTPs inhibits UV-induced enhancement of COX-2 expression and PGE2 production in wild-type mice but less effective in IL-12 KO mice
UVB-induced COX-2 expression and a subsequent increase in the production of PG metabolites in the skin is a characteristic response of keratinocytes to acute or chronic exposure to UVB radiation. In addition, increased expression of COX-2 and PG metabolites has been observed in squamous and basal cell carcinomas of the skin (Mukhtar and Elmets, 1996; Vanderveen et al., 1986). We therefore compared the effects of GTPs on the levels of COX-2 and PGE2 in skin samples from wild-type and IL-12 KO mice that had been chronically exposed to UVB light. Immunohistochemical analysis indicated that chronic exposure of the skin to UV radiation resulted in enhanced expression of COX-2 in the skin of IL-12 KO and wild-type mice as compared to the non-UVB-exposed mouse skin (Fig. 1A, Supplementary data). Administration of GTPs in the drinking water significantly reduced the UVB-induced expression of COX-2 in the skin of the wild-type mice but was less effective in reducing the levels of COX-2 in the IL-12 KO mouse skin (Fig. 1A, Supplementary data). These data were further confirmed by western blot analysis which showed higher expression levels of COX-2 protein in IL-12 KO than wild-type mouse skin (Fig. 2A), and that the GTPs were more effective in inhibiting the expression levels of COX-2 in the wild-type mouse skin than in IL-12 KO mouse skin.
Figure 2.
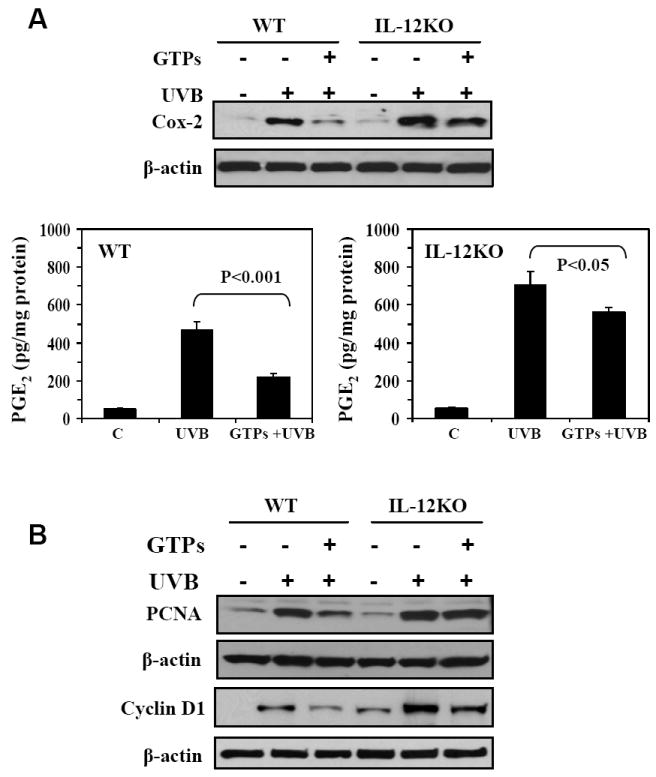
Administration of GTPs in drinking water inhibits UVB-induced increases in inflammatory responses, their mediators and markers of proliferation in the epidermis of WT mouse skin, but is less effective in inhibiting these responses in IL-12 KO mouse skin. A, Epidermal COX-2 was determined by western blotting and PGE2 was determined in the epidermal homogenates using an enzyme-linked immunosorbent assay. The concentration of PGE2 is expressed in terms of pg/mg protein as a mean ± SD, n = 10. B, The levels of epidermal PCNA and cyclin D1 were determined employing western blot analysis. Representative western blots are shown from three independent experiments with identical observations. In each experiment, epidermis was pooled from 2-3 mice for preparation of lysates, and equivalent protein loading was checked by probing stripped blots for β-actin as shown.
As increased expression of COX-2 results in the formation of greater amounts of PG metabolites, we determined the levels of PG metabolites in the skin of the mice from the different treatment groups with a particular emphasis on PGE2. PGE2 appears to be pivotal in the reciprocal regulation of IL-10 and IL-12 production in addition to its role in tumor promotion. As shown in Figure 2A, the levels of PGE2 in UVB-irradiated skin were significantly higher in both IL-12 KO and wild-type mouse skin (p<0.001) compared to non-UVB-exposed epidermis. The administration of GTPs significantly inhibited (59%, p<0.001) the UVB-induced increases in the levels of PGE2 in wild-type mouse skin but was less effective (22%, p<0.05) in inhibiting the UVB-induced increases observed in the skin of IL-12 KO mice.
(ii) Administration of GTPs is more effective in inhibiting UVB-induced increases in the levels of PCNA and cyclin D1 in the skin of wild-type mice than IL-12 KO mice
We then determined the proliferation potential of epidermal cells (i.e., the hyperplastic response) as another marker of the UVB-induced inflammatory reaction in the skin. For this purpose, the levels of PCNA and cyclin D1 were determined using immunohistochemical detection (Figure 1B, Supplementary data) and western blot analysis. As shown in Fig. 2B, the western blot analysis indicated that the levels of PCNA were higher in the UVB-irradiated mouse skin than the non-UVB-irradiated mouse skin. Administration of GTPs in the drinking water reduced the UVB-induced increases in the levels of PCNA in the skin of wild-type mice but oral administration of GTPs did not exert a similar effect in IL-12 KO mice. Similarly, we determined the levels of cyclin D1, another marker of cell proliferation. As shown in Figure 2B, the levels of cyclin D1 were higher in UVB-exposed skin than non-UVB-exposed mouse skin, and oral administration of GTPs inhibited the expression levels of cyclin D1 to a greater extent in wild-type mice than in IL-12 KO mouse skin.
(iii) The inhibitory effect of GTPs on UVB-induced pro-inflammatory cytokines is higher in wild-type mouse skin than IL-12-deficient mouse skin
It is well known that exposure of skin to UVB radiation induces inflammatory responses, including the induction of pro-inflammatory cytokines. We therefore compared the effects of chronic UVB-exposure on the secretion or synthesis of proinflammatory cytokines in the skin of IL-12 KO mice and their wild-type counterparts as well as the effects of oral administration of GTPs on the levels of proinflammatory cytokines in the UVB-exposed skin of the mice. The levels of TNF-α, IL-1β and IL-6 in skin homogenates were determined using commercially available ELISA kits. As shown in Fig. 3, chronic exposure of the skin to UVB radiation resulted in significantly higher synthesis or accumulation of TNF-α (p<0.005), IL-1β (p<0.01) and IL-6 (p<0.01-p<0.001) cytokines in the skin of both IL-12 KO and wild-type mice as compared with their non-UVB-exposed counterparts; however, this effect was significantly greater (p<0.05–0.01) in the IL-12 KO mice than the wild-type mice. As shown in Figure 3, provision of GTPs in the drinking water of wild-type mice was associated with a significant inhibition of the levels of TNF-α (57%, p<0.005), IL-1β (53%, p<0.005) and IL-6 (50%, p<0.005) in the UVB-exposed skin whereas provision of GTPs in the drinking water of IL-12 KO mice was markedly less effective in inhibiting the production of these cytokines (inhibition; TNFα-25%, IL-1β-29% and IL-6-10%) in the UVB-exposed skin.
Figure 3.
