

SUMMARY
Tyrosine sulfation of the chemokine receptor CXCR4 enhances its interaction with the chemokine SDF-1α. Given similar post-translational modification of other receptors, including CCR5, CX3CR1 and CCR2b, tyrosine sulfation may be of universal importance in chemokine signaling. N-terminal domains from 7-transmembrane chemokine receptors have been employed for structural studies of chemokine-receptor interactions, but never in the context of proper post-translational modifications known to affect function. A CXCR4 peptide modified at position 21 by expressed tyrosylprotein sulfotransferase-1 and unmodified peptide are both disordered in solution, but bind SDF-1α with low micromolar affinities. NMR and fluorescence polarization measurements showed that the CXCR4 peptide stabilizes dimeric SDF-1α, and that sulfotyrosine 21 binds a specific site on the chemokine that includes arginine 47. We conclude that SDF-1α dimer preferentially interacts with receptor peptide, and residues beyond the extreme N-terminal region of CXCR4, including sulfotyrosine 21, make specific contacts with the chemokine ligand.
Keywords: GPCR, tyrosine sulfonation, NMR, post-translational modification, HIV coreceptor
INTRODUCTION
The CXCR4 receptor and its chemokine ligand SDF-1α (CXCL12) are crucial for embryonic development, and have been implicated in various pathologic conditions, including cancer metastasis and HIV-1 infection. Unlike other chemokine/receptor pairs, deletion of the genes for SDF-1α or CXCR4 in mice is absolutely lethal in the perinatal period.1-3 The SDF-1α-/- or CXCR4-/- phenotypes are characterized by abnormal B-cell lymphopoiesis, bone marrow myelopoiesis and vascularization of the gut, and by defects in cerebellar and cardiac ventricular septum formation.1-3 Furthermore, injured cardiac and central nervous system tissues produce SDF-1α and can recruit CXCR4-expressing stem cells derived from bone marrow or neuronal stem cells, respectively, that are thought to produce regenerative signals at the site of tissue damage.4-6 Cancer progression appears to be similarly dependent on SDF-1α/CXCR4 signaling 7. Increased CXCR4 expression in metastatic breast cancer cells causes them to seed secondary tumors by migrating into tissues and organs that express SDF-1α constitutively,8; 9 and other types of cancer likely exploit the same mechanism.10-15
CXCR4 is also the co-receptor for cellular entry of X4 tropic HIV-1, and SDF-1α has been shown to inhibit infection of host cells through binding to CXCR4. Another chemokine receptor CCR5 is the analogous co-receptor for R5 tropic HIV-1 strains. X4 tropic or dual R5X4 tropic HIV-1 stains are rarely transmitted; however, emergence of these highly pathogenic strains correlates with the onset of immunodeficiency and depletion of CD4+ T-helper cells. Interestingly, a CXCR4 Y21A mutant is a less effective co-receptor for a X4 (III-B) or a dual R5X4 tropic (89.6) HIV-1 strain.16 Systematic mutagenesis of CXCR4 residues to alanine in the N-terminus correspondingly identified Y21 as a key residue for high affinity SDF-1α binding16; 17
CXCR4 is subject to several post-translational modifications, in particular tyrosine sulfation, that are important for its interaction with SDF-1α.18; 19 Three potential sites of tyrosine sulfation are present in the CXCR4 N-terminal domain (tyrosines 7, 12, and 21, Figure 1). Metabolic labeling studies with CXCR4 mutants suggest that a sulfotyrosine at position 21 is involved in the interaction with SDF-1α.18; 20 Tyrosine sulfation, catalyzed by tyrosylprotein sulfotransferase (TPST) enzymes in the trans-Golgi network, is also important for the interactions of CCR2b, CCR5 and CX3CR1 with their respective chemokine ligands.21-23 Activation of the G protein-coupled receptor CXCR4 by SDF-1α is hypothesized to occur through a “two-step / two-site” mechanism.24 According to this model, the core of the chemokine initially binds to the N-terminus of the receptor. Subsequently, the N-terminus of the chemokine interacts with a secondary site on the receptor, which leads to high affinity binding and receptor activation.24 However, a detailed structural understanding of SDF-1α/CXCR4 interactions is lacking, especially the role of sulfotyrosine recognition.
