

Abstract
Most forms of Parkinson’s Disease (PD) are sporadic in nature, but some have genetic causes as first described for the α-synuclein gene. The α-synuclein protein also accumulates as insoluble aggregates in Lewy bodies in sporadic PD as well as in most inherited forms of PD. The focus of the present study is the modulation of synaptic plasticity in the corticostriatal pathway of transgenic (Tg) mice that over express the human α-synuclein protein throughout the brain (ASOTg). Paired-pulse facilitation was detected in vitro by activation of corticostriatal afferents in ASOTg mice, consistent with a presynaptic effect of elevated human α-synuclein. However basal synaptic transmission was unchanged in ASOTg, suggesting that human α-synuclein could impact paired-pulse facilitation via a presynaptic mechanism not directly related to the probability of neurotransmitter release. Mice lacking α-synuclein or those expressing normal and A53T human α-synuclein in tyrosine hydroxylase-containing neurons showed, instead, paired-pulse depression. High-frequency stimulation induced a presynaptic form of long-term depression solely in ASOTg striatum. A presynaptic, NMDA receptor-independent form of chemical long-term potentiation induced by forskolin (FSK) was enhanced in ASOTg striatum, while FSK-induced cAMP levels were reduced in ASOTg synaptoneurosome fractions. Overall the results suggest that elevated human α-synuclein alters presynaptic plasticity in the corticostriatal pathway, possibly reflecting a reduction in glutamate at corticostriatal synapses by modulation of adenylyl cyclase signaling pathways. ASOTg mice may recapitulate an early stage in PD during which over expressed α-synuclein dampens corticostriatal synaptic transmission and reduces movement.
Keywords: paired-pulse facilitation, presynaptic, long-term depression, forskolin, long-term potentiation, glutamate, cAMP
Most forms of Parkinson’s Disease (PD) are sporadic in nature, but a small percentage have genetic causes as first described for dominant, single base pair changes in the α-synuclein gene (Polymeropoulos et al., 1997; Kruger et al., 1998; Zarranz et al., 2004). The α-synuclein gene encodes a 140 amino acid, mostly unfolded protein that is expressed ubiquitously throughout the brain and is enriched in presynaptic terminals at synapses (Maroteaux and Scheller, 1991; Hsu et al., 1998; Clayton and George, 1998). The α-synuclein protein accumulates as insoluble aggregates in Lewy bodies in sporadic forms of PD (Spillantini et al., 1997; Braak et al., 2003) and in most inherited forms of PD including the most common forms with leucine-rich repeat kinase 2 (LRRK2) gene mutations (Paisan-Ruiz et al, 2004; Zimprich et al., 2004; Giasson et al., 2006; Bonifati, 2007). The identification of α-synuclein gene triplications as well as promoter variants in additional families with inherited PD suggest that not only mutant forms of α-synuclein but over expression of normal α-synuclein is a major contributing factor in PD (Farrer et al., 2001; Holzman et al., 2003; Singleton et al., 2003; Pals et al., 2004). Thus, there is both genetic and neuropathological evidence to support an important role for elevated levels of α-synuclein in PD.
In light of α-synuclein’s prominent role in many forms of PD, understanding its presynaptic function is paramount and can suggest potential mechanisms for intervention. Studies of α-synuclein ”knock-out” mice and over-expressing mice suggest that one function of α-synuclein is to negatively regulate synaptic vesicles (reserve pool, “primed”) leading to decreased release of dopamine or glutamate (Abeliovich et al., 2000; Steidl et al., 2003; Yavich et al., 2004, 2005; Larsen et al., 2006). In contrast, studies in a separate α-synuclein “knock-out” mice (Cabin et al., 2002) suggest that α-synuclein maintains the reserve synaptic vesicle pool in glutamatergic terminals, consistent with a previous report using antisense approaches to lower α-synuclein (Murphy et al, 2000). There are also reports supporting a positive role for α-synuclein in promoting long-lasting increases in presynaptic glutamate release underlying hippocampal synaptic plasticity (Liu et al., 2004; Liu et al., 2007). Together, these studies suggest a presynaptic role for α-synuclein in the modulation of synaptic vesicle pools of multiple neurotransmitter release pathways. However, mice with double “knock-outs” of α-synuclein and β-synuclein genes showed no detectable presynaptic changes in vesicle pools, neurotransmitter release or synaptic plasticity (Chandra et al., 2004). Thus, a gain of function rather than a loss of function for α-synuclein may be more relevant as a model for PD.
The focus of the present study is the modulation of synaptic plasticity in the corticostriatal pathway of adult Tg mice in which levels of human α-synuclein are over-expressed under control of the Thy-1 promoter (ASOTg)(Rockenstein et al., 2002). Alterations in striatal synaptic plasticity are relevant to PD, since persistent forms of striatal synaptic plasticity are thought to underlie motor learning (Graybiel et al., 1994; Haber, 2003; Picconi et al., 2005). Furthermore ASOTg mice are excellent models to study synaptic function early in PD, since they accumulate protease-resistant α-synuclein aggregates and exhibit motor behavioral deficits in the absence of detectable neuron cell loss (Fleming et al., 2004, 2006; Fernagut et al., 2007). The present study examined both short-term and long-term forms of synaptic plasticity in vitro in corticostriatum from ASOTg relative to WT controls. Results suggest that elevated human α-synuclein alters presynaptic plasticity in the corticostriatal pathway, possibly reflecting a reduction in glutamate at corticostriatal synapses.
EXPERIMENTAL PROCEDURES
Animals
WT and ASOTg littermate mice over-expressing human α-synuclein under the control of the mouse Thy-1 promoter were generated previously on the C57BL/6 X DBA2 genetic background (Rockenstein et al., 2002). Non-Tg α-synuclein control mice (Snca+/+) and “knock-out” (Snca-/-) littermates lacking α-synuclein gene expression were generated previously in the 129 X SvEv genetic background (Cabin et al., 2002). Tg mice expressing either normal (wt) or mutant A53T human α-synuclein in tyrosine-hydroxylase (TH)-containing neurons (THwt Tg, THmutA53T Tg) were generated previously in the C57BL/6-DBA2 X Swiss Webster background and maintained separately as homozygous lines (Matsuoka et al., 2001). For experiments, mice (mostly male) ranged in age from 2-6 months. Groups of 3-4 mice were maintained in cages on a 12 h light cycle at room temperature (21°C) and were fed food and water ad libitum. All efforts were made to minimize the number of animals used and their suffering. Studies were carried out according to guidelines of the National Institutes of Health Guide for Care and Use of Laboratory Animals (NIH Publications No.80-23), “Guidelines for the Use of Animals in Neuroscience Research” (Society for Neuroscience), and with approval from the Institutional Animal Care and Use Committee at UCLA.
Electrophysiology
Brain hemispheres were obtained from halothane-anesthetized mice and corticostriatal coronal slices (400 μm thick) were cut in oxygenated (95% O2, 5%CO2), ice cold modified artificial cerebrospinal fluid (ACSF) with low calcium and high magnesium (in mM: 130 NaCl, 3 KCl, 26 NaHCO3, 1.25 NaH2PO4, 10 glucose, 5 MgCl2, 1 CaCl2, pH 7.2-7.4, 290-300 mOsm) using a Leica VT1000S vibratome. Slices were submerged and allowed to recover in oxygenated normal ACSF (in mM: 124 NaCl, 5 KCl, 26 NaHCO3, 1.25 NaH2PO4, 10 glucose, 2 MgSO4, 2 CaCl2, pH 7.2-4, 290-300 mOsm) for 2 h at room temperature. For each recording, a single slice was transferred to a Haas chamber, fully submerged by constant perfusion with ACSF (1-2 ml/min, 31-32°C), a bipolar tungsten stimulating electrode was placed in the corpus callosum, and extracellular field potentials were recorded in the dorsolateral striatum with glass microelectrodes filled with ACSF (3-5 MΩ). To minimize variability in striatal extracellular field potentials between individual experiments as well as between mouse strains, strict criteria were followed in recording each field potential response. First careful attention was paid to record field potentials only in the dorsolateral striatum of slices from adult mice (≥2 months of age) and at a distance ≤0.5 mm from the stimulating electrode placed exclusively in white matter. Second the amplitude (-mV) of a field potential was obtained by measuring the change in voltage, starting at the recording baseline and ending at the inward most negative inflection point as described previously (Spencer and Murphy, 2000; Smith et al., 2001; Ade and Lovinger, 2007). Third only slices with field potentials of maximum amplitudes between 1-3.5 mV were used for experiments. The highest stimulation intensities were mostly near 1 mA, but were never higher than 2 mA.
To generate input-output relationships, pulses of presynaptic fiber stimulation (100 μsec in duration) were delivered once every 15 sec in a series of increasing intensities to obtain a range of mV responses. To standardize responses, the % maximum response (where 100% occurred at the highest stimulus intensity) was plotted against stimulus intensity. For paired-pulse ratio (PPR) studies, the stimulation intensity was set to evoke a response at 50% of maximum throughout the experiment. PPRs were obtained by delivering two stimulation pulses with interstimulus intervals of 25, 50, or 100 msec and calculating the ratio of the amplitude of the 2nd to the 1st field potential response.