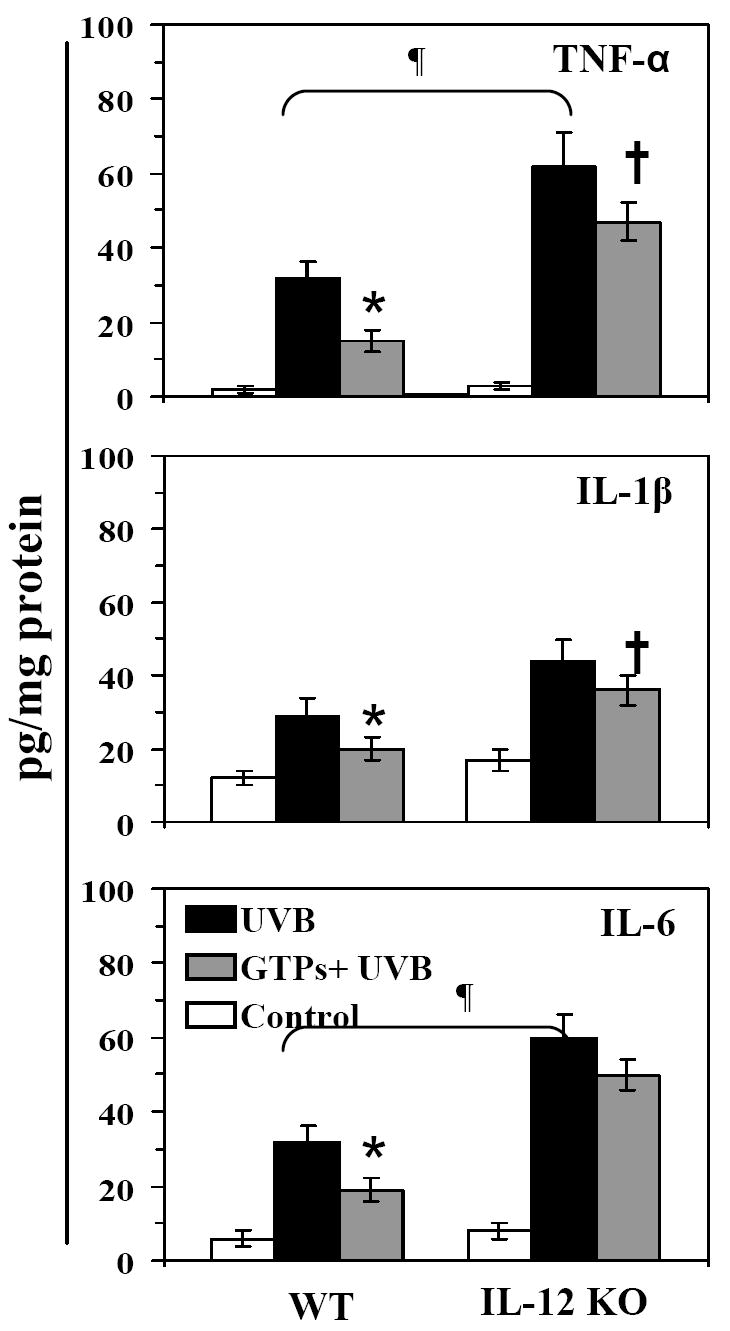
Oral administration of GTPs inhibits chronic UVB-induced increases in pro-inflammatory cytokines in the skin of WT mice but has a less inhibitory effect in the skin of IL-12 KO mice. At the termination of the photocarcinogenesis experiment, epidermal homogenates were prepared for the analysis of the levels of TNF-α, IL-1β and IL-6 using ELISA. The concentration of each cytokine is reported in terms of pg/mg protein as a mean ± SD, n = 10.
Significant inhibition versus UVB-irradiated group, *p<0.005,
Significant increase in IL-12 KO mice versus UVB-irradiated WT mice, ¶p<0.005.
Significant inhibition versus UVB-irradiated IL-12 KO mice, †p<0.05
Administration of GTPs inhibits the UVB-induced enhancement of expression of COX-2, PGE2, PCNA and cyclin D1, and proinflammatory cytokines in the tumors of wild-type mice but is less effective in inhibiting the expression of these markers in tumors of IL-12 KO mice
We further determined the effects of oral administration of GTPs on the expression of these biomarkers of inflammatory reactions in the UVB-induced skin tumors. Western blot analysis indicated that the level of COX-2 protein was higher in the tumors of IL-12 KO mice than in the tumors of wild-type mice (Fig. 4A). These data were further confirmed by immunohistochemical analysis of COX-2 expression in the tumors of these treatment groups, and shown in Supplementary data (Fig. 2A). The concentration of PGE2 was also higher in the tumors of IL-12 KO mice than in the tumors of wild-type mice (Fig. 4B). As shown in Figures 4A and 4B, administration of GTPs in the drinking water inhibited the expression of COX-2 and PGE2 more efficiently in the tumors of wild-type mice than the tumors of IL-12-deficient mice. Analysis of the levels of TNF-α, IL-6 and IL-1β by ELISA indicated that the levels of these proinflammatory cytokines were higher in the tumors of IL-12 KO mice than the tumors of wild-type mice. The administration of GTPs in the drinking water inhibited the levels of TNF-α (45%, p<0.01), IL-6 (48%, p<0.01) and IL-1β (35%, p<0.01) in the tumors of wild-type mice but had a less pronounced effect in the tumors of IL-12 KO mice (Fig. 4B). Administration of GTPs in the drinking water was associated with a greater reduction in the levels of UVB-induced expression of PCNA in the tumors of wild-type mice than in the tumors of IL-12 KO mice (Fig. 4C). The levels of cyclin D1, another marker of cellular proliferation, were also higher in the tumors of IL-12 KO mice than in the tumors of wild-type mice (Fig. 4C), and administration of GTPs in the drinking water markedly inhibited the levels of cyclin D1 in the tumors of wild-type mice but was less effective in inhibiting the levels in the skin tumors of IL-12 KO mice. The western blot data of COX-2 and PCNA in tumor samples were further confirmed using immunohistochemical analysis (Supplementary data, Fig. 2).
Figure 4.
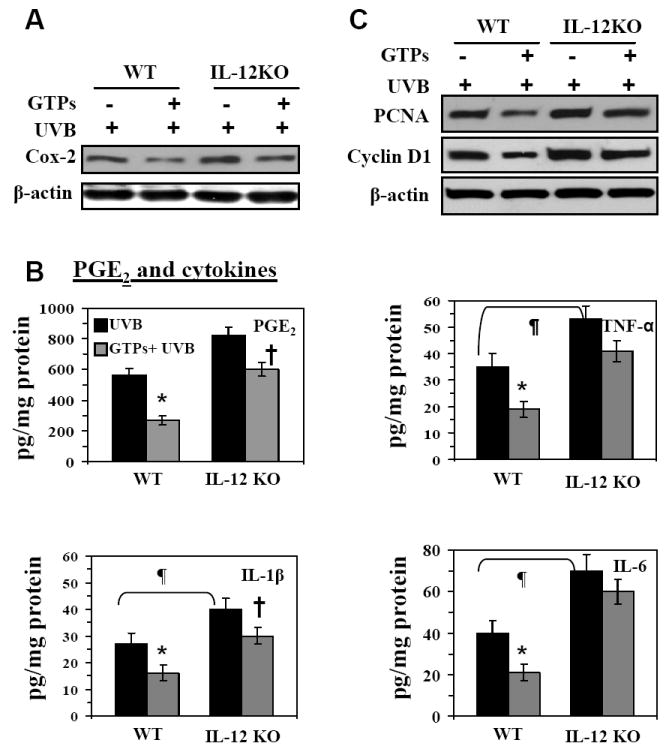
The UVB-induced tumors that develop in IL-12 KO mice express higher levels of inflammatory mediators, including COX-2, PGE2, TNF-α, IL-6 and IL-1β (Panels A & B), and markers of proliferation, including PCNA and cyclin D1 (Panel C) as compared to WT mice. Administration of GTPs inhibits the expression levels of these UVB-induced inflammatory mediators in the tumors of WT mice but the inhibitory effect is less pronounced in the tumors of IL-12 KO mouse. The expression levels of COX-2, PCNA and cyclin D1 were determined using western blot analysis. The representative blots are shown from three independent experiments, and in each experiment the tumor samples were pooled from at least two mice. The levels of PGE2, TNF-α, IL-1β and IL-6 were determined in tumor homogenates using ELISA kits and data are presented as the mean± SD in terms of pg/mg protein. Experiments were repeated in tumor samples from at least 8 mice. Significant difference versus non-GTPs-treated UVB-exposed controls, *p<0.01; Significant increase versus WT mouse, ¶p<0.01. Significant inhibition versus UVB-irradiated IL-12 KO mice, †p<0.05
The inhibition of UV-induced inflammation and skin tumorigenesis in mice by GTPs is linked to their repair of UV-induced DNA damage