Figure 1.
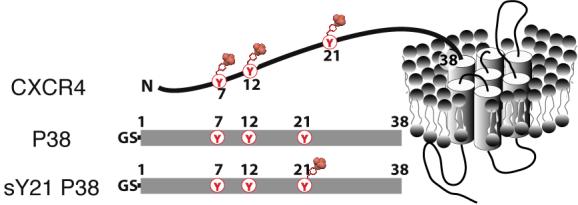
Sulfotyrosines in the N-terminal domain of CXCR4. Three tyrosine residues in the N-terminus of CXCR4 are potential modification sites. Unmodified P38 peptide [Gly-Ser CXCR4(1-38) C28A] and sulfotyrosine-containing sY21 P38 are shown schematically.
Here, we investigated the interaction of SDF-1α with a peptide corresponding to residues 1-38 of CXCR4, which comprises its predicted extracellular N-terminal region. Previously, we have shown that SDF-1α exists in a salt- and pH-dependent monomer-dimer equilibrium that is shifted toward dimer in the presence of glycosaminoglycans (GAGs) such as heparin.25 Our results show that the N-terminal CXCR4 peptide shifts the SDF-1α monomer-dimer equilibrium toward dimeric SDF-1α. To investigate the role of Y21 sulfation in the interaction of CXCR4 with SDF-1α, we prepared the corresponding sulfotyrosine peptide by enzymatic sulfation using recombinant TPST-1.20 Using NMR we found a putative binding site for sulfotyrosine 21 in the vicinity of SDF-1α residue R47. These findings provide a structural basis for understanding the role of tyrosine sulfation in chemokine receptor interaction that may ultimately enable new strategies for pharmacological intervention in disease conditions involving chemokine receptor systems.
RESULTS
SDF-1α recognition by residues of the CXCR4 N-terminal tail
In order to identify specific interactions between CXCR4 and SDF-1α we produced a peptide corresponding to the predicted extracellular N-terminal domain of CXCR4 (residues 1-38) as a recombinant fusion protein (Figure 1). Residue C28 was replaced with alanine to prevent oxidative peptide dimer formation and an N-terminal Gly-Ser dipeptide remained after proteolysis of the fusion protein. We assigned the 1H, 15N and 13C chemical shifts of GS-CXCR4(1-38) C28A (P38) using [U-15N, 13C] peptide and standard 3D NMR methods. It was apparent from 15N-1H heteronuclear NOE values (which ranged from -3 to 0; data not shown) and the absence of medium- or long-range 1H-1H NOEs that free P38 is dynamically disordered with no stable secondary or tertiary structure.
We monitored 15N/1H HSQC spectra of 15N/13C P38 with incremental additions of unlabeled SDF-1α to identify CXCR4 residues involved in chemokine binding. Backbone amide 1H/15N signals are very sensitive to changes in the local environment, and binding to SDF-1α should induce chemical shift perturbations in the spectrum of P38. Combined 1H/15N shift perturbations were mapped onto the P38 sequence (Figure 2(a)). In agreement with previous mutagenesis investigations, the effects of SDF-1α binding are concentrated in the N-terminal half of P38 (residues 3-18),16; 17 with the largest shifts observed for residues 5-7; however Y21, K25, E26, and R30 also display significant perturbations.
Figure 2.