The following drugs were used: 6-cyano-7-nitroquinoaxaline-2, 3-dione (CNQX) to block 2-amino-3-hydroxy-5-methyl-4-isoxazolepropionate (AMPA)/kainate glutamate receptors, 2-amino-5-phosphovalerate (APV D-L isomer) to block N-methyl D-aspartate (NMDA) receptors, picrotoxin to block GABAA receptors, quinpirole and sulpiride to activate and block D2 dopamine receptors respectively, WIN55,212-2 and AM251 to activate and block endocannabinoid CB1 receptors respectively, and forskolin (FSK) to constitutively activate adenylyl cyclase. All drugs were purchased from Sigma-Aldrich (St. Louis, MO) except AM251 and WIN55,212-2, which were from Tocris (Ellisville, MO).
Corticostriatal Synaptic Plasticity: Long-Term Depression and Long-Term Potentiation
High frequency stimulation (HFS) of the corpus callosum dorsolateral to the striatum was used to induce long-term depression (LTD). Synaptic field potentials were recorded at 50 % of maximum stimulus intensity every 15 sec for a period of 10 min before, and for a period of 30 min after HFS in ACSF. The HFS induction protocol consisted of 3 X 1 sec trains at 100 Hz (200 μsec duration per pulse) with an intertrain interval of 6 sec. This paradigm was shown previously to induce a persistent NMDA receptor-independent form of LTD in mouse dorsal striatum over a 60 min time period post-HFS (Geracitano et al., 2003). To maximize the number of recordings in both WT and ASOTg slices on the same day of an experiment, a 30 min period of recording was chosen post-HFS as a more convenient time frame. N values are shown for each synaptic plasticity study and represent the number of mice from which one or more slices were recorded. Results are expressed as the mean ± standard error of mean (SEM) percent (%) of the amplitude of average baseline field potentials. For each set of HFS experiments, the PPR at the 50 msec interstimulus interval was recorded over the same time course before and after the HFS.
Chemical long-term potentiation (chemLTP) was induced by FSK using a modification of previous protocols (Makhinson et al., 1999; Spencer and Murphy, 2002; Chotiner et al., 2003). Field potentials were recorded every 15 sec in dorsolateral striatum of slices, stimulated at 50 % maximum intensity as described above. Synaptic transmission was monitored for 10 min in ACSF followed by 10 min in 50 μM FSK and then for 60 min in ACSF. The PPR at the 50 msec interstimulus interval was also recorded over the same time course before and after FSK. In separate experiments, 50 μM APV was added to ACSF to block NMDA receptors for 10 min prior to and during FSK perfusion and afterwards for an additional 20 min.
Data Analyses of Electrophysiology
For electrophysiology studies, acquisition and analyses of data were performed with pCLAMP 8.2 software (Axon Instruments Inc., Foster City, CA). For statistical analyses, Student’s t-tests (unpaired or paired) were used for two group comparisons, while appropriately designed ANOVAs followed by Bonferroni t-tests were used for multiple comparisons.
Immunoblot Analysis
Whole-cell homogenates were prepared from pre-frozen mouse forebrain (mostly cortex, striatum) and analyzed by SDS-PAGE (5% stacking/12% resolving) and Western immunoblotting as described previously (Watson et al., 2002; 2006). For some experiments, olfactory bulbs were used because of their enrichment in TH-containing neurons. For immunoblotting, nitrocellulose membranes (0.2 μm) were subdivided into smaller segments corresponding to proteins within the range of the molecular weight of a desired target protein and incubated overnight at 4°C with appropriate primary antisera diluted in BLOTTO (4% dried non-fat milk in phosphate-buffered saline). The antibody for α-synuclein (rabbit polyclonal, 1:500 to 1:1000 dilution) was purchased from Chemicon/Millipore (Temescula, CA). The β-actin antibody (monoclonal, 1:1000 dilution) was purchased from Sigma-Aldrich (St. Louis, MO). Although the α-synuclein antibody was generated against amino acid residues 111-131 of the human protein, it was observed to cross-immunoreact with the endogenous mouse protein.
Membranes were incubated with horseradish peroxidase-conjugated secondary antibodies diluted 1:10,000 in BLOTTO at room temperature for 1-2 h. Bound antibodies were visualized on nitrocellulose by enhanced chemiluminescence (Immun-star HRP detection kit, Bio-Rad, Hercules, CA, USA). Chemiluminescent images were acquired using a cooled CCD camera-based image acquisition and analysis system (Chemi-Doc and Quantity One software package from Bio-Rad)(Watson et al., 2006). Density values for a protein band of interest were normalized to density values for β-actin controls obtained in the same lane of the identical blot. Values for Student’s t-tests were used to determine statistical significance.
cAMP Assay
The basal and FSK-induced accumulation of cAMP was measured in synaptoneurosome (SN) membrane-enriched fractions, prepared from frozen mouse forebrain as described previously (Johnson et al., 1997; Watson et al., 2006). The cAMP assays were performed according to previously described protocols with modifications (Watabe et al., 2000; Lobo et al., 2007). SNs (1μg/μl protein) were incubated in 96-well plates containing 10mM imidazole (pH 7.4), 1mM IBMX, 6mM MgSO4, 0.6mM EGTA, 1.5mM ATP, 0.01 mM GTP, with or without 10μM FSK. FSK, IBMX and cAMP-binding protein were purchased from Sigma-Aldrich (St. Louis, MO). Plates were incubated at 30°C for 10 min and boiled to stop the reaction. The cAMP levels were measured by radioimmunoassay by incubating samples with 3H cAMP (30.1Ci/mmol, Perken Elmer, Waltham, MA) at ~12,000 DPM/well and 80 μg/ml cAMP binding protein supplemented with 50 picomoles ADP for 3 h on ice. Samples were incubated with charcoal dextran and centrifuged at 3,100 RPM for 15 min to generate a supernatant in which pmol cAMP/μg protein was determined. Data were analyzed with a two-way ANOVA with repeated measures (one factor repetition) followed by Bonferroni t-tests.
RESULTS
Normal glutamatergic synaptic transmission in ASOTg striatum
Elevated expression of human α-synuclein protein in ASOTg relative to WT brain was confirmed by Western immunoblotting using an antibody that recognizes both exogenous human and endogenous mouse α-synuclein (Fig. 1A). Based on density values (mean ± SEM) for α-synuclein normalized to β-actin, human α-synuclein is elevated many fold relative to endogenous mouse α-synuclein in ASOTg forebrain (WT, 0.05 ± 0.0, N = 4 mice; ASOTg, 1.19 ± 0.03, N = 4 mice; P < 0.05) consistent with an initial report (Rockenstein et al., 2002).
Figure 1. Basal synaptic transmission is unchanged in corticostriatal slices from ASOTg mice and is mainly glutamatergic.

A. Western immunoblotting confirmed elevated expression of human α-synuclein in ASOTg forebrain. Density values (mean ± SEM) for α-synuclein were normalized to β-actin in the same lane (N = 4 each, Student t-test, P < 0.05). B. To generate input-output relationships, the amplitudes of field potentials (-mV) were measured in response to a series of increasing intensities in slices. To standardize responses, the % maximum response (where 100% occurred at the highest stimulus intensity) was plotted against stimulus intensity. No significant differences were evident between WT and ASOTg responses (P = 0.588). C. Basal synaptic transmission recorded at 50% maximum stimulus intensity was significantly reduced in both WT and ASOTg slices treated with CNQX (10 μM, 5 min) to inhibit AMPA/kainate glutamate receptors (* P = 0.02). There was no difference in % change in responses for CNQX-treated WT relative to CNQX-treated ASOTg slices (P = 0.41). Responses shown are averaged synaptic field potentials recorded in WT and ASOTg slices either untreated or treated with CNQX (dashed line).
To determine whether over expression of human α-synuclein alters synaptic transmission, we recorded field potentials at an increasing series of stimulus intensities in corticostriatal slices from WT and ASOTg mice. The dorsolateral region of the striatum was chosen for recordings, because it receives afferent fibers mainly from sensorimotor cortex (Haber, 2003) and has been well characterized for synaptic plasticity (Partridge et al., 2000; Smith et al., 2001). Input-output relationships were similar for slices from WT and ASOTg mice (WT, N = 25 mice; ASOTg, N = 20 mice; two-way ANOVA with repeated measures, one factor repetition, P = 0.588) (Fig. 1B). No difference in synaptic transmission was detected between WT and ASOTg slices as a function of gender or age (2-3 months versus 4-6 months)(P > 0.05). Responses were slightly reduced in ASOTg slices at the lowest intensity (0.2 mA) but the difference was not statistically significant (P = 0.188). Because few responses were obtained below 0.2 mA, thresholds for synaptic responses in ASOTg relative to WT striatum could not be accurately compared at lower intensities. Data from all subsequent experiments including PPR and LTP studies were obtained using a stimulus intensity that generated a 50% maximum response for each slice. Because there were no differences in input-output function, corticostriatal synaptic transmission is not significantly altered when human α-synuclein is elevated. This finding is consistent with previous studies showing that α-synuclein protein levels do not alter synaptic transmission in hippocampus (Cabin et al., 2002; Steidl et al., 2003; Gureviciene et al., 2007).
Medium-sized spiny neurons (MSSNs) are the predominant neurons in the neostriatum and their synaptic responses generate most of the synaptic field potential (Colwell and Levine, 1995; Klapstein et al., 2001). While MSSNs receive most of their glutamatergic presynaptic input from the cortex and thalamus, they also receive GABAergic input from striatal interneurons and other MSSNs (Graybiel et al., 1981; Wilson, 1987; Dube et al., 1988). Interneurons also provide cholinergic input, while dopaminergic inputs derive from the substantia nigra. To examine if there were differences in these inputs in ASOTg field potentials, basal synaptic transmission was examined in WT and ASOTg slices treated with a variety of receptor-selective drugs.