Taken together with previous studies, the results of the experiments described above clearly established that IL-12-deficiency exacerbates inflammatory responses in UV-irradiated skin (Meeran et al., 2008) and also enhances the development of skin tumors (Fig. 1). As UV-induced inflammatory responses and UV-induced skin tumor development both are causally related to UV-induced DNA damage, we undertook additional experiments to determine whether the inhibitory effect of GTPs on UV-induced inflammation and skin tumor development is due to a GTP-mediated enhancement of the repair or removal of UVB-induced DNA damage from the UVB-exposed skin sites. In these experiments, the IL-12 KO mice and their wild-type counterparts were exposed to an acute UVB exposure (90 mJ/cm2). The mice were randomly divided into groups that were provided either regular drinking water or drinking water containing GTPs (0.2%, w/v) starting at least 14 days before UVB exposure and continuing until the termination of the experiment. The mice were sacrificed either immediately [within 30 min] after UVB exposure or at 24, 48 and 72 h after UVB exposure. Skin samples were then collected and subjected to the analysis of CPDs, as a marker of UVB-induced DNA damage, and the levels of PGE2 and TNF-α, as markers of inflammation and/or inflammatory mediators. Frozen skin sections (5 μm thick) were subjected to immunohistochemical detection of CPD+ cells using an antibody directed against CPD (Kamiya Biomedical Co.). In skin samples obtained immediately after UVB exposure, no differences in the staining pattern of CPDs were observed between GTPs-treated and non-GTPs-treated mouse skin samples from either wild-type or IL-12 KO mice (Fig. 5A). Similarly, no difference in the staining pattern of CPD-positive cells were observed in both wild-type and IL-12 KO mice whether treated or not with GTPs at 24 h time point after UVB exposure (data not shown). However, in samples obtained 48 h after UVB exposure, there were significantly fewer CPD+ cells (p<0.01) in the skin of wild-type mice that were treated with GTPs than those that were not treated with GTPs. In contrast, although the numbers of CPD+ cells in the skin of IL-12 KO mice treated with GTPs had decreased at 48 h after UVB exposure, this decrease was not significantly different from the number of CPD+ cells detected in the skin of IL-12 KO mice that had not been treated with GTPs at the same time point. This effect of the GTPs on the DNA repair was more pronounced (p<0.001) at 72 h after UVB exposure of the wild-type mice, but was less pronounced in the skin of IL-12 KO mice at this time point. The results of the analysis of CPD+ cells in the skin of the mice in the different treatment groups are summarized in Figure 5B. These data suggest that the difference in the GTPs-associated DNA repair capacity between wild-type and IL-12 KO mice may be due to the absence of IL-12 in the IL-12 KO mice. We also determined the levels of epidermal PGE2 in the same skin samples and found that the levels of PGE2 were higher at 48 h after the UVB exposure of the skin of wild-type mice but had decreased at 72 h with the spontaneous decrease in DNA damage. Administration of GTPs significantly reduced the levels of PGE2 (p<0.01) at 48 and 72 h after UVB exposure of the wild-type mice and this effect paralleled the enhancement of CPDs repair in wild-type mouse skin (Fig. 5C). However, GTPs-induced reduction in the levels of PGE2 was less pronounced in the skin of IL-12 KO mice. To further correlate the effect of UVB-induced DNA damage and its repair with the induction of or inhibition of inflammation in the mouse skin, we also determined the levels of TNF-α in the same skin samples as markers of inflammation. As shown in Fig. 5D, the levels of TNF-α were significantly reduced in the skin samples of GTPs-treated wild-type mice as compared with the skin samples of non-GTPs-treated wild-type mice, and the reduction in this proinflammatory cytokine paralleled the reduction in DNA damage induced by the GTPs treatment. In contrast, this chemopreventive effect of GTPs on the levels of TNF-α was not statistically significant in the skin of IL-12 KO mice.
Figure 5.
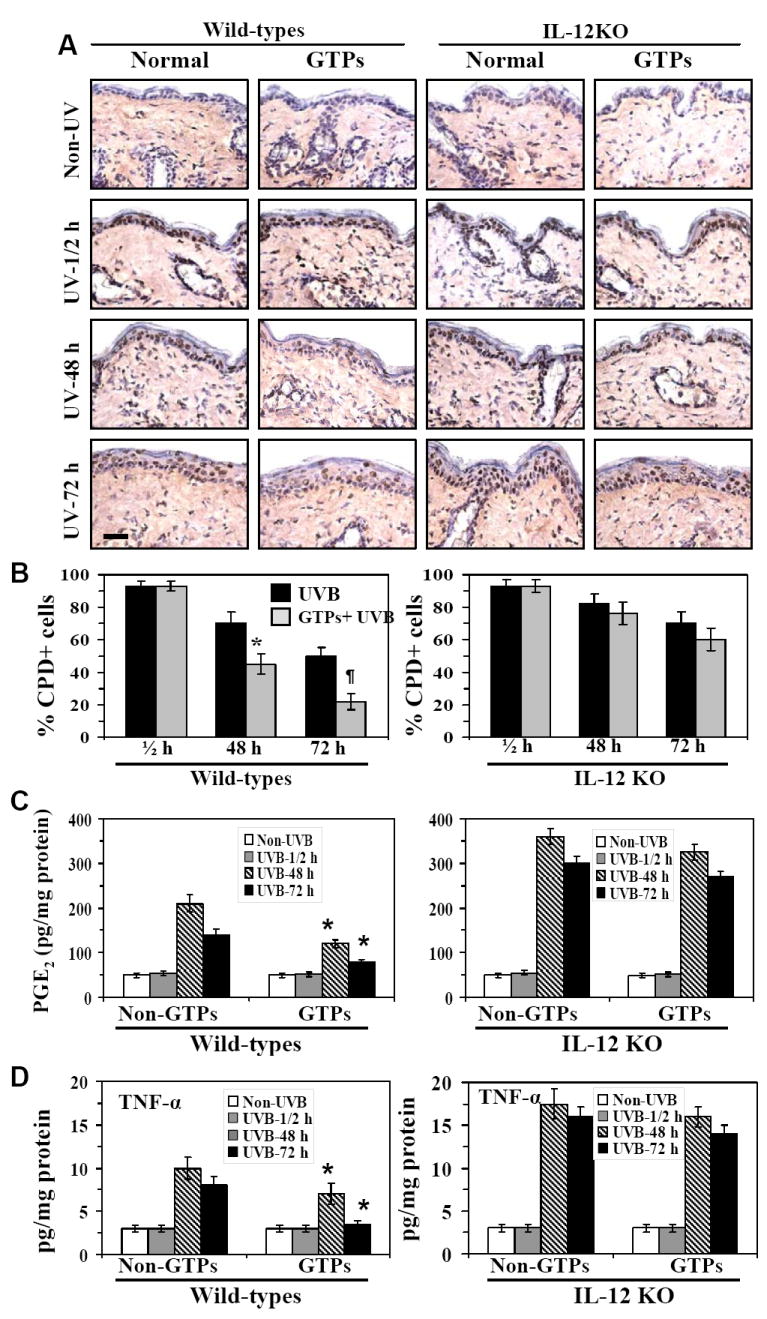
Administration of GTPs removes or repairs UV-induced CPDs more rapidly in the skin of WT mice than in the skin of IL-12 KO mouse and the resolution of CPDs parallels the subsequent reduction in the levels of PGE2 and TNF-α. Mice were exposed to acute UVB (90 mJ/cm2) radiation and thereafter sacrificed at different time points (1/2, 48 and 72 h). GTPs (0.2%, w/v) were given ad libitum in drinking water from at least 14 days before UVB exposure and continuing until the termination of the experiment. Skin samples were obtained and analyzed for CPDs, PGE2 and TNF-α. Panel A, Frozen sections (5 μm thick) were subjected to immunoperoxidase staining to detect CPD+ cells that are dark brown. CPDs were not detected in non-UV-exposed skin whether treated or not treated with GTPs. Panel B, The numbers of CPD+ cells in each treatment group are summarized in terms of percent positive cells and the data are expressed as the mean ± SD. Panel C, Epidermal PGE2 was determined as a marker of inflammation using an immunoassay kit as described in Materials and methods. Panel D, The levels of epidermal TNF-α was determined using ELISA. Experiments were conducted and repeated separately in 5 animals in each group with identical results. Results of enumeration of CPD+ cells and the levels of PGE2 and TNF-α are expressed as mean ± SD. Scale bar = 50 μm. Significant inhibition versus control (non-GTPs-treated UVB-exposed group) group, *p<0.01; ¶p<0.001.