Characterization of sY21 P38 by NMR. (a) The sequence of P38 with combined 1H/15N chemical shift perturbations from titration with SDF-1α as indicated: red, > 0.5 ppm; purple, 0.5-0.3 ppm; green, 0.3-0.2 ppm; and blue, 0.2-0.15 ppm. HSQC spectra of 15N P38 (250 μM in 20 mM MES at pH 6.8) were monitored during the course of a titration with SDF-1α to a final ratio of 1:1. Extensive line broadening was observed at higher SDF-1α concentrations. (b) Aromatic 13C-1H HSQC spectrum of P38 (left) and sY21 P38 (right). Assignments for 1Hδ-13Cδ and 1Hε-13Cε cross peaks are indicated, and peaks corresponding to sY21 are highlighted in red. No changes in the aromatic 1H and 13C shifts are observed for Y12 and Y7. (c) Assigned portion of a 15N-1H HSQC spectrum of P38 (black contours) overlaid with the spectrum of sY21 P38 (red contours) showing large chemical shift differences for Y21 and D22 upon sulfation of Y21, while Y7 and Y12 are unperturbed. (d) Overlaid portions of 15N-1H HSQC spectra of sY21 P38 with 0 (black), 0.25 (gray), 0.5 (light red) and 1 (red) equivalents of SDF-1α. As with P38 N-terminal residues, like I4 and I6, showed extensive line broadening at and above 1:1 peptide to SDF-1α. (e) The sequence of sY21 P38 with chemical shift perturbations from titration with SDF-1α illustrated as in panel (b). Residues of sY21 P38 that display larger shift perturbations than P38 upon SDF-1α binding are boxed.
On the basis of our NMR data and studies suggesting that sulfation of Y21 enhances the affinity of CXCR4 for SDF-1α,18 we sought to address the physiological importance of Y21 sulfation by investigating its structural role in the SDF-1α/CXCR4 complex. CXCR4 sulfation at Y21 could enhance SDF-1α binding by stabilizing a specific tertiary structure in the N-terminal extracellular domain with higher affinity for the chemokine. Alternatively, the sulfate modification may promote binding through a direct interaction at a specific site on SDF-1α.
Enzymatic sulfation of Y21
To enable the structural analysis of Y21 sulfation, we used recombinant TPST-1 to generate milligram quantities of unlabeled, [U-15N] and [U-15N/13C] P38 containing sulfotyrosine at position 21 (sY21) by methods described previously for sulfation of a CCR5 N-terminal peptide.20 Using proteolytic digestion and mass spectrometry we demonstrated that Y21 was sulfated but not Y12 or Y7 (data not shown). Incorporation of a sulfonate should alter chemical shifts in the aromatic ring, and we observed large changes in the 1Hα, 13Cα, 1Hβand 13C’ resonances of Y21 in 13C-1H HSQC spectra of P38 and sY21 P38 (Figure 2(b)). Chemical shifts of Y7 and Y12 were unperturbed, thus verifying the proteolytic mass spectrometry results.
Impact of Y21 sulfation on P38 structure
To determine whether sY21 P38 adopts a conformation distinct from unmodified P38, we compared the 15N-1H HSQC spectra of P38 and sY21 P38 (Figure 2(c)). If the global structure of P38 is altered by sulfation, substantial shift differences should be observed in the 15N-1H HSQC spectrum for residues distant from Y21. However, only residues Y21 and D22 were perturbed, corresponding to the two backbone amides nearest to the modified side chain. As with the unstructured P38, we examined 3D 15N- and 13C-edited NOESY spectra for evidence of stable secondary or tertiary structure in sY21 P38, but only weak intraresidue or sequential NOEs were observed. Also, 15N-1H heteronuclear NOE values (which ranged from -3 to 0; data not shown) did not differ from those of P38 suggesting sY21 P38 is also dynamically disordered. Hence, at least in the context of a soluble CXCR4 fragment, sulfation of Y21 does not enhance the interaction with SDF-1α by stabilizing a specific receptor conformation.
An HSQC titration of [U-15N/13C] sY21 P38 with unlabeled SDF-1α also exhibited chemical shift perturbations (Figure 2(d)) and revealed a pattern of shift perturbations (Figure 2(e)) similar to that obtained with unsulfated P38 (Figure 2(a)). The only notable differences involved residues adjacent to sY21 (D20, D22 and S23). Increased shift perturbations in the vicinity of sY21 may be a sign that sulfation strengthens the interaction between that portion of CXCR4 and a specific sulfotyrosine binding site on the surface of SDF-1α. To locate the sY21 binding site, we first examined the interaction of unlabeled P38 and [U-15N/13C] SDF-1α by NMR.