Blockade of non-NMDA, AMPA/kainate glutamate receptors selectively with CNQX (10 μM, 5 min) inhibited most synaptic transmission at 50% maximum stimulus intensity (Fig. 1C). Values for field potentials for WT were: untreated control, -0.72 ± 0.17 mV, + CNQX, -0.21 ± 0.03 mV, N = 3 mice, P = 0.02. This result is consistent with a previous study (Spencer and Murphy, 2000) which showed that extracellular field potential and intracellular fEPSP recordings, evoked in tandem, were both blocked by CNQX. Slices treated with APV (50 μM, 10 min) to inhibit non-NMDA glutamate receptors were unaffected (WT, untreated, -0.76 ±0.08 mV, N = 4; WT +APV, -0.72 ± 0.07 mV, N = 4, P = 0.69), supporting a primary role for AMPA/kainate glutamate receptors in basal synaptic transmission (also see Fig. 6C). Synaptic transmission in ASOTg slices was also inhibited by CNQX (untreated control, -0.75 ± 0.05 mV; + CNQX, -0.41 ± 0.09 mV, N = 4 mice, P = 0.02). However there was no significant difference in % decrease in responses for CNQX-treated WT (29.1 ± 9.8 % of untreated control) relative to CNQX-treated ASOTg slices (43.3 ± 11.3 % of untreated control, P = 0.41).
Figure 6. FSK-induced chemLTP is enhanced in ASOTg striatum.

A Slices were perfused with FSK (50 μM) for 10 min to induce chemLTP followed by wash-out with ACSF for 60 min. ChemLTP is enhanced in ASOTg relative to WT striatum (* P = 0.05). Responses shown are averaged synaptic field potentials recorded pre-FSK (5 min) and post-FSK (55-60 min) treatment. Calibrations (0.5mV/5 msec) are shown for each set of traces. B. A 2nd field potential response was recorded at a 50 msec interstimulus interval to obtain PPRs during the time course of chemLTP. The PPR drops significantly below 1.0 after FSK induction of chemLTP only in ASOTg striatum, indicative of paired-pulse depression and presynaptic maintenance of LTP (* P = 0.006). C. Corticostriatal chemLTP is NMDA receptor-independent based on insensitivity to APV post-FSK treatment (25-30 min) but is not different from control (P = 0.51). D. Basal and FSK-induced accumulation of cAMP were measured in SN membrane-enriched subcellular fractions prepared from WT and ASOTg forebrain. The cAMP levels (picomole/μg SN protein) were significantly lower in ASOTg compared to WT SN groups (Two-Way ANOVA, P = 0.048). Pair-wise comparisons showed that FSK-induced cAMP levels in WT SNs to a greater degree than in ASOTg SNs (* Bonferoni t-test, P = 0.015)
WT and ASOTg slices treated with picrotoxin (100 μM, 5 min) to inhibit GABAA receptors had no significant effect on synaptic transmission (WT: untreated control, -0.90 ± 0.23 mV; + picrotoxin, -1.09 ± 0.22 mV, N = 3 mice, P = 0.60)(ASOTg, untreated control, -0.97 ± 0.11mV; + picrotoxin, -0.93 ± 0.14 mV, N = 6 mice, P = 0.85). Activating (10 μM quinpirole, 5 min) or blocking (10 μM sulpiride, 5 min) D2 dopamine receptors also had no effect on synaptic transmission (P > 0.05). Activating (3 μM WIN55, 212-2, 5 min) or blocking (3 μM AM251, 5 min) endocannabinoid CB1 receptors had no significant effects as well. However a longer time of CB1 drug treatments may be required to produce an effect on field potentials, since a previous report showed that a 20 min application of WIN55,212-2 was sufficient to depress synaptic transmission, supporting a role for corticostriatal presynaptic CB1 receptors in glutamate release (Ade and Lovinger, 2007). Overall the drug treatments confirmed that field potentials in the corticostriatal pathways are mediated mainly by activation of AMPA/kainate glutamate receptors, but they were not altered by elevated levels of human α-synuclein.
Paired-pulse facilitation occurs in ASOTg striatum
Since α-synuclein has been reported to modulate presynaptic vesicle pools of neurotransmitters, the effect of over expression of human α-synuclein on short-term synaptic plasticity was examined by recording PPRs. The PPR was measured at 25, 50 and 100 msec interstimulus intervals. Figure 2 shows that PPR values > 1.0, indicative of facilitation, are detected in the striatum in ASOTg and are significantly different from WT (WT, N = 16 mice; ASOTg, N = 20 mice, Two-Way ANOVA, P = 0.001). The PPR data demonstrate paired-pulse facilitation in the corticostriatal pathway of ASOTg mice, consistent with a presynaptic effect of elevated human α-synuclein on short-term synaptic plasticity (Zucker and Regehr, 2002; Stevens, 2003).
Figure 2. Paired-pulse facilitation is detected in ASOTg striatum.

A. PPRs > 1.0 were detected in ASOTg striatum at 25, 50 and 100 msec interstimulus intervals and were significantly different from WT (WT, N = 16 mice; ASOTg, N = 20 mice, Two-Way ANOVA, * P = 0.001). The PPR data demonstrate paired-pulse facilitation in the corticostriatal pathway of ASOTg mice, consistent with a presynaptic effect of over-expressed human α-synuclein. Responses shown are averaged 1st and 2nd field potentials recorded at a 50 msec interstimulus interval.
No paired-pulse facilitation in striatum from α-synuclein “knock-out” mice
The PPR was also examined in the corticostriatal pathway in slices obtained from Snca-/- mice lacking expression of the endogenous mouse α-synuclein gene. Western immunoblotting (Fig. 3A) confirmed the loss of endogenous mouse α-synuclein protein in Snca-/- mouse olfactory bulb. Density values (mean ± SEM) for α-synuclein normalized to β-actin were: Snca+/+, 0.12 ± 0.03, N = 3 mice; Snca-/-, 0.0, P < 0.05, N =3 mice). Lack of expression in forebrain was also confirmed (data not shown). Human α-synuclein was elevated in expression in ASOTg olfactory bulb resolved on the same blot with Snca+/+ and Snca-/- brain samples (WT, 0.21 ± 0.155, N = 4 mice; ASOTg, 1.1 ± 0.04, N = 4 mice, P = 0.002).
Figure 3. Paired-pulse facilitation is not detected in striatum from Snca-/- mice.

A. Western immunoblotting confirmed loss of expression of endogenous mouse α-synuclein protein in Snca-/- olfactory bulb compared to Snca+/+ non-Tg control (N = 3 each, P< 0.05). B. The PPR recorded in corticostriatal slices from Snca-/- mice was not different from Snca+/+ controls and did not show paired-pulse facilitation (Snca+/+, N = 4; Snca-/-, N = 4,Two-Way ANOVA, P = 0.179). Responses shown are averaged 1st and 2nd field potentials recorded at a 50 msec interstimulus interval.
Similar to the hippocampal CA1 region (Cabin et al., 2002), synaptic transmission was unchanged in the Snca-/- corticostriatal pathway based on responses recorded at 0.2 mA stimulus intensity (Snca+/+, 37.0 ± 21.0 % of max response, N = 4; Snca-/-, 52.3 ± 19.4 % of max response, N = 4; P = 0.62). In contrast to ASOTgs, PPR values < 1.0 at most interstimulus intervals were obtained for Snca-/- mice indicating paired-pulse depression. However the values were not statistically different from non-Tg controls (Fig. 3B)(Snca+/+, N = 4 mice, Snca-/-, N = 4 mice, Two-Way ANOVA, P = 0.179). Interestingly, the paired-pulse depression in striatum was different from studies of the same mice in hippocampal CA1, which is known to exhibit a robust form of paired-pulse facilitation at glutamatergic synapses (Cabin et al., 2002; Gureviciene et al., 2007). The differences may reflect multiple synaptic inputs (GABAergic, dopaminergic, cholinergic) on MSSNs in the striatum in addition to the corticostriatal glutamatergic terminals.
Paired-pulse depression in striatum from THwt Tg mice
As additional controls, the PPR was also examined in corticostriatal slices from Tg mice over expressing either normal (wt) or mutant A53T human α-synuclein in TH-containing neurons (THwt Tg, THmutA53T Tg)(Matsuoka et al., 2001). Levels of normal human α-synuclein appear higher than mutant A53T human α-synuclein in TH-containing olfactory bulb by immunoblotting but are not significantly different (Fig. 4A)(THwt Tg, 1.87 ± 0.69, N = 3 mice; THmutA53T Tg, 1.07 ± 0.20, N = 3 mice, P = 0.33). Because both mice were bred as homozygous Tgs, non-Tg WT littermate controls were not available for comparisons. As expected, human α-synuclein is detected at elevated levels in ASOTg relative to WT non-Tg olfactory bulb resolved on the same blot as the TH Tg sample (WT, 1.45 ± 0.30, N = 3 mice; ASOTg, 4.70 ± 0.56, N = 3 mice, P = 0.007).
Figure 4. Paired-pulse depression occurs in striatum of THwt Tg mice.