Removal or repair of UVB-induced DNA damage leads to a reduction in UVB-induced inflammation
We have shown previously that the repair of UV-induced DNA damage in the form of CPDs requires IL-12 (Meeran et al., 2006). We therefore determined the effect of subcutaneous injection of rIL-12 at UV-irradiated skin sites of GTPs-treated IL-12 KO mice. Immunohistochemical analysis indicated that this resulted in rapid repair or removal of the CPD+ cells (Fig. 6A, left panels), suggesting that GTPs-mediated repair or removal of UVB-induced CPDs requires IL-12. Similar results were obtained when dot-blot analysis was performed to detect the levels of UVB-induced DNA damage (Fig. 6B, left panel). For this purpose epidermal genomic DNA was isolated from the different treatment groups of IL-12 KO mice and subjected to southwestern dot-blot analysis. On analysis of the levels of epidermal PGE2 in the same skin samples, we also found a significant reduction in the levels of PGE2 (61%, p<0.001) in the rIL-12-treated UVB-irradiated skin sites of the GTPs-treated IL-12 KO mice. Using a converse approach, we treated the UVB-irradiated and GTPs-treated wild-type mice i.p. with anti-IL-12 antibodies. The administration of the anti-IL-12 antibodies resulted in a failure in the repair or removal of the UVB-induced CPD+ cells in the skin of these mice (Fig. 6A, right panels) and the levels of PGE2 were significantly higher (p<0.01) as compared to the levels in UVB-irradiated skin sites of GTPs-treated wild-type mice that were not treated with anti-IL-12 antibodies (Fig. 6C, right panels). Similar DNA repair observations were obtained when the epidermal genomic DNA was subjected to dot-blot analysis (Fig. 6B, right panel). Analysis of the levels of cyclin D1, as a marker of proliferation, indicated that the levels of cyclin D1 were reduced in the rIL-12/IL-12 KO mouse model and increased in the anti-IL-12/wild-type mouse model (Fig. 6D).
Figure 6.
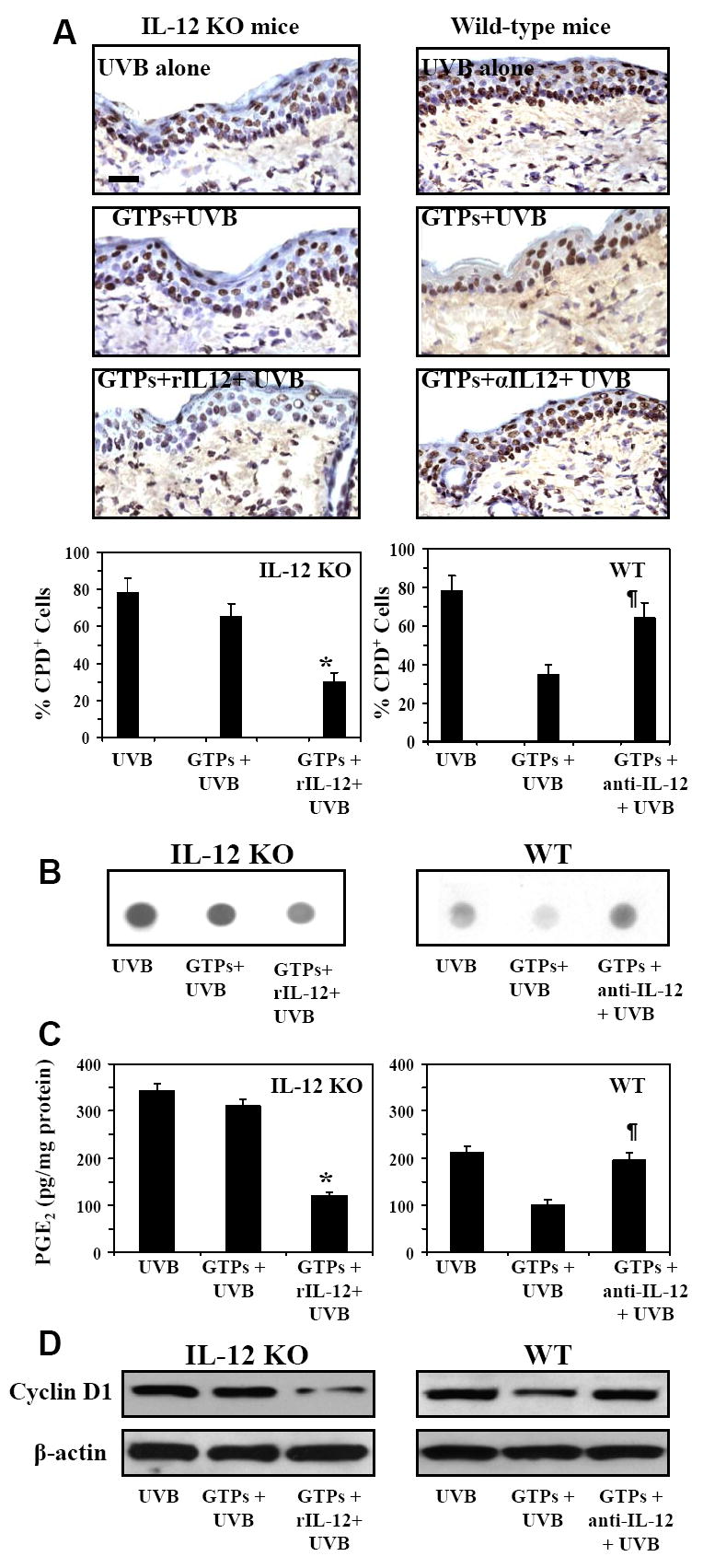
CPDs are removed or repaired more rapidly in the skin of UVB-exposed GTPs-treated IL-12 KO mice that have been treated with rIL-12 than in the skin of UVB-exposed GTPs-treated IL-12 KO mice that have not been treated with rIL-12. Conversely, CPDs are not removed or repaired rapidly in UVB-exposed GTPs-treated WT mice that have been treated with neutralizing anti-IL-12 antibody as compared to UVB-exposed GTPs-treated WT mice not treated with neutralizing anti-IL-12 antibody. Mice were acutely exposed to UVB (90 mJ/cm2) and GTPs were provided in the drinking water as detailed under Figure 5. One group of IL-12 KO mice was injected s.c. with murine rIL-12 (50 ng/mouse) 3 h before exposure of UVB. One group of WT mice was injected i.p. with anti-IL-12 antibodies (500 ng/mouse/100 μl PBS) 24 and 3 h before UVB exposure. Mice were sacrificed either immediately or 48 h after the UVB exposure, and skin samples were obtained and analyzed for CPDs using immunohistochemistry (Panel A), and dot-blot analysis (Panel B). Frozen sections (5 μm thick) were subjected to immunoperoxidase staining to detect CPD+ cells, which appear dark brown in the photomicrograph. Epidermal genomic DNA was subjected to Southwestern dot blot analysis to detect UV-induced damaged DNA using an antibody specific for CPD. Panel C, Epidermal PGE2 was determined as a marker of inflammation using an immunoassay kit as described in the Materials and Methods. Panel D, The levels of cyclin D1 were determined as a marker of cellular proliferation in epidermal lysate samples using western blot analysis. Experiments were conducted and repeated separately using samples obtained from 5 animals in each group with almost identical observations. Equal loading of protein samples was confirmed using β-actin.
Scale bar = 50 μm.
Significantly less versus GTPs+ UVB-treated IL-12 KO mice, *p<0.001
Significantly increased versus GTPs+ UVB-treated WT mice, ¶p<0.01.