Characterization of SDF-1α binding to P38
From a series of HSQC spectra for [U-15N/13C]-SDF-1α with increasing amounts of P38 (Figure 3(a)), we identified signals for many residues that shift significantly upon P38 binding and other residues that are unaffected. These perturbations result from a change in chemical environment that reflects either P38 binding near the nuclei with altered shifts, or an SDF-1α structural rearrangement. Non-linear fitting of chemical shift changes as a function of peptide concentration yielded an apparent P38/SDF-1α dissociation constant (Kd) of 4.5 ± 2.2 μM. It should also be noted that the apparent Kd value obtained in this manner may reflect contributions from coupled equilibria corresponding to P38 binding and SDF-1α dimer formation, as discussed below.
Figure 3.

P38 binds SDF-1α and promotes SDF-1α dimer formation. (a) HSQC spectra of SDF-1α (50 μM) in the absence of P38 (black contours) and after additions of 0.25, 0.5, and 0.75 equivalents (gray) or 2 equivalents of P38 (green) are overlaid. (b) Combined 1H/15N chemical shift perturbations from P38 binding plotted as a function of SDF-1α residue. Missing values correspond to prolines (2, 10, 32, and 53) or SDF-1α residues not observed at the beginning of the titration with P38 (4, 17, 26, 30 and 44). At the end of the titration, residues 58 and 61 were absent due to line broadening, but chemical shift perturbations obtained from the last observed signals are shown (gray). SDF-1α residues in the β1 strand and α helix that contribute to the SDF-1α dimer interface are shown in blue. (c) Self-association of SDF-1α observed by fluorescence polarization reveals an apparent SDF-1α dimer dissociation constant of 49 ± 15 μM in the presence of 2 mM P38 (○) and ∼5 mM in the absence of P38 (λ).
Large 1H/15N shift perturbations are distributed throughout the SDF-1α sequence (Figure 3(b)), including residues from the ‘N-loop’ (R12 and F14), β1 (23, 25, 28, and 29), β2 (37, 39, 40, and 41), β3 (48, 49, and 50), and the α-helix (57, 58, 60, 61, 62, 64, 65, and 66). Previous mutagenesis studies have identified the 12RFFESH17 sequence and the SDF-1α N-terminus as distinct functional epitopes with important roles in CXCR4 binding and activation, respectively.24 For example, a SDF-1α R12A mutant binds CXCR4 with significantly reduced affinity,26 while removal of two residues at the N-terminus converts the chemokine into a potent CXCR4 antagonist. Our results show that R12 and F14 are clearly affected by P38 binding, but the SDF-1α N-terminus does not participate in the interaction. Residues in β1 and the α-helix display large shift perturbations (Figure 3(b)), and these regions contribute to the SDF-1α dimer interface observed in two different crystal structures.26; 27 Our previous studies have shown that, while SDF-1α is predominantly monomeric in the solution conditions used here (MES buffer at pH 6.8), ligand binding can promote SDF-1α dimerization.25 For example, fluorescence polarization (FP) measurements of the SDF-1α monomer-dimer Kd showed that binding of a heparin disaccharide increases the stability of the chemokine dimer 50-fold. Consequently, we speculated that binding to the CXCR4 N-terminus could similarly influence the monomer-dimer equilibrium of SDF-1α.