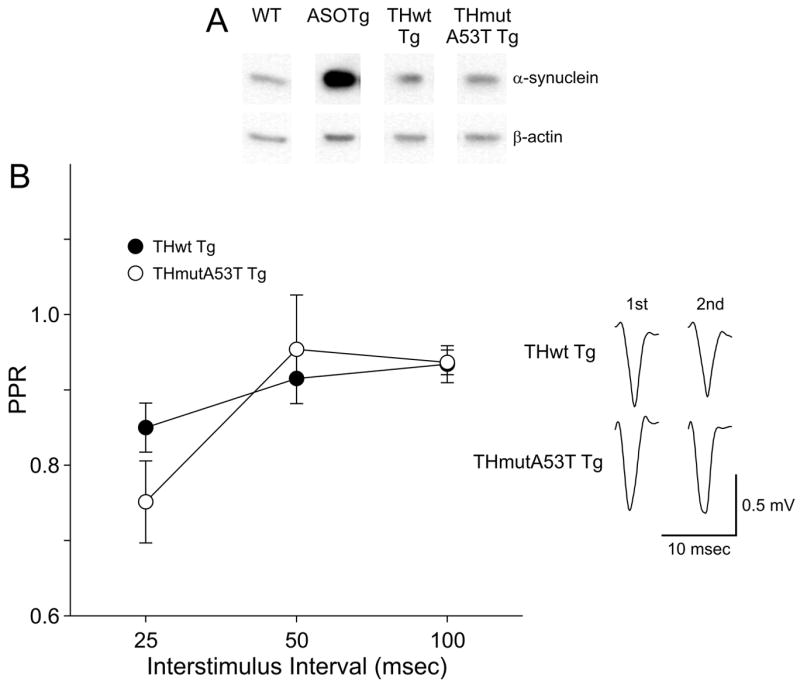
A. Westerns blots show similar levels of normal and A53T human α-synuclein in TH-containing olfactory bulb from THwt and THmutA53T Tg mice (P = 0.33). As positive control, human α-synuclein is detected at elevated levels in ASOTg relative to WT olfactory bulb (P = 0.007) resolved on the same blot as the TH Tg samples. B. The PPRs recorded in corticostriatal slices from THwt Tg mice revealed paired-pulse depression (PPR < 1.0) at all interstimulus intervals but was not different from THmutA53T Tg (N = 4 each, Two-Way ANOVA, P = 0.619). Responses shown are averaged 1st and 2nd field potentials recorded at a 50 msec interstimulus interval.
Synaptic transmission was similar in the THwt Tg and THmutA53T Tg corticostriatal pathways based on responses recorded at 0.4 mA stimulus intensity (THwt, 40.3 ± 6.3 % max response, N = 4; THmutA53T, 52.2 ± 15.9 % max response, N = 4, P = 0.51). Similar to Snca-/- mice, PPR values < 1.0 denoting paired-pulse depression were obtained in slices from THwt Tg mouse at all interstimulus intervals but were not different from THmutA53T Tg (Fig. 4B)(N = 4 each, Two-Way ANOVA, P = 0.619). The data show that selective expression of either normal or mutant A53T human synuclein in TH-containing nigrostriatal terminals does not confer paired-pulse facilitation in the corticostriatal pathway.
HFS-LTD occurs in ASOTg striatum
Overall the PPR data from multiple α-synuclein Tg and “knock-out” mice show that significant short-term synaptic plasticity in the form of paired-pulse facilitation occurs only in ASOTg mice. This observation raises the possibility that elevated human α-synuclein might also alter long-term forms of synaptic plasticity. To test this hypothesis further, LTD was induced in vitro in WT and ASOTg mouse corticostriatal slices by HFS using multiple trains of 100 Hz stimulation as first described in studies using rats (Calabresi et al., 1992a, b; Lovinger et al., 1993; Walsh, 1993; Wickens et al., 1996). For these experiments, we used an HFS paradigm previously shown to induce LTD in slices from non-Tg control mice (Geracitano et al., 2003). Surprisingly significant LTD was detected in the ASOTg but not in the WT corticostriatal pathway (Fig. 5A)(WT, post-HFS, 90.1 ± 2.0 %, N = 4 mice, 6 slices, P = 0.09)(ASOTg, post-HFS, 54.2 ± 10.1 %, N = 6 mice, 7 slices, P = 0.004). Post-HFS corresponded to the 25-30 min period after HFS. Consistent with previous reports (Partridge et al., 2000; Spencer and Murphy, 2000; Akopian et al., 2000; Mahon et al., 2004; Fino et al., 2007; reviewed in Berretta et al., 2008), the same HFS protocol in some experiments was also able to induce LTP in both WT and ASOTg slices. For subsequent LTP studies, FSK was used to induce a presynaptic form of chemLTP (see below).
Figure 5. HFS-LTD is detected in ASOTg striatum.

A. HFS induced significant LTD in ASOTg (* P =0.004) but not in WT striatum (P = 0.13). Responses shown are averaged synaptic field potentials recorded pre-HFS (5 min) and post-HFS (25-30 min). B. Paired-pulse facilitation increases in WT and ASOTg slices after HFS induction of LTD. A 2nd field potential response was recorded at a 50 msec interstimulus interval to obtain a PPR before and after HFS induction. The PPR remained above 1.0 pre-HFS and increased significantly post-HFS, indicative of alterations in paired-pulse facilitation when ASOTg LTD was induced (*P = 0.001).
The experiments using HFS showed that LTD was preferentially induced in ASOTg striatum. Corticostriatal HFS-LTD is also accompanied by increased paired-pulse facilitation after HFS, supportive of a presynaptic mechanism for LTD (Choi and Lovinger, 1997). Using the HFS-LTD protocol of the present study, PPRs with a 50 msec interstimulus interval were also measured before and after the HFS. As shown in Figure 5B, PPRs > 1.0 (indicating paired-pulse facilitation) increased over 30 min only when LTD is induced in ASOTg striatum (pre-HFS, 1.26 ± 0.03; post-HFS, 1.44 ± 0.03, P = 0.001). Conversely the PPRs did not change significantly in WT striatum (P = 0.13). Taken together with the LTD data, it is concluded that over-expressed human α-synuclein facilitates a persistent presynaptic effect in the corticostriatal pathway leading to long-term decreases in synaptic strength.
FSK-induces chemLTP coincident with paired-pulse depression in ASOTg striatum
Because striatal HFS-LTD has been shown to be NMDA receptor-independent (Lovinger et al., 1993; Geracitano et al., 2003), an NMDA receptor-independent form of LTP was also examined. For these experiments, a modified FSK protocol was used to induce chemLTP by constitutive activation of adenylyl cyclase (Spencer and Murphy, 2002). Slices were perfused with FSK (50 μM) for 10 min followed by wash-out with ACSF for 60 min. As shown in Figure 6A, chemLTP is detected in both WT and ASOTg striatum at 55-60 min post-FSK (WT, 159.2 ± 9.4 %, N = 7 mice, P = 0.001)(ASOTg, 202.1 ± 19.6 %, N = 4 mice, P = 0.01). Interestingly striatal chemLTP is enhanced in ASOTg relative to WT mice (P = 0.05), suggesting that there are synaptic alterations in the adenylyl cyclase signaling pathway in the ASOTg striatum.
To further examine this hypothesis, the PPR with a 50 msec interstimulus interval was examined in the same experiments assessing chemLTP (Fig. 6B). After FSK application, the PPR drops below 1.0, indicating paired-pulse depression, in both WT and ASOTg striatum but is significantly different only in ASOTg (WT: pre-FSK, 0.99 ± 0.04; post-FSK, 0.89 ± 0.04, P = 0.13) (ASOTg: pre-FSK, 1.23 ± 0.07; post-FSK, 0.88 ± 0.04, P = 0.006). The decrease in the PPR after FSK treatment confirms the presynaptic maintenance of this form of chemLTP in ASOTg striatum (Spencer and Murphy, 2002) In contrast to FSK-induced chemLTP in the hippocampus (Makhinson et al., 1999; Chotiner et al., 2003), chemLTP in the striatum is NMDA receptor-independent based on insensitivity to APV (50 μM) (Fig. 6C)(25-30 min post FSK, 169.1 ± 29.9 %, N= 4 mice) but is not different from untreated controls (P = 0.51).
FSK’s enhancement of the PPR in ASOTg striatum could reflect an alteration in the adenylyl cyclase signaling pathway at presynaptic terminals. This idea was examined further by measuring the FSK-induced accumulation of cAMP in SN membrane-enriched fractions prepared from WT and ASOTg forebrain (Fig. 6D). The cAMP levels (picomole/μg SN protein) were significantly lower in ASOTg compared to WT groups (WT: basal cAMP, 7.7 ± 1.6; FSK-cAMP, 39.3 ± 4.3, N = 12)(ASOTg: basal cAMP, 5.0 ± 0.7; FSK-cAMP; 29.4 ± 3.0, N = 12, Two-Way ANOVA, P = 0.048). Pair-wise comparisons showed that FSK induced cAMP levels in WT SNs to a greater degree than in ASOTg SNs (Bonferoni t-test, P = 0.015), pointing to an adenylyl cyclase pathway as a potential target of modulation by elevated human α-synuclein.
DISCUSSION
Short-term presynaptic plasticity is altered in the corticostriatal pathway of ASOTg
The central finding from this study is that ubiquitous over expression of human α-synuclein in mouse brain significantly alters both short-term and long-term presynaptic plasticity in the corticostriatal pathway. Results from short-term synaptic plasticity studies of corticostriatal slices from both α-synuclein over-expressing mice and “knock-out” mice showed that only elevated amounts of human α-synuclein reliably induced paired-pulse facilitation in the dorsolateral region of the striatum. In contrast, previous reports detected paired-pulse depression or reduced paired-pulse facilitation in either the dentate gyrus perforant pathway or the mossy fiber-CA3 pathway of hippocampus from Tg mice expressing human α-synuclein or a variant α-synuclein with the A30P mutation (Steidl et al., 2003; Gureviciene et al., 2007). The cumulative results suggest that there are differential effects of α-synuclein on short-term synaptic plasticity, dependent on the specific brain region sampled and thus not necessarily applicable to all brain regions.