DISCUSSION
Green tea is consumed as a popular beverage world-wide (Katiyar and Mukhtar, 1996). In the present study we demonstrate a new mechanism by which drinking active ingredients of green tea, i.e., green tea polyphenols (GTPs), prevents photocarcinogenesis in mice. That is, we show that the GTPs when taken orally inhibit UVB-induced inflammatory responses, which promote carcinogenesis, through the rapid repair or removal of UVB-induced DNA damage in mice. This DNA repair process is mediated through the enhancement of IL-12 levels by GTPs in vivo in UVB-exposed animals. The chemopreventive effect of drinking GTPs on photocarcinogenesis was less pronounced in IL-12 KO mice but significant inhibitory effect was observed in their wild-type (IL-12 proficient) mice, suggesting that the prevention of photocarcinogenesis by GTPs is mediated primarily through IL-12 induction. The treatment of IL-12 KO mice with GTPs did result in some inhibition of UVB-induced tumor development and repair of UVB-induced DNA damage, although these effects were not of sufficient magnitude to be statistically significant in these experiments. Our data that suggest a role for GTPs-mediated induction of IL-12 in their anti-carcinogenic activity are consistent with the demonstrated anti-tumor activity of IL-12 against established cutaneous deposits and experimental metastases (Brunda et al., 1993; Nastala et al., 1994; Colombo et al., 1996; Wigginton et al., 1996; Maeda et al., 2006; Meeran et al., 2006). As we observed that treatment of wild-type mice with GTPs increased the levels of IL-12 in lymph nodes of UV-exposed mice (data not shown), it is possible that this enhancement of the levels of IL-12 is involved in the inhibition of growth of tumors. Thus, our present study indicates that effective chemoprevention of photocarcinogenesis by provision of GTPs in the drinking water of mice requires IL-12 activity. It is worth mentioning that we could not find any significant difference in the development of UVB-induced skin tumors between IL-12p40 KO mice and IL-12p35 KO mice (Meeran et al., 2006).
Chronic exposure of the skin to solar UV radiation is considered as a major etiologic factor for the development of melanoma and non-melanoma skin cancers. In addition to other adverse effects of UV radiation, UV-induced chronic and sustained inflammation and their mediators are considered as potent regulators of skin tumor promotion. We have shown previously using IL-12 KO mice that a deficiency of IL-12 promotes photocarcinogenesis as well as exacerbating UVB-induced inflammatory responses in mouse skin (Meeran et al., 2006; Meeran et al., 2008, in press), which seems to be a contributing factor in the early and more rapid development of skin tumors in UV-exposed skin. Using this mouse model as a mechanistic tool, we designed experiments to determine whether the chemopreventive effects of GTPs against photocarcinogenesis is mediated, at least in part, through the inhibition of UVB-induced inflammatory responses, and whether inflammatory responses are inhibited through the rapid repair or removal of UVB-induced DNA damage. One of the most important enzymes in the process of inflammation and cancer development is inducible COX-2. COX-2 is a rate-limiting enzyme for the generation of PG metabolites from arachidonic acid (Langenbach et al., 1999). COX-2 overexpression has been linked to the pathophysiology of inflammation and cancer (Chapple et al., 2000) due to enhanced synthesis of PG metabolites which have been shown to be a potential contributing factor in UV-induced nonmelanoma skin cancers (Marks et al., 1999; Williams et al., 1999). A number of studies have demonstrated overexpression of COX-2 in chronically UVB-irradiated skin, as well as in UVB induced premalignant lesions and squamous cell carcinomas (Buckman et al., 1998; Athar et al., 2001). A role for COX-2 in photocarcinogenesis is also supported by several studies that demonstrate that inhibition of COX-2 activity by specific inhibitors can partially block carcinogenesis induced by long-term UVB exposure (Wilgus et al., 2003; Pentland et al., 1999). The induction of COX-2 enhances the formation of PG metabolites, wherein PGE2 is a potential mediator of inflammatory reactions. In this study, we found that administration of GTPs in the drinking water of wild-type mice significantly inhibited UVB-induced inflammatory responses in terms of inhibition of COX-2 expression, PGE2 production, and reduction in the levels of epidermal PCNA and cyclin D1, which are potent markers of cellular proliferation. Less pronounced chemopreventive effects of GTPs were observed in similarly treated IL-12-deficient mice. These results suggest that the inhibition of photocarcinogenesis by GTPs is mediated by an inhibition of inflammatory responses and that this inhibition requires the presence or reactivity of the immunoregulatory cytokine IL-12. The inhibition of proliferating potential of epidermal keratinocytes by GTPs, which is indicated by the reduced expression of PCNA and cyclin D1, may also be a contributing factor for the inhibitory effects of GTPs on development of tumors in UV-exposed wild-type mice but, this inhibitory effect was less pronounced in IL-12 KO mice.
The role of IL-12 in ameliorating the UVB-induced inflammatory responses is further suggested by the finding of higher levels of pro-inflammatory cytokines, such as TNF-α, IL-1β and IL-6, in the UVB-exposed skin of IL-12 KO mice than the UVB-exposed skin of wild-type mice. The higher levels of these inflammatory cytokines would be expected to contribute to the tumor promotion process and thus the development of tumors would be expected to occur earlier and progress more rapidly; this trend was observed in IL-12 KO mice in which the development of UVB-induced skin tumors occurred earlier and their growth was faster than UVB-induced tumor in their wild-type counterparts (Fig. 1). These data suggest a positive relationship between UVB-induced inflammation and UVB-induced enhanced skin tumorigenesis in IL-12 KO mice. Our data indicated that administration of GTPs significantly inhibited the expression of proinflammatory cytokines in the wild-type mice but this inhibitory effect of GTPs was less pronounced in the IL-12 KO mice. Elevated levels of proinflammatory cytokines have been implicated in skin cancer risk (Mukhtar and Elmets, 1996; Scott et al., 2003; Tron et al., 1988). COX-2-induced PGE2 has been shown to be a potent inhibitor of IL-12 and proinflammatory cytokines can induce COX-2 expression (Pang et al., 1997), which in turn may increase the production of PG metabolites which would stimulate tumor development. The differences in the patterns of expression of the pro-inflammatory cytokines and mediators of inflammation observed in the UVB-exposed skin of the different mice, also held true for their expression in skin tumors. The levels of these biomarkers of the inflammatory process and mediators of inflammation (PCNA, COX-2, PGE2, cyclin D1, TNF-α, IL-6 and IL-1β) were higher in the UVB-induced tumors of IL-12 KO mice than the UVB-induced tumor in their wild-type counterparts. The administration of GTPs significantly decreased the levels of UVB-induced biomarkers of inflammation in the wild-type mice but this chemopreventive effect of the GTPs was significantly lower in the tumors of the IL-12 KO mice. Thus our findings suggest that IL-12-deficiency exacerbates UVB-induced inflammatory responses in mouse skin and that this may be a contributing factor to the early occurrence and rapid development of skin tumors in these mice. Notably, it is also suggested that the chemopreventive effect of GTPs on UV-induced skin tumor development is mediated, at least in part, through IL-12-dependent inhibition of UVB-induced inflammatory responses.
Both UVB-induced inflammatory responses and UVB-induced tumorigenesis are causally related to UVB-induced DNA damage. Thus, we explored the effects of the GTPs-stimulated enhancement of the levels of IL-12 on UV-induced DNA damage and its relationship with the development of skin inflammation in our model. Following acute UVB exposure, it was observed that the UV-induced DNA damage in the form of CPDs was repaired or removed more rapidly in wild-type mice that had been administered GTPs in the drinking water than in similarly treated IL-12 KO mice. Recently, Schwarz et al. reported that green tea phenol extracts reduce UVB-induced DNA damage in vitro in human cells through induction of IL-12 (Schwarz et al., 2008), and thus support our in vivo observations. Additionally, we found that in the GTPs-treated wild-type mice the levels of inflammatory biomarkers (PGE2 and TNF-α) were elevated after acute UVB exposure when the levels of UV-induced DNA damage were highest and then declined as the levels of DNA damage declined. The role of IL-12 in this process was further supported by the data obtained after subcutaneous injection of the rIL-12 at the UV-irradiated skin site of GTPs-treated IL-12 KO mice which resulted in more rapid removal or repair of UV-induced CPDs than that in skin sites that were not treated with rIL-12 in GTPs-treated IL-12 KO mice. This treatment also reduced the intensity of skin inflammation in parallel with the temporal decline in the number of CPDs. Conversely, treatment of wild-type mice that had been administered GTPs in the drinking water with neutralizing antibodies to IL-12 blocked the ability of the GTPs to protect against UV-induced DNA damage, and the inflammatory responses in terms of PGE2 and cyclin D1 were significantly higher in the skin of the wild-type mice that were treated with anti-IL-12 than those were not. Our in vivo findings with GTPs on repair of UV-induced DNA damage are supported by the observations of Schwarz et al. who found that IL-12 reduces or repairs UV-induced DNA damage in vitro and in vivo mouse skin, and that this repair is mediated through a nucleotide excision repair mechanism (Schwarz et al., 2002). These data provide further support for the concept that UV-induced DNA damage and inflammatory responses are causally related with the increased risk of photocarcinogenesis. In addition, this study indicates that the ability of GTPs administered in the drinking water to prevent photocarcinogenesis is mediated through inhibition of UVB-induced inflammation, which in turn is mediated, at least in part, through IL-12-dependent DNA repair. The outcome of this study therefore suggests that endogenous enhancement of IL-12-mediated DNA repair by GTPs may be considered as an effective strategy for the prevention of inflammation-associated skin diseases including skin cancers.