SDF-1α dimer formation in the presence of P38
Equilibrium dissociation constants for the SDF-1α dimer were determined as previously described25 from FP measurements of the single SDF-1α tryptophan at protein concentrations ranging from 0.003-1 mM in the presence of 0, 1, and 2 mM P38. In the absence of P38, FP values increased only slightly at high SDF-1α concentrations, indicating that under these solution conditions (20 mM HEPES, pH 7.4) the protein is predominantly monomeric, with a dimer Kd ∼ 5 mM (Figure 3(c)). However, in the presence of 1 mM P38, the FP values were strongly dependent on SDF-1α concentration, yielding an apparent dimer Kd of 150 ± 65 μM (data not shown). A higher P38 concentration of 2 mM further stabilized the SDF-1α dimer (Kd = 49 ± 15 μM) (Figure 3(c)). The decrease in the SDF-1α dimer Kd as peptide concentration is increased is consistent with a thermodynamic linkage between the equilibria governing peptide binding and SDF-1α self-association, suggesting that the chemokine may be subject to ligand-induced dimerization.28
Evidence of P38-induced SDF-1α oligomerization prompted us to consider the stoichiometry of the complex, and we examined the NMR titration data for evidence of a specific binding model. A complex with 2:1 chemokine: peptide stoichiometry would show a more complex SDF-1α HSQC spectrum with distinct signals from each chemokine monomer as a result of the loss of dimer symmetry. However, the number of signals in the SDF-1α HSQC spectra remains constant during the titration with P38 (Figure 3(a)), consistent with the formation of a symmetric 2:2 dimer complex. In contrast, 15N-1H HSQC spectra of P38 and sY21 P38 in the presence of saturating concentrations of SDF-1α displayed extensive line broadening suggesting conformational heterogeneity. The apparent conformational heterogeneity of P38 is likely the result of peptide being bound to SDF-1α exchanging between monomer and dimer.
The FP results indicate a shift in the SDF-1α monomer-dimer equilibrium towards dimer upon addition of the CXCR4 peptide. P38-induced shift perturbations in β1 and the α-helix of SDF-1α are likely to arise from a combination of ligand binding and dimer formation. We recognize the difficulty in parsing the contributions from peptide ligand binding and dimer formation. However, shift differences due to protein-protein contacts at the SDF-1α dimer interface can be suppressed by directly comparing the results of separate titration experiments with P38 and sY21 P38. Since the peptides differ only in the addition of a single sulfonate moiety, differences in the chemical shift perturbations they induce should reveal the site of interaction of sulfotyrosine 21 with SDF-1α.
The SDF-1α binding site for CXCR4 sulfotyrosine 21
As with P38, unlabeled sY21 P38 elicited chemical shift perturbations when titrated into [U-15N/13C]-SDF-1α, and nonlinear fitting of the NMR data yielded an apparent Kd of 1.3 ± 0.5 μM (data not shown). While SDF-1α residues like E15, S16, K27, K43, or I51 displayed identical perturbations in response to P38 and sY21 P38, a subset of residues, including Q48, V49 and C50, were clearly affected differently as the result of Y21 sulfation (Figure 4(a)). To identify the SDF-1α residues most sensitive to sulfation of P38 at Y21, we calculated amide 1H/15N chemical shift differences between the extrapolated endpoints of the two peptide titrations, δ[P38:SDF-1α]-δ[sY21 P38:SDF-1α] (Figure 4(b)). Eight SDF-1α residues (F14, V18, T31, A40, L42, Q48, V49, and C50) display chemical shifts differences that appear significant (>0.25 ppm), and we mapped their locations on to the SDF-1α dimer structure (Figure 4(c)).
Figure 4.

Sulfotyrosine-induced SDF-1α chemical shift perturbations. (a) 15N-1H HSQC spectra of SDF-1α in 20 mM MES pH 6.8 in the absence (black) or presence of 2 equivalents of P38 (green) or sY21 P38 (red). SDF-1α residues E15 and S16 are among those unaffected by peptide binding, while K27, K43, and I51 shift upon peptide binding, but have the same final chemical shift irrespective of Y21 sulfation. In contrast, Q48, V49, and C50 respond differently to P38 and sY21 P38, reflected by differences in the positions of red and green cross peaks. (b) SDF-1α 1H/15N shift perturbations due to sulfation of Y21 calculated as the difference between the chemical shifts of SDF-1α bound to sY21 P38 and SDF-1α bound to P38. Residues with sulfotyrosine-induced chemical shift differences >0.25 ppm are indicated with orange bars. Basic residues in SDF-1α are indicated with blue bars. Missing values are the same as in Figure 3(c). (c) Residues with sulfotyrosine-induced chemical shift differences >0.25 ppm are highlighted on the structure of the SDF-1α dimer (PDB ID 1QG7) in orange. Nearby basic residues R47 and R41 are shown in blue.