Since alterations in paired-pulse facilitation are commonly associated with presynaptic forms of synaptic plasticity, human α-synuclein likely exerts its functional effects primarily at the presynaptic terminal in ASOTg mice. Paired-pulse facilitation has also been interpreted to reflect a low probability of release of neurotransmitter from ready-releasable synaptic vesicles docked at terminals (Zucker and Regehr, 2002; Stevens, 2003; Garcia-Junco-Clemente et al., 2005). Since CNQX was shown to block synaptic transmission mediated by AMPA/kainate glutamate receptors in the ASOTg corticostriatal pathway, one possibility is that elevated α-synuclein reduces the release of glutamate from corticostriatal terminals. However input-output relationships were not significantly different between WT and ASOTg mice, suggesting that basal synaptic transmission is unchanged. Thus elevated human α-synuclein could impact paired-pulse facilitation via a presynaptic mechanism not directly related to the probability of neurotransmitter release. For example, the correlation between paired-pulse facilitation and a low probability of release does not always prove true as shown for the D2 dopamine receptor-expressing MSSNs in the striatum (Cepeda et al., 2008). Possibly over-expressed human α-synuclein interacts with a reserve pool of vesicles upstream of release as suggested by previous studies of short-term glutamatergic synaptic plasticity in the hippocampus (Cabin et al., 2002; Steidl et al., 2003). A more remote possibility is that paired-pulse facilitation in the corticostriatal pathway of ASOTg is influenced by postsynaptic mechanisms (Wang and Kelly, 1997). Lastly field potentials can be influenced by both presynaptic neurotransmitter release and the properties of postsynaptic membranes, so a presynaptic effect of over-expressed human α-synuclein on basal glutamate release may have been diminished and gone undetected during input-output studies. For example, ASOTgs appeared to have reduced responses at the lowest stimulus intensity of 0.2 mA, possibly reflecting a higher threshold for synaptic responses in ASOTg relative to WT striatum at lower intensities.
Nonetheless significant paired-pulse facilitation is detected across multiple interstimulus intervals solely in the ASOTg corticostriatal pathway, when compared to multiple WT mouse strains as well as to α-synuclein “knock-out” mice and TH Tg mice. Thus it remains a possibility that ubiquitous expression of human α-synuclein could impact evoked release of glutamate and multiple other neurotransmitters from terminals in the striatum. Supporting this finding, previous studies of evoked dopamine release in nigrostriatal terminals from α-synuclein ”knock-out” mice, as well as in neuronal cultures from Tg mice over expressing α-synuclein, suggest that human α-synuclein negatively regulates reserve pools of synaptic vesicles and “primed” synaptic vesicles required for dopamine release (Abeliovich et al., 2000; Yavich et al., 2004, 2005; Larsen et al., 2006). Because dopamine-rich axon terminals from the substantia nigra remain in the coronal corticostriatal sections used in the present study, it remains possible that nigral dopamine release is also reduced in ASOTg striatum. Surprisingly paired-pulse depression was primarily detected in the corticostriatal pathway in α-synuclein “knock-out” Snca-/- mice and also in Thwt/THmutA53 Tg mice expressing human α-synuclein selectively in TH-containing brain regions. These differences could simply be a reflection of the different mouse strains used. However, they also suggest that ubiquitous over expression of human α-synuclein in brain, as is the case in PD, may be a more accurate model for the disease.
Long-term presynaptic plasticity is altered in the corticostriatal pathway of ASOTg
Long-term synaptic plasticity was altered in ASOTg mouse striatum. Paradoxically striatal LTD was detected in ASOTg striatum but not in WT striatum using a paradigm shown previously to induce LTD in control non-Tg mice. Possibly the lack of LTD induction in WT mice in the present study is due to the use of a different strain of mice or perhaps different experimental conditions. Interestingly paired-pulse facilitation increased after HFS induction of LTD in the ASOTg striatum, suggesting that α-synuclein may impact long-term persistent forms of synaptic plasticity by preferentially reducing presynaptic neurotransmitter (mostly glutamate) release from corticostriatal terminals (Choi and Lovinger, 1997). The protocol used to induce striatal HFS-LTD was also shown previously not to require activation of the NMDA subtype of glutamate receptors. Consistent with this observation, elevated human α-synuclein in the ASOTg mice enhanced an NMDA receptor-independent form of striatal chemLTP induced by FSK application. Moreover short-term facilitation was lost and replaced by paired-pulse depression over the same one hour period, confirming the presynaptic maintenance of chemLTP when human α-synuclein is over-expressed.
Results using chemLTP demonstrated that constitutive activation of adenylyl cyclases by FSK induces an NMDA receptor-independent form of presynaptic LTP that is enhanced in ASOTg striatum. Since FSK-induced cAMP levels are reduced in synaptic subcellular fractions from ASOTg brain, the enhancement of chemLTP in ASOTg striatum was unexpected. Possibly this reflects a relative ceiling effect, where LTP saturates in WT well before it levels off in ASOTg due to differences in cAMP levels at corticostriatal terminals. However it is possible that postsynaptic adenylyl cyclase pathways that enhance glutamate receptor functions in MSSNs are also impacted when chemLTP is induced in ASOTg striatum. For example, FSK can enhance short-term glutamate excitatory synaptic transmission in the corticostriatal pathway (Colwell and Levine, 1995). FSK can also induce a protein kinase A (PKA) phosphorylation of the GluR1 subunit required for priming postsynaptic membrane insertion of AMPA receptors during hippocampal LTP induction in CA1 (Malinow and Malenka, 2002).
There are conflicting reports supporting a positive role for α-synuclein in promoting long-lasting increases in presynaptic glutamate release (Liu et al., 2004; Liu et al., 2007). These studies differed in that synaptic plasticity was examined in hippocampal neuronal cultures, either from mice lacking rodent α-synuclein or cultures in which the presynaptic neuron was injected with α-synuclein. The hippocampal cell culture approach may have unmasked unique conditions for synaptic plasticity, not accessible when recording in corticostriatal slices prepared from Tg mice.
What are the presynaptic targets of elevated human α-synuclein?
Striatal chemLTP is reminiscent of previous reports showing that NMDA receptor-independent forms of LTP in the mossy fiber pathway of hippocampus, the lateral amygdala, and the granule cell-Purkinje cell pathway in cerebellum require presynaptic activation of adenylyl cyclase and downstream cAMP/PKA signaling pathways (Weisskopf et al., 1994; Huang and Kandel, 1998; Linden and Ahn, 1999; Lonart et al., 2003). Enhancement of chemLTP in ASOTg striatum further suggests that elevated human α-synuclein may interact with adenylyl cyclase signaling pathways that underlie potential glutamate release from corticostriatal terminals. An attractive candidate is the synaptic vesicle scaffold containing the Rim1α protein, whose PKA phosphorylation is closely associated with presynaptic forms of LTP (Castillo et al., 2002; Lonart et al., 2003).
Other potential targets are presynaptic cAMP signaling pathways triggered by G-protein coupled receptors (GPCR) on corticostriatal terminals particularly D2 dopamine GPCRs coupled to Gi, inhibition of adenylyl cyclase and cAMP reduction (Fisher et al., 1994; Bamford et al., 2004). For example over-expressed α-synuclein has been shown in vitro in co-transfected cultures to activate D2 dopamine receptor signaling and to reduce cAMP levels (Kim et al., 2006). Interestingly synaptic plasticity studies in a DJ-1 “knock-out” mouse model for PD reported a loss of HFS-LTD in the corticostriatal pathway of these mice and the loss was rescued by the D2 dopamine receptor agonist quinpirole (Goldberg et al., 2005). Future synaptic plasticity studies using single cell recordings in MSSNs selectively tagged with green fluorescent protein in ASOTg mice, for either the direct D1 dopamine receptor pathway or indirect D2 dopamine receptor pathway, will be required to examine this question more precisely in mice over expressing human α-synuclein (Wang et al., 2006; Kreitzer and Malenka, 2007; Cepeda et al., 2008).
ASOTg mice are models for early synaptic dysfunction in PD
The signature events of PD are the loss of dopaminergic neurons of the pars compacta of the substantia nigra (Dauer and Przedborski, 2003), but earlier pathological changes may occur involving α-synuclein accumulation in Lewy bodies in the olfactory bulb and brain stem nuclei (Braak et al., 2003). The present study utilized the ASOTg mouse as a model to study synaptic function early in PD. ASOTg mice over express human α-synuclein, accumulate protease-resistant α-synuclein aggregates, and exhibit motor behavioral deficits in the absence of detectable neuron cell loss (Fleming et al., 2004; Fernagut et al., 2007). Our results here show that ubiquitous over-expression of α-synuclein in the brains of ASOTg mice alters both short-term and long-term presynaptic forms of synaptic plasticity, consistent with a potential decrease in glutamate release from corticostriatal terminals. Because α-synuclein is expressed ubiquitously throughout the brain in this model, we cannot completely rule out similar α-synuclein alterations at other neurotransmitter terminals including dopaminergic nigrostriatal terminals (Wu et al., 2005; Fleming et al., 2006).
How would a reduction in glutamatergic synaptic plasticity in the corticostriatal pathway impact PD? The net effect of persistent reduction in cortical glutamate is to decrease long-term excitatory synaptic transmission between the cortex and striatum, thereby blocking the main inhibitory outputs from the striatum to the substantia nigra and globus pallidus (Graybiel et al., 1981; Wilson, 1987; Haber, 2003). This ultimately would lead to disinhibition of the striatal-thalamo-cortical loop, which would be expected to produce hypokinetic effects and reduce overall movement, so characteristic of PD. Thus ASOTg mice may recapitulate an early stage in PD during which over-expressed α-synuclein dampens corticostriatal synaptic transmission and reduces movement.