MATERIALS AND METHODS
Animals and IL-12 genotyping
The female C3H/HeN mice (6-7 weeks old) used in these studies were purchased from Charles River Laboratory (Wilmington, MA). The IL-12p40 KO mice on a C3H/HeN background were generated in our Animal Resource Facility. Briefly, male IL-12p40-/- (knock-out) mice on a C57BL/6J genetic background were obtained from The Jackson Laboratory (Bar Harbor, ME). Male IL-12-/- and female C3H/HeN mice were mated to obtain male and female IL-12+/- mice. We then mated male IL-12-/- mice with IL-12+/- female or IL-12+/- males with IL-12+/- females and genotyped the progeny using a new rapid method of backcrossing for the generation of IL-12p40 KO mice onto a C3H/HeN mice background (Markel et al., 1997). Mice that are homozygous for IL-12 deletion mutation are viable and their viability was not reduced compared to their wild-type counterparts (C3H/HeN). All mice were maintained under standard conditions of a 12-h dark/12-h light cycle, a temperature of 24 ± 2°C, and relative humidity of 50 ± 10%. The animal protocol used in this study was approved by the Institutional Animal Care and Use Committee of the University of Alabama at Birmingham.
Antibodies, chemicals and real-time PCR primers
Endotoxin-free mouse recombinant IL-12 (rIL-12) was purchased from eBioscience (San Diego, CA). Immunostaining-specific COX-2 antibody and a kit for PGE2 analysis were obtained from Cayman Chemicals (Ann Arbor, MI). The antibodies used to detect proliferating cell nuclear antigen (PCNA) and cyclin D1 and secondary antibodies were obtained from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). The antibody specific for CPDs was obtained from Kamiya Biomedical Co. (Seattle, WA). ELISA kits specific for mouse TNF-α, IL-1β and IL-6 were obtained from BioSource International (Camarillo, CA). The trypsin, DNase and all other chemicals of analytical grade were purchased from Sigma Chemical Co. (St. Louis, MO).
Green tea polyphenols (GTPs)
The purified mixture of green tea polyphenols was obtained from Mitsui Norin Co. Ltd., Japan, and contains primarily 5 major epicatechin derivatives, such as, (-)-epigallocatechin-3-gallate, (-)-epigallocatechin, (-)-epicatechin gallate, (-)-epicatechin and gallocatechin gallate (Mantena et al., 2005), as shown in Table 1. This mixture of GTPs was given in the normal drinking water ad libitum. Fresh GTPs-containing water was provided every third day. The polyphenolic constituents of samples of the three-day-old GTPs-containing drinking water were analyzed by HPLC and this confirmed that the chemical composition of the GTPs in the drinking water was not significantly altered during this time period when compared with the fresh samples (Table 1).
UVB irradiation and photocarcinogenesis protocol
Female mice of 6-7 weeks of age were used in this study. The IL-12 KO mice on a C3H/HeN background and their wild-type (C3H/HeN) counterparts were divided randomly into three separate treatment groups with 20 mice in each group. Mice that were not UVB-exposed served as controls (Group 1). The remaining mice were exposed to photocarcinogenesis protocol and were provided regular drinking water (Group 2) or drinking water containing GTPs (0.2%, w/v) (Group 3). The provision of drinking water containing GTPs was started at least 14 days before the start of UVB exposure and continued until the end of experiments. The backs of the mice were shaved with electric clippers before UVB exposure. The groups of mice that were not exposed to UVB also were shaved to maintain a similar treatment protocol. Mice were UVB-irradiated as described earlier (Meeran et al., 2006; Mantena et al., 2005). Briefly, the shaved dorsal skin was exposed to UV radiation from a band of four FS24T1 UVB lamps (Daavlin, UVA/UVB Research Irradiation Unit, Bryan, OH) equipped with an electronic controller to regulate UV dosage. The UVB lamps primarily emit UVB (290-320 nm, >80% of total energy) and UVA (320-375 nm, <20% of total energy) radiation with peak emission at 314 nm as monitored (Meeran et al., 2006). Under the standard photocarcinogenesis protocol, mice were UVB irradiated (180 mJ/cm2) three times a week until the end of the protocol. The backs of the mice were shaved using clippers if hairs grew on the skin during the photocarcinogenesis experiment.
Evaluation of tumor growth
The UVB-irradiated dorsal skin of the mice was examined once a week for papillomas or tumor appearance. Growths that were >1 mm in diameter and that persisted for at least 2 weeks were defined as tumors and recorded. Tumor data on each mouse were recorded until the yield and size of the tumors had stabilized. The dimensions of all the tumors on each mouse were recorded at the termination of the experiment. Tumor volumes were calculated using the hemiellipsoid model formula: tumor volume = 1/2 (4π/3) (l/2) (w/2) h, where l = length, w = width and h = height. When tumor yield and growth stabilized, the photocarcinogenesis experiment was terminated and skin and tumor samples were collected for the analysis of various biomarkers of interest.
Pathological evaluation of skin tumors
At the termination of the experiment, representative biopsies from all the skin tumors were collected, fixed in 10% formaldehyde and embedded in paraffin. Sections (5 μm thick) were stained with H&E for pathological evaluation. Tumors were analyzed in a blinded fashion, and the discrimination between real tumors and non-neoplastic lesions was performed according to the following major criteria: loss of keratinization or keratinized centers, presence of horn pearls and atypical cells.
In vivo treatment of recombinant IL-12 and anti-IL-12 antibody
To verify that IL-12-deficiency is associated with enhanced inflammation in UV-exposed skin, IL-12 KO mice were treated with endotoxin-free murine rIL-12 (50 ng/mouse/100 μL PBS, eBioscience, San Diego, CA), or an equal volume of PBS, that was administered as a subcutaneous injection on the shaved back of the mice at least 3 h before UVB exposure (90 mJ/cm2). Conversely, wild-type mice were treated with anti-IL-12 antibody, which was diluted in sterile endotoxin-free saline and injected i.p. The mice received two doses of anti-IL-12 antibodies (500 ng each) 24 and 3 h before UVB exposure. Control mice were injected i.p. with equal volumes of rat IgG1 (isotype control of anti-IL-12) in saline. After exposure to UVB the mice were sacrificed at the desired time point and skin samples collected for the analysis.
Immunohistochemical detection of CPDs
Immunohistochemical detection of CPD+ cells in the skin samples was performed using a procedure described previously (Katiyar et al., 2000). Briefly, frozen skin sections (5 μm thick) were thawed, and kept in 70 mM NaOH in 70% ethanol for 2 min to denature nuclear DNA, followed by neutralization for 1 min in 100 mM Tris-HCl (pH 7.5) in 70% ethanol. The sections were washed with PBS buffer and incubated with 10% goat serum in PBS to prevent non-specific binding prior to incubation with a monoclonal antibody specific for CPDs, or its isotype control (IgG1). Bound anti-CPD antibody was detected by incubation with biotinylated goat-anti-mouse IgG1 followed by peroxidase-labeled streptavidin. After washing, sections were incubated with diaminobenzidine and counterstained with H & E.
Southwestern dot-blot analysis
Genomic DNA from the epidermal skin or tumor samples was isolated following standard procedures and dot-blot analysis was performed as detailed previously (Meeran et al., 2006). Briefly, genomic DNA (500 ng) was transferred to a positively-charged nitrocellulose membrane by vacuum dot-blotting (Bio-Dot Apparatus, Bio-Rad, Hercules, CA) and fixed by baking the membrane for 30 min at 80°C. After blocking the non-specific binding sites in blocking buffer (5% non-fat dry milk, 1% Tween 20 in 20 mM TBS, pH 7.6), the membrane was incubated with the antibody specific for CPDs for 1 h at room temperature. After washing, the membrane was incubated with HRP-conjugated secondary antibody. Detection of bound antibody was accomplished by chemiluminescence using the ECL detection system. Genomic DNA from at least 5 mice in each group was tested independently.