Acknowledgments
Supported by The Center for Gene Environment Studies in Parkinson’s Disease (CGEP) at UCLA (NIH U54 ES012078)(MSL, JBW), The UDALL Center for Excellence in the Study of Parkinson’s Disease (NIH NS 38367)(MSL, JBW), and a Faculty Research Grant from the UCLA Academic Senate (JBW). Many thanks to Gloria Klapstein, Damian Cummings, Carlos Cepeda, Véronique André, Emily Jocoy, Nanping Wu and other members of Mike Levine’s lab as well as Tom O’Dell for their critical feedback. Special thanks to Mike Levine and Marie-Francoise Chesselet for their kind support.
ABBREVIATIONS
- ACSF
artificial cerebrospinal fluid
- AMPA
2-amino-3-hydroxy-5-methyl-4-isoxazolepropionate
- APV
2-amino-5-phosphovalerate
- ASOTg
transgenic mice over expressing human α-synuclein
- BLOTTO
4% dried non-fat milk in phosphate-buffered saline
- chemLTP
chemical long-term potentiation
- CNQX
6-cyano-7-nitroquinoaxaline-2, 3-dione
- FSK
forskolin
- GPCR
G-protein coupled receptors
- HFS
high-frequency stimulation
- LRRK2
leucine-rich repeat kinase 2
- LTD
long-term depression
- MSSN
medium-sized spiny neuron
- NMDA
N-methyl D-aspartate
- PD
Parkinson’s Disease
- PKA
protein kinase A
- PPR
paired-pulse ratio
- SEM
standard error of mean
- SN
synaptoneurosome
- Snca+/+
α-synuclein control
- Snca-/-
α-synuclein “knock-out”
- Tg
transgenic
- TH
tyrosine-hydroxylase
- THwt
normal human α-synuclein in TH-containing neurons
- THmutA53T
mutant A53T human α-synuclein in TH-containing neurons
- WT
wildtype
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abeliovich A, Schmitz Y, Farinas I, Choi-Lundberg D, Ho WH, Castillo PE, Shinsky N, Verdugo JM, Armanini M, Ryan A, Hynes M, Phillips H, Sulzer D, Rosenthal A. Mice lacking alpha-synuclein display functional deficits in the nigrostriatal dopamine system. Neuron. 2000;25:239–252. doi: 10.1016/s0896-6273(00)80886-7. [DOI] [PubMed] [Google Scholar]
- Ade KK, Lovinger DM. Anandamide regulates postnatal development of long-term synaptic plasticity in the rat dorsolateral striatum. J Neurosci. 2007;27:2403–2409. doi: 10.1523/JNEUROSCI.2916-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akopian G, Musleh W, Smith R, Walsh JP. Functional state of corticostriatal synapses determines their expression of short- and long-term plasticity. Synapse. 2000;38:271–280. doi: 10.1002/1098-2396(20001201)38:3<271::AID-SYN6>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- Bamford NS, Zhang H, Schmitz Y, Wu N-P, Cepeda C, Levine MS, Schmauss C, Zakharenko SS, Zablow L, Sulzer D. Heterosynaptic dopamine neurotransmission selects sets of corticostriatal terminals. Neuron. 2004;42:653–663. doi: 10.1016/s0896-6273(04)00265-x. [DOI] [PubMed] [Google Scholar]
- Berreta N, Nisticò, Bernardi G, Mercuri NB. Synaptic plasticity in the basal ganglia: A similar code for physiological and pathological conditions. Prog Neurobiol. 2008;84:343–362. doi: 10.1016/j.pneurobio.2007.12.004. [DOI] [PubMed] [Google Scholar]
- Bonifati V. LRRK2 low-penetrance mutations (Gly201Ser) and risk alleles (Gly2385Arg)-linking familial and sporadic Parkinson’s disease. Neurochem Res. 2007;32:1700–1708. doi: 10.1007/s11064-007-9324-y. [DOI] [PubMed] [Google Scholar]
- Braak H, Del Tredici K, Rub U, de Vos RA, Steur ENJ, Braak E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging. 2003;24:197–211. doi: 10.1016/s0197-4580(02)00065-9. [DOI] [PubMed] [Google Scholar]
- Cabin DE, Shimazu K, Murphy D, Cole NB, Gottschalk W, McIlwain KL, Orrison B, Chen A, Ellis CE, Paylor R, Lu B, Nussbaum RL. Synaptic vesicle depletion correlates with attenuated synaptic responses to prolonged repetitive stimulation in mice lacking α-synuclein. J Neurosci. 2002;22:8797–8807. doi: 10.1523/JNEUROSCI.22-20-08797.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabresi P, Maj R, Pisani A, Mercuri NB, Bernardi G. Long-term synaptic depression in the striatum: physiological and pharmacological characterization. J Neurosci. 1992a;12:4224–4233. doi: 10.1523/JNEUROSCI.12-11-04224.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabresi P, Pisani A, Mercuri NB, Bernardi G. Long-term potentiation in the striatum is unmasked by removing the voltage-dependent magnesium block of the NMDA receptor channels. Eur J Neurosci. 1992b;4:929–935. doi: 10.1111/j.1460-9568.1992.tb00119.x. [DOI] [PubMed] [Google Scholar]
- Castillo PE, Schoch S, Schmitz F, Südhof TC, Malenka RC. RIM1alpha is required for presynaptic long-term potentiation. Nature. 2002;415:327–330. doi: 10.1038/415327a. [DOI] [PubMed] [Google Scholar]
- Cepeda C, André VM, Yamazaki I, Wu N, Kleiman-Weiner M, Levine MS. Differential electrophysiological properties of dopamine D1 and D2 receptor-containing striatal medium-sized spiny neurons. Eur J Neurosci. 2008;27:671–82. doi: 10.1111/j.1460-9568.2008.06038.x. [DOI] [PubMed] [Google Scholar]
- Chandra S, Fornai F, Kwon H-B, Yazdani U, Atasoy D, Liu X, Hammer RE, Battaglia G, German DC, Castillo PE, Südhof TC. Double-knockout mice for α- and β-synucleins: Effect on synaptic functions. Proc Natl Acad Sci. 2004;101:14966–14971. doi: 10.1073/pnas.0406283101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi S, Lovinger DM. Decreased probability of neurotransmitter release underlies striatal long-term depression and postnatal development of corticostriatal synapse. Proc Natl Acad Sci USA. 1997;94:2665–2670. doi: 10.1073/pnas.94.6.2665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chotiner JK, Khorasani H, Nairn AC, O’Dell TJ, Watson JB. Adenylyl cyclase-dependent form of chemical long-term potentiation triggers translational regulation at the elongation step. Neuroscience. 2003;116:743–752. doi: 10.1016/s0306-4522(02)00797-2. [DOI] [PubMed] [Google Scholar]
- Clayton DF, George JM. The synucleins: a family of proteins involved in synaptic function, plasticity, neurodegeneration, and disease. Trends Neurosci. 1998;21:249–254. doi: 10.1016/s0166-2236(97)01213-7. [DOI] [PubMed] [Google Scholar]
- Colwell CS, Levine MS. Excitatory synaptic transmission in neostriatal neurons: Regulation by cAMP-dependent mechanisms. J Neurosci. 1995;15:1704–1713. doi: 10.1523/JNEUROSCI.15-03-01704.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dauer W, Przedborski S. Parkinson’s disease: mechanisms and models. Neuron. 2003;39:889–909. doi: 10.1016/s0896-6273(03)00568-3. [DOI] [PubMed] [Google Scholar]
- Dube L, Smith AD, Bolam JP. Identification of synaptic terminals of thalamic or cortical origin in contact with distinct medium-size spiny neurons in the rat neostriatum. J Comp Neurol. 1988;267:455–471. doi: 10.1002/cne.902670402. [DOI] [PubMed] [Google Scholar]
- Farrer M, Maraganore DM, Lockhart P, Singleton A, Lesnick TG, de Andrade M, West A, de Silva R, Hardy J, Hernandez D. α-Synuclein gene haplotypes are associated with Parkinson’s disease. Hum Mol Genet. 2001;10:1847–1851. doi: 10.1093/hmg/10.17.1847. [DOI] [PubMed] [Google Scholar]
- Fernagut PO, Hutson CB, Fleming SB, Tetreaut NA, Salcedo J, Masliah E, Chesselet MF. Behavioral and histopathological consequences of paraquat intoxication in mice: Effects of α-synuclein over-expression. Synapse. 2007;61:991–1001. doi: 10.1002/syn.20456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fino E, Glowinski J, Venance L. Bidirectional activity-dependence at corticostriatal synapses. J Neurosci. 2005;25:11279–11287. doi: 10.1523/JNEUROSCI.4476-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher RS, Levine MS, Sibley DR, Ariano MA. D2 dopamine receptor protein location: Golgi impregnation-gold toned and ultrastructural analysis of the rat neostriatum. J Neurosci Res. 1994;38:551–564. doi: 10.1002/jnr.490380508. [DOI] [PubMed] [Google Scholar]
- Fleming SM, Salcedo J, Fernagut PO, Rockenstein E, Masliah E, Levine MS, Chesselet MF. Early and progressive sensorimotor anomalies in mice overexpressing wild-type human α-synuclein. J Neurosci. 2004;24:9434–9440. doi: 10.1523/JNEUROSCI.3080-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming SM, Salcedo J, Hutson CB, Rockenstein E, Masliah E, Levine MS, Chesselet MF. Behavioral effects of dopaminergic agonists in transgenic mice overexpressing human wildtype α-synuclein. Neuroscience. 2006;142:1245–1253. doi: 10.1016/j.neuroscience.2006.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Junco-Clemente P, Linares-Clemente P, Fernández-Chacón R. Active zones for presynaptic plasticity in the brain. Molecular Psychiatry. 2005;10:185–200. doi: 10.1038/sj.mp.4001628. [DOI] [PubMed] [Google Scholar]
- Geracitano R, Paolucci E, Prisco S, Guatteo E, Zona C, Longone P, Ammassari-Teule M, Bernardi G, Berretta N, Mercuri NB. Altered long-term corticostriatal synaptic plasticity in transgenic mice overexpressing human Cu/Zn superoxide dismutase (Gly93→Ala) mutation. Neuroscience. 2003;118:399–408. doi: 10.1016/s0306-4522(02)00809-6. [DOI] [PubMed] [Google Scholar]
- Giasson BI, Covy JP, Bonini NM, Hurtig HI, Farrer MJ, Trojanowski JQ, Van Deerlin VM. Biochemical and pathological characterization of Lrrk2. Ann Neurol. 2006;59:315–322. doi: 10.1002/ana.20791. [DOI] [PubMed] [Google Scholar]
- Goldberg MS, Pisani A, Haburcak M, Vortherms TA, Kitada T, Costa C, Tong Y, Martella G, Tscherter A, Martins A, Bernardi G, Roth BI, Pothos EN, Calabresi P, Shen J. Dopaminergic deficits report and hypokinesia caused by inactivation of the familial Parkinsonism-linked gene DJ-1. Neuron. 2005;45:489–496. doi: 10.1016/j.neuron.2005.01.041. [DOI] [PubMed] [Google Scholar]
- Graybiel AM, Ragsdale CW, Jr, Yoneoka ES, Elde RP. An immunohistochemical study of enkephalins and other neuropeptides in the striatum of the cat with evidence that the opiate peptides are arranged to form mosaic patterns in register with the striosomal compartments visible by acetylcholinesterase staining neurons and their striatal targets. Neuroscience. 1981;6:377–397. doi: 10.1016/0306-4522(81)90131-7. [DOI] [PubMed] [Google Scholar]
- Graybiel AM, Aosaki T, Flaherty A, Kimura M. The basal ganglia and adaptive motor control. Science. 1994;265:826–31. doi: 10.1126/science.8091209. [DOI] [PubMed] [Google Scholar]
- Gureviciene I, Gurevicius K, Tanila H. Role of α-synuclein in synaptic glutamate release. Neurobiol Dis. 2007;28:83–89. doi: 10.1016/j.nbd.2007.06.016. [DOI] [PubMed] [Google Scholar]
- Haber SN. Review The primate basal ganglia: parallel and integrative networks. JCN. 2003;26:317–330. doi: 10.1016/j.jchemneu.2003.10.003. [DOI] [PubMed] [Google Scholar]
- Holzmann C, Kruger R, Saecker AM, Schmitt I, Schols L, Berger K, Riess O. Polymorphisms of the α-synuclein promoter: Expression analyses and association studies in Parkinson’s disease. J Neural Transm. 2003;110:67–76. doi: 10.1007/s00702-002-0769-5. [DOI] [PubMed] [Google Scholar]
- Huang YY, Kandel ER. Postsynaptic induction and PKA-dependent expression of LTP in the lateral amygdala. Neuron. 1998;21:169–178. doi: 10.1016/s0896-6273(00)80524-3. [DOI] [PubMed] [Google Scholar]
- Hsu LJ, Mallory M, Xia Y, Veinbergs I, Hashimoto M, Yoshimoto M, Thal LJ, Saitoh T, Masliah E. Expression patters of synucleins (non-Abeta component of Alzheimer’s disease amyloid precursors protein/alpha-synuclein) during murine brain development. J Neurochem. 1998;71:338–344. doi: 10.1046/j.1471-4159.1998.71010338.x. [DOI] [PubMed] [Google Scholar]
- Johnson MW, Chotiner JK, Watson JB. Isolation and characterization of synaptoneurosomes from single rat hippocampal slices. J Neurosci Methods. 1997;77:151–156. doi: 10.1016/s0165-0270(97)00120-9. [DOI] [PubMed] [Google Scholar]
- Kim SJ, Kima SY, Naa Y-S, Leeb HJ, Chung KC, Baika J-H. α-Synuclein enhances dopamine D2 receptor signaling. Brain Res. 2006;1124:5–9. doi: 10.1016/j.brainres.2006.09.079. [DOI] [PubMed] [Google Scholar]
- Klapstein GJ, Fisher RS, Zanjani H, Cepeda C, Jokel ES, Chesselet M-F, Levine MS. Electrophysiological and morphological changes in striatal spiny neurons in R6/2 Huntington’s Disease transgenic mice. J Neurophysiol. 2001;86:2667–2677. doi: 10.1152/jn.2001.86.6.2667. [DOI] [PubMed] [Google Scholar]
- Kreitzer AC, Malenka RC. Endocannabinoid-mediated rescue of striatal LTD and motor deficits in Parkinson’s disease models. Nature. 2007;445:643–647. doi: 10.1038/nature05506. [DOI] [PubMed] [Google Scholar]
- Kruger R, Kuhn W, Muller T, Woitalla D, Graeber M, Kosel S, Przuntek H, Epplen JT, Schols L, Riess O. Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson’s disease. Nat Genet. 1998;18:106–108. doi: 10.1038/ng0298-106. [DOI] [PubMed] [Google Scholar]
- Larsen KE, Schmitz Y, Troyer MD, Mosharov E, Dietrich P, Quazi AZ, Savalle M, Nemani V, Chaudhry FA, Edwards RH, Stefanis L, Sulzer D. α-synuclein overexpression in PC12 and chromaffin cells impairs catecholamine release by interfering with a late step in exocytosis. J Neurosci. 2006;26:11915–11922. doi: 10.1523/JNEUROSCI.3821-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linden DJ, Ahn S. Activation of presynaptic cAMP-dependent protein kinase is required for induction of cerebellar long-term potentiation. J Neurosci. 1999;19:10221–10227. doi: 10.1523/JNEUROSCI.19-23-10221.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Ninan I, Antonova I, Battaglia F, Trinchese F, Narasanna A, Kolodilov N, Dauer W, Hawkins RD, Arancio O. Alpha-synuclein produces a long-lasting increase in neurotransmitter release. EMBO J. 2004;23:4506–4516. doi: 10.1038/sj.emboj.7600451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Fa M, Ninan I, Trinchese F, Dauer W, Arancio O. α-Synuclein involvement in hippocampal synaptic plasticity: role of NO, cGMP, cGK and CaMKII. Eur J Neurosci. 2007;25:3583–3596. doi: 10.1111/j.1460-9568.2007.05569.x. [DOI] [PubMed] [Google Scholar]
- Lobo MK, Cui Y, Ostlund SB, Balleine BW, Yang XW. Genetic control of instrumental conditioning by striatopallidal neuron-specific S1P receptor Gpr6. Nat Neurosci. 2007;10:1395–1397. doi: 10.1038/nn1987. [DOI] [PubMed] [Google Scholar]
- Lonart G, Schoch S, Kaeser PS, Larkin CJ, Sudhof TC, Linden DJ. Phosphorylation of RIM1alpha by PKA triggers presynaptic long-term potentiation at cerebellar parallel fiber synapses. Cell. 2003;115:49–60. doi: 10.1016/s0092-8674(03)00727-x. [DOI] [PubMed] [Google Scholar]
- Lovinger DM, Tyler EC, Merritt A. Short- and long-term synaptic depression in rat neostriatum. J Neurophysiol. 1993;70:1937–1949. doi: 10.1152/jn.1993.70.5.1937. [DOI] [PubMed] [Google Scholar]
- Mahon S, Deniau JM, Charpier S. Corticostriatal plasticity: life after the depression. Trends Neurosci. 2004;27:460–467. doi: 10.1016/j.tins.2004.06.010. [DOI] [PubMed] [Google Scholar]
- Makhinson M, Chotiner JK, Watson JB, O’Dell TJ. Adenylyl cyclase activation modulates activity-dependent changes in synaptic strength and Ca2+/calmodulin-dependent kinase II autophosphorylation. J Neurosci. 1999;19:2500–2510. doi: 10.1523/JNEUROSCI.19-07-02500.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malinow R, Malenka RC. AMPA receptor trafficking and synaptic plasticity. Annu Rev Neurosci. 2002;25:103–126. doi: 10.1146/annurev.neuro.25.112701.142758. [DOI] [PubMed] [Google Scholar]
- Maroteaux L, Scheller RH. The rat brain synucleins; family of proteins transiently associated with neuronal membrane. Brain Res Mol Brain Res. 1991;11:335–343. doi: 10.1016/0169-328x(91)90043-w. [DOI] [PubMed] [Google Scholar]
- Matsuoka Y, Villa M, Lincoln S, McCormack A, Picciano M, LaFrancois J, Yu X, Dickson D, Langston WJ, McGowan E, Farrer M, Hardy J, Duff K, Przedborski S, Di Monte DA. Lack of nigral pathology in transgenic mice expressing human α-synuclein driven by the tyrosine hydroxylase promoter. Neurobiol Dis. 2001;8:535–539. doi: 10.1006/nbdi.2001.0392. [DOI] [PubMed] [Google Scholar]