Enzyme immuno-assay for PGE2
Skin or tumor samples were homogenized in 100 mM phosphate buffer, pH 7.4 containing 1 mM ethylenediamine tetraacetic acid and 10 μM indomethacin using a polytron homogenizer (PT3100, Fisher Scientific, GA). The supernatants were collected and the concentration of PGE2 was determined in supernatants using the Cayman PGE2 Enzyme Immunoassay Kit (Cayman Chemical, Ann Arbor, MI) following the manufacturer’s protocol.
Cytokine assay by ELISA
Epidermal or tumor homogenates from each treatment group were used for the analysis of cytokines, such as, TNF-α, IL-1β and IL-6 using ELISA kits (BioSource International, Camarillo, CA, USA) following the manufacturer’s protocol.
Western blot analysis
Epidermal or tumor lysates for western blot analysis were prepared as described previously (Mantena et al., 2005). Epidermis was separated from the whole skin as described earlier (Katiyar et al., 1999). The epidermis or tumor tissue samples were pooled from at least two mice in each group, and five sets of pooled samples from each treatment group were used to prepare lysates, thus n = 10. Proteins (25-50 μg) were resolved on 10-12% Tris-glycine gel and transferred onto nitrocellulose membranes. Membranes were incubated in blocking buffer for 1 h at room temperature and then incubated with the primary antibodies in blocking buffer overnight at 4°C. The membrane was then washed with PBS and incubated with HRP-conjugated secondary antibody. Protein bands were visualized using the enhanced chemiluminescence detection reagents. To verify equal protein loading and transfer of proteins from gel to membrane, the blots were stripped and re-probed for ß-actin using an anti-actin rabbit polyclonal antibody.
Statistical analysis
Statistical analysis of tumor data was performed at the termination of the experiment. Tumor incidence in the UVB alone and GTPs + UVB groups was compared using the χ2 test. Tumor multiplicity data were analyzed using the Wilcoxon rank sum test. The results of cytokine levels, PGE2, and CPDs are expressed as means ± SD. The statistical significance of difference between the values of control and treatment groups was determined by analysis of variance followed by post hoc test.
Supplementary Material
Acknowledgments
This work was supported by grants from the National Center for Complementary and Alternative Medicine/NIH (1 RO1 AT002536, S.K.K.) and the Veterans Administration Merit Review Award (S.K.K.).
Abbreviations
- CPD
cyclobutane pyrimidine dimers
- COX-2
cyclooxygenase-2
- GTPs
green tea polyphenols
- IL
interleukin
- PG
prostaglandins
- PCNA
proliferating cell nuclear antigen
- TNF-α
tumor necrosis factor-α
- UV
ultraviolet
Footnotes
CONFLICT OF INTEREST No conflict of interest.
References
- Applegate LA, Ley RD, Alcalay J, Kripke ML. Identification of molecular targets for the suppression of contact hypersensitivity by ultraviolet radiation. J Exp Med. 1989;170:1117–31. doi: 10.1084/jem.170.4.1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Athar M, An KP, Morel KD, Kim AL, Aszterbaum M, Longley J, et al. Ultraviolet B (UVB)-induced COX-2 expression in murine skin: an immunohistochemical study. Biochem Biophys Res Commun. 2001;280:1042–7. doi: 10.1006/bbrc.2000.4201. [DOI] [PubMed] [Google Scholar]
- Black AK, Greaves MW, Hensby CN, Plummer NA. Increased prostaglandins E2 and F2alpha in human skin at 6 and 24 h after ultraviolet B irradiation (290- 320 nm) Br J Clin Pharmacol. 1978;5:431–36. doi: 10.1111/j.1365-2125.1978.tb01650.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunda MJ. Interleukin-12. J Leukocyte Biol. 1994;55:280–8. doi: 10.1002/jlb.55.2.280. [DOI] [PubMed] [Google Scholar]
- Brunda MJ, Luistro L, Warrier RR, Wright RB, Hubbard BR, Murphy M, et al. Antitumor and antimetastatic activity of interleukin-12 against murine tumors. J Exp Med. 1993;178:1223–30. doi: 10.1084/jem.178.4.1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckman SY, Gresham A, Hale P, Hruza G, Anast J, Masferrer J, et al. COX-2 expression is induced by UVB exposure in human skin: implications for the development of skin cancer. Carcinogenesis. 1998;19:723–9. doi: 10.1093/carcin/19.5.723. [DOI] [PubMed] [Google Scholar]
- Chapple KS, Cartwright EJ, Hawcroft G, Tisbury A, Bonifer C, Scott N, et al. Localization of cyclooxygenase-2 in human sporadic colorectal adenomas. Am J Pathol. 2000;156:545–53. doi: 10.1016/S0002-9440(10)64759-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Chen D, Bloack E, O’Donnell M, Kufe DW, Clinton SK. Eradication of murine bladder carcinoma by intratumor injection of a bicistronic adenoviral vector carrying cDNAs for the IL-12 heterodimer and its inhibition by the p40 subunit homodimer. J Immunol. 1997;159:351–9. [PubMed] [Google Scholar]
- Colombo MP, Vagliani M, Spreafico F, Parenza M, Chiodoni C, Melani C, et al. Amount of interleukin 12 available at the tumor site is critical for tumor regression. Cancer Res. 1996;56:2531–34. [PubMed] [Google Scholar]
- Katiyar SK. Oxidative stress and photocarcinogenesis: Strategies for prevention. In: Singh KK, editor. Oxidative stress disease and cancer. London: Imperial College Press; 2006. pp. 933–64. [Google Scholar]
- Katiyar SK. Interleukin-12 and photocarcinogenesis. Toxicol Appl Pharmacol. 2007;224:220–7. doi: 10.1016/j.taap.2006.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katiyar SK, Challa A, McCormick TS, Cooper KD, Mukhtar H. Prevention of UVB-induced immunosuppression in mice by green tea polyphenol (-)-epigallocatechin-3-gallate may be associated with alterations in IL-10 and IL-12 production. Carcinogenesis. 1999;20:2117–24. doi: 10.1093/carcin/20.11.2117. [DOI] [PubMed] [Google Scholar]
- Katiyar S, Elmets CA, Katiyar SK. Green tea and skin cancer: Photoimmunology, angiogenesis and DNA repair. J Nutr Biochem. 2007;18:287–96. doi: 10.1016/j.jnutbio.2006.08.004. [DOI] [PubMed] [Google Scholar]
- Katiyar SK, Matsui MS, Mukhtar H. Kinetics of UV light-induced cyclobutane pyrimidine dimers in human skin in vivo: An immunohistochemical analysis of both epidermis and dermis. Photochem Photobiol. 2000;72:788–93. doi: 10.1562/0031-8655(2000)072<0788:koulic>2.0.co;2. [DOI] [PubMed] [Google Scholar]
- Katiyar SK, Meeran SM. Obesity increases the risk of UV radiation-induced oxidative stress and activation of MAPK and NF-κB signaling. Free Rad Biol Med. 2007;42:299–310. doi: 10.1016/j.freeradbiomed.2006.10.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katiyar SK, Mukhtar H. Tea in chemoprevention of cancer: Epidemiologic and experimental studies. Int J Oncol. 1996;8:221–38. doi: 10.3892/ijo.8.2.221. [DOI] [PubMed] [Google Scholar]
- Kripke ML, Cox PA, Alas LG, Yarosh DB. Pyrimidine dimers in DNA initiated systemic immunosuppression in UV-irradiated mice. Proc Natl Acad Sci USA. 1992;89:7516–20. doi: 10.1073/pnas.89.16.7516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langenbach R, Loftin CD, Lee C, Tiano H. Cyclooxygenase-deficient mice. A summary of their characteristics and susceptibilities to inflammation and carcinogenesis. Ann N Y Acad Sci. 1999;889:52–61. doi: 10.1111/j.1749-6632.1999.tb08723.x. [DOI] [PubMed] [Google Scholar]
- Maeda A, Schneider SW, Kojima M, Beissert S, Schwarz T, Schwarz A. Enhanced photocarcinogenesis in interleukin-12-deficient mice. Cancer Res. 2006;66:2962–69. doi: 10.1158/0008-5472.CAN-05-3614. [DOI] [PubMed] [Google Scholar]
- Mantena SK, Meeran SM, Elmets CA, Katiyar SK. Orally administered green tea polyphenols prevent ultraviolet radiation-induced skin cancer in mice through activation of cytotoxic T cells and inhibition of angiogenesis in tumors. J Nutr. 2005;135:2871–77. doi: 10.1093/jn/135.12.2871. [DOI] [PubMed] [Google Scholar]
- Markel P, Shu P, Ebeling C, Carlson GA, Nagle DL, Smutko JS, et al. Theoretical and empirical issues for marker-assisted breeding of congenic mouse strains. Nature Genetics. 1997;17:280–4. doi: 10.1038/ng1197-280. [DOI] [PubMed] [Google Scholar]
- Marks F, Fürstenberger G, Müller-Decker K. Metabolic targets of cancer chemoprevention: interruption of tumor development by inhibitors of arachidonic acid metabolism. Recent Results Cancer Res. 1999;151:45–67. doi: 10.1007/978-3-642-59945-3_4. [DOI] [PubMed] [Google Scholar]
- Meeran SM, Mantena SK, Meleth S, Elmets CA, Katiyar SK. Interleukin-12-deficient mice are at greater risk of ultraviolet radiation-induced skin tumors and malignant transformation of papillomas to carcinomas. Mol Cancer Ther. 2006;5:825–32. doi: 10.1158/1535-7163.MCT-06-0003. [DOI] [PubMed] [Google Scholar]
- Meeran SM, Punathil T, Katiyar SK. Interleukin-12-deficiency exacerbates inflammatory responses in UV-irradiated skin and skin tumors. J Invest Dermatol. 2008 doi: 10.1038/jid.2008.140. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhtar H, Elmets CA. Photocarcinogenesis: Mechanisms, models and human health implications. Photochem Photobiol. 1996;63:355–447. doi: 10.1111/j.1751-1097.1996.tb03040.x. [DOI] [PubMed] [Google Scholar]
- Nastala CL, Edington HD, McKinney TG, Tahara H, Nalesnik MA, Brunda MJ, et al. Recombinant IL-12 administration induces tumor-regression in association with IFN-γ production. J Immunol. 1994;153:1697–706. [PubMed] [Google Scholar]
- Pang L, Knox AJ. Effect of interleukin-1 beta, tumour necrosis factor-alpha and interferon-gamma on the induction of cyclo-oxygenase-2 in cultured human airway smooth muscle cells. Br J Pharmacol. 1997;121:579–87. doi: 10.1038/sj.bjp.0701152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pentland AP, Schoggins JW, Scott GA, Khan KN, Han R. Reduction of UV-induced skin tumors in hairless mice by selective COX-2 inhibition. Carcinogenesis. 1999;20:1939–44. doi: 10.1093/carcin/20.10.1939. [DOI] [PubMed] [Google Scholar]
- Rivas JM, Ullrich SE. The role of IL-4, IL-10, and TNF-alpha in the immune suppression induced by ultraviolet radiation. J Leukoc Biol. 1994;56:769–75. doi: 10.1002/jlb.56.6.769. [DOI] [PubMed] [Google Scholar]
- Robertson MJ, Ritz J. Interleukin-12: basic biology and potential applications in cancer treatment. Oncologist. 1996;1:88–97. [PubMed] [Google Scholar]
- Schwarz A, Maeda A, Gan D, Mammone T, Matsui MS, Schwarz T. Green tea phenol extracts reduce UVB-induced DNA damage in human cells via interleukin-12. Photochem Photobiol. 2008;84:350–5. doi: 10.1111/j.1751-1097.2007.00265.x. [DOI] [PubMed] [Google Scholar]
- Schwarz A, Ständer S, Berneburg M, Böhm M, Kulms D, van Steeg H, et al. Interleukin-12 suppresses ultraviolet radiation-induced apoptosis by inducing DNA repair. Nat Cell Biol. 2002;4:26–31. doi: 10.1038/ncb717. [DOI] [PubMed] [Google Scholar]
- Scott KA, Moore RJ, Arnott CH, East N, Thompson RG, Scallon BJ, et al. An anti-tumor necrosis factor-alpha antibody inhibits the development of experimental skin tumors. Mol Cancer Ther. 2003;2:445–51. [PubMed] [Google Scholar]
- Siders W, Wright P, Hixon JA, Alvord WG, Back TC, Wiltrout RH, et al. T cell-and NK cell-independent inhibition of hepatic metastases by systemic administration of an IL-12-expressing recombinant adenovirus. J Immunol. 1998;160:5465–74. [PubMed] [Google Scholar]
- Trinchieri G. Interleukin-12 a cytokine produced by antigen-presenting cells with immunoregulatory functions in the generation of T-helper cells type 1 and cytotoxic lymphocytes. Blood. 1994;84:4008–27. [PubMed] [Google Scholar]
- Tron VA, Rosenthal D, Sauder DN. Epidermal interleukin-1 is increased in cutaneous T-cell lymphoma. J Invest Dermatol. 1988;90:378–81. doi: 10.1111/1523-1747.ep12456433. [DOI] [PubMed] [Google Scholar]
- Vanderveen EE, Grekin RC, Swanson NA, Kragballe K. Arachidonic acid metabolites in cutaneous carcinomas. Arch Dermatol. 1986;122:407–12. doi: 10.1001/archderm.122.4.407. [DOI] [PubMed] [Google Scholar]
- Wang ZY, Huang MT, Ferraro T, Wong CQ, Lou YR, Reuhl K, et al. Inhibitory effect of green tea in the drinking water on tumorigenesis by ultraviolet light and 12-O-tetradecanoylphorbol-13-acetate in the skin of SKH-1 mice. Cancer Res. 1992;52:1162–70. [PubMed] [Google Scholar]
- Wigginton JM, Komschlies KL, Back TC, Franco JL, Brunda MJ, Wiltrout RH. Administration of interleukin 12 with pulse interleukin 2 and the rapid and complete eradication of murine renal carcinoma. J Natl Cancer Inst. 1996;88:38–43. doi: 10.1093/jnci/88.1.38. [DOI] [PubMed] [Google Scholar]
- Wilgus TA, Koki AT, Zweifel BS, Kusewitt DF, Rubal PA, Oberyszyn TM. Inhibition of cutaneous ultraviolet light B-mediated inflammation and tumor formation with topical celecoxib treatment. Mol Carcinog. 2003;38:49–58. doi: 10.1002/mc.10141. [DOI] [PubMed] [Google Scholar]
- Williams CS, Mann M, DuBois RN. The role of cyclooxygenases in inflammation, cancer, and development. Oncogene. 1999;18:7908–16. doi: 10.1038/sj.onc.1203286. [DOI] [PubMed] [Google Scholar]
- Yang CS, Maliakal P, Meng X. Inhibition of carcinogenesis by tea. Annu Rev Pharmacol Toxicol. 2002;42:25–54. doi: 10.1146/annurev.pharmtox.42.082101.154309. [DOI] [PubMed] [Google Scholar]
- Yarosh D, Alas LG, Yee V, Oberyszyn A, Kibitel JT, Mitchell D, et al. Pyrimidine dimer removal enhanced by DNA repair liposomes reduces the incidence of UV skin cancer in mice. Cancer Res. 1992;52:4227–31. [PubMed] [Google Scholar]
- Yarosh D, Klein J, O’Connor A, Hawk J, Rafal E, Wolf P. Effect of topically applied T4 endonuclease V in liposomes on skin cancer in xeroderma pigmentosum: a randomised study. Xeroderma Pigmentosum Study Group. Lancet. 2001;357:926–9. doi: 10.1016/s0140-6736(00)04214-8. [DOI] [PubMed] [Google Scholar]
- Zou JP, Yamatato N, Fuzii T, Zou JP, Yamamoto N, Fujii T, et al. Systemic administration of rIL-12 induces complete tumor regression and protective immunity: response is correlated with a striking reversal of suppressed IFN-γ production by anti-tumor T cells. Int Immunol. 1995;7:1135–45. doi: 10.1093/intimm/7.7.1135. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.