- Murphy DD, Rueter SM, Trojanowski JQ, Lee VM. Synucleins are developmentally expressed and α-synuclein regulates the size of the presynaptic vesicular pool in primary hippocampal neurons. J Neurosci. 2000;20:3214–3220. doi: 10.1523/JNEUROSCI.20-09-03214.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paisan-Ruiz C, Jain S, Evans EW, Gilks WP, Simon J, van der Brug M, Lopez de Munain A, Aparicio S, Gil AM, Khan N, Johnson J, Martinez JR, Nicholl D, Carrera IM, Pena AS, de Silva R, Lees A, MartiMasso JF, Perez-Tur J, Wood NW, Singleton AB. Cloning of the gene containing mutations that cause PARK8-linked Parkinson’s disease. Neuron. 2004;44:595–600. doi: 10.1016/j.neuron.2004.10.023. [DOI] [PubMed] [Google Scholar]
- Pals P, Lincoln S, Manning J, Heckman M, Skipper L, Hulihan M, Van den Broeck M, De Pooter T, Cras P, Crook J, Van Broeckhoven C, Farrer MJ. α-Synuclein promoter confers susceptibility to Parkinson’s disease. Ann Neurol. 2004;56:591–595. doi: 10.1002/ana.20268. [DOI] [PubMed] [Google Scholar]
- Partridge JG, Tang KC, Lovinger DM. Regional and postnatal heterogeneity of activity-dependent long-term changes in synaptic efficacy in the dorsal striatum. J Neurophysiol. 2000;84:1422–1429. doi: 10.1152/jn.2000.84.3.1422. [DOI] [PubMed] [Google Scholar]
- Picconi B, Pisani A, Barone I, Bonsi P, Centonze D, Bernardi G, Calabresi P. Review Pathological synaptic plasticity in the striatum: Implications for Parkinson’s Disease. NeuroToxicology. 2005;26:779–783. doi: 10.1016/j.neuro.2005.02.002. [DOI] [PubMed] [Google Scholar]
- Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, Di Iorio G, Golbe LI, Nussbaum RL. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science. 1997;276:2045–2047. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- Rockenstein E, Mallory M, Hashimoto M, Song D, Shults CW, Lang I, Masliah E. Differential neuropathological alterations in transgenic mice expressing alpha-synuclein from the platelet-derived growth factor and Thy-1 promoters. J Neurosci Res. 2002;68:568–578. doi: 10.1002/jnr.10231. [DOI] [PubMed] [Google Scholar]
- Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, Hulihan M, Peuralinna T, Dutra A, Nussbaum R, Lincoln S, Crawley A, Hanson M, Maraganore D, Adler C, Cookson MR, Muenter M, Baptista M, Miller D, Blancato J, et al. Alpha-synuclein locus triplication causes Parkinson’s disease. Science. 2003;302:841. doi: 10.1126/science.1090278. [DOI] [PubMed] [Google Scholar]
- Smith R, Musleh W, Akopian G, Buckwalter G, Walsh JP. Regional differences in the expression of corticostriatal synaptic plasticity. Neuroscience. 2001;106:95–101. doi: 10.1016/s0306-4522(01)00260-3. [DOI] [PubMed] [Google Scholar]
- Spencer JP, Murphy KPSJ. Bi-directional changes in synaptic plasticity induced at corticostriatal synapses in vitro. Exp Brain Res. 2000;135:497–503. doi: 10.1007/s002210000523. [DOI] [PubMed] [Google Scholar]
- Spencer JP, Murphy KPSJ. Activation of cyclic AMP-dependent protein kinase is required for long-term enhancement at corticostriatal synapses in rats. Neurosci Lett. 2002;329:217–221. doi: 10.1016/s0304-3940(02)00659-6. [DOI] [PubMed] [Google Scholar]
- Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. α-Synuclein in Lewy bodies. Nature. 1997;388:839–840. doi: 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- Steidl JV, Gomez-Isla T, Mariash A, Ashe KH, Boland LM. Altered short-term hippocampal synaptic plasticity in mutant α-synuclein transgenic mice. NeuroReport. 2003;14:219–223. doi: 10.1097/00001756-200302100-00012. [DOI] [PubMed] [Google Scholar]
- Stevens CF. Neurotransmitter release at central synapses. Neuron. 2003;40:381–388. doi: 10.1016/s0896-6273(03)00643-3. [DOI] [PubMed] [Google Scholar]
- Walsh JP. Depression of excitatory input in rat striatal neurons. Brain Res. 1993;608:123–128. doi: 10.1016/0006-8993(93)90782-i. [DOI] [PubMed] [Google Scholar]
- Wang JH, Kelly PT. Attenuation of paired-pulse facilitation associated with synaptic potentiation mediated by postsynaptic mechanisms. J Neurophysiol. 1997;78:2707–2716. doi: 10.1152/jn.1997.78.5.2707. [DOI] [PubMed] [Google Scholar]
- Wang Z, Kai L, Day M, Ronesi J, Yin HH, Ding J, Tkatch T, Lovinger DM, Surmeier DJ. Dopaminergic control of corticostriatal long-term synaptic depression in medium spiny neurons is mediated by cholinergic interneurons. Neuron. 2006;50:443–452. doi: 10.1016/j.neuron.2006.04.010. [DOI] [PubMed] [Google Scholar]
- Watabe AM, Zaki PA, O’Dell TJ. Coactivation of β-adrenergic and cholinergic receptors enhances the induction of long-term potentiation and synergistically activates mitogen-activated protein kinase in the hippocampal CA1 region. J Neurosci. 2000;20:5924–5931. doi: 10.1523/JNEUROSCI.20-16-05924.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson JB, Khorasani H, Persson A, Huang K-P, Huang FI, O’Dell TJ. Age-related deficits in long-term potentiation are insensitive to hydrogen peroxide: coincidence with enhanced autophosphorylation of Ca2+/calmodulin-dependent protein kinase II. J Neurosci Res. 2002;70:298–308. doi: 10.1002/jnr.10427. [DOI] [PubMed] [Google Scholar]
- Watson JB, Arnold MM, Ho Y-S, O’Dell TJ. Age-dependent modulation of hippocampal long-term potentiation by antioxidant enzymes. J Neurosci Res. 2006;84:1564–1574. doi: 10.1002/jnr.21040. [DOI] [PubMed] [Google Scholar]
- Weisskopf MG, Castillo PE, Zalutsky RA, Nicholl RA. Mediation of hippocampal mossy fiber long-term potentiation by cyclic AMP. Science. 1994;265:1878–1882. doi: 10.1126/science.7916482. [DOI] [PubMed] [Google Scholar]
- Wickens JR, Begg AJ, Arbuthnott GW. Dopamine reverses the depression of rat corticostriatal synapses which normally follows high-frequency stimulation of cortex in vitro. Neuroscience. 1996;70:1–5. doi: 10.1016/0306-4522(95)00436-m. [DOI] [PubMed] [Google Scholar]
- Wilson CJ. Morphology and synaptic connections of crossed corticostriatal neurons in the rat. J Comp Neurol. 1987;263:567–580. doi: 10.1002/cne.902630408. [DOI] [PubMed] [Google Scholar]
- Wu N-P, Cepeda C, Masliah E, Levine MS. Abnormal glutamate and dopamine receptor function in the striatum of a-synuclein overexpressing mice. Soc Neurosci Abs 85.12 2005 [Google Scholar]
- Yavich L, Tanila H, Vepsalainen S, Jakala P. Role of α-synuclein in presynaptic dopamine recruitment. J Neurosci. 2004;24:11165–11170. doi: 10.1523/JNEUROSCI.2559-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yavich L, Oksman M, Tanila H, Kerokoski P, Hiltunen M, van Groen T, Puolivali J, Mannisto PT, Garcia-Horsman A, MacDonald E, Beyreuther K, Hartmann T, Jakala P. Locomotor activity and evoked dopamine release are reduced in mice overexpressing A30P-mutated human alpha-synuclein. Neurobiol Dis. 2005;20:303–313. doi: 10.1016/j.nbd.2005.03.010. [DOI] [PubMed] [Google Scholar]
- Zalutsky RA, Nicoll RA. Comparison of two forms of long-term potentiation in single hippocampal neurons. Science. 1990;248:1619–1624. doi: 10.1126/science.2114039. [DOI] [PubMed] [Google Scholar]
- Zarranz JJ, Alegre J, Gomez-Esteban JC, Lezcano E, Ros R, Ampuero I, Vidal L, Hoenicka J, Rodriguez O, Atares B, Llorens V, Gomez Tortosa E, del Ser T, Munoz DG, de Yebenes JG. The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann Neurol. 2004;55:164–173. doi: 10.1002/ana.10795. [DOI] [PubMed] [Google Scholar]
- Zimprich A, Biskup S, Leitner P, Lichtner P, Farrer M, Lincoln S, Kachergus J, Hulihan M, Uitti RJ, Calne DB, Stoessl AJ, Pfeiffer RF, Patenge N, Carbajal IC, Vieregge P, Asmus F, Muller-Myhsok B, Dickson DW, Meitinger T, Strom TM, Wszolek ZK, Gasser T. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron. 2004;44:601–607. doi: 10.1016/j.neuron.2004.11.005. [DOI] [PubMed] [Google Scholar]
- Zucker RS, Regehr WG. Short-term synaptic plasticity. Annu Rev Physiol. 2002;64:355–405. doi: 10.1146/annurev.physiol.64.092501.114547. [DOI] [PubMed] [Google Scholar]