Crystal structures of complexes involving sulfotyrosines show that in each case the sulfonate group forms an extensive hydrogen bonding network and participates in a salt bridge with a positively charged sidechain.29; 30 For example, the sulfotyrosine sulfonate in hirugen bound to α-thrombin is within 4 Å of a Lys ε-amino group,29 and sulfotyrosines 7 and 10 of P-selectin glycoprotein ligand-1 (PSGL-1) pair with a His and an Arg residue from the lectin and EGF domains, respectively, of P-selectin.30 SDF-1α residues A40 (colored, but not labeled in figure 4(c)), L42, Q48, V49 and C50 are adjacent to the basic residues R47 and R41, and F14 and V18 are also nearby (Figure 4(c)). Only T31 is outside this cluster of residues. Chemical shift perturbation data therefore implicate R47 or R41 as a potential element of a sulfotyrosine 21 binding site.
To resolve R47 or R41 as the most likely candidate for forming a salt bridge with CXCR4 sulfotyrosine 21, we surveyed 15N-1H HSQC and 13C-1H HSQC spectra for sulfotyrosine induced chemical shift differences in the side chains closest to R47 or R41 (Figure 5(a) and (b)). The side chains of H25, N46, and Q48 are all ∼ 4Å from R41, but neither the side chain NH2 or the aromatic CH resonances for those residues shift as a result of Y21 sulfation (Figure 5(a)). In contrast, aliphatic 13C-1H HSQC spectra for SDF-1α reveal clear differences in chemical shift for a CγH3 signal from V49 (Figure 5(b)) and the CδH3 signals from L42 when bound to sY21 P38 versus P38 (V49 and L42 are ∼ 4Å from R47). Hence, we conclude that the guanidinium group of R47 in SDF-1α forms a salt bridge with the sulfonate of CXCR4 sY21 (Figure 5(c)).
Figure 5.

Identifcation of a CXCR4 sulfotyrosine 21 binding site on SDF-1α. Comparisons of 13C-1H HSQC spectra of SDF-1α in 20 mM MES pH 6.8 in the absence (black) or presence of 2 equivalents of P38 (green) or sY21 P38 (red). (a) Neither the H17 nor H25 Hε1-Cε1 signals show significant sulfation-dependent changes in chemical shift. (b) Among the methyl groups shown, only V49 is noticeably different in the presence of sY21 P38 (red) relative to the P38 complex (green). (c) Positively charged R47 from SDF-1α interacts with sY21 from CXCR4. Residues with sulfation-dependent shift perturbations >0.25 ppm (Figure 4(b)) are highlighted in orange as in Figure 4(c). Sidechains surrounding R41, including H25, N46 and Q48, are unaffected by sulfation (cyan). A sidechain methyl group of V49 (magenta sphere) adjacent to R47 is sensitive to sulfation of Y21 (shown in panel b).
DISCUSSION
Sulfation of tyrosine residues has been found in an increasing number of secreted and integral membrane proteins of multicellular organisms.31 While the precise number of proteins containing sulfotyrosines is not known yet, it has been estimated that up to 1% of all tyrosines in the eukaryotic proteome may be sulfated.32 The high occurrence of this post-translational modification suggests it may play a critical role in many types of extracellular protein-protein interactions. In particular, it has been shown for a number of chemokine receptors and other GPCRs that sulfotyrosine residues are involved in receptor-ligand recognition. In addition to the CXCR4/SDF-1α system, sulfotyrosines in CCR5 are important for binding to natural CCR5 ligands and for aiding in HIV-1 co-receptor function through binding to gp120.22; 33; 34 Likewise, a sulfotyrosine in the CX3CR1 N-terminus increases affinity for its ligand fractalkine (CX3CL1).23 While the importance of tyrosine sulfation has been appreciated in these and other systems, the molecular basis for sulfotyrosine recognition is largely unknown. In this study we identify for the first time a putative sulfotyrosine binding site for a chemokine, thereby adding to the general understanding of the role of sulfotyrosines in protein-protein interactions.
Known CXCR4 post-translational modifications include tyrosine sulfation and N and O-linked gylcosylation.18; 19 Because sulfation at Y21 is thought to play an important role in SDF-1α signaling,18 our initial structural investigation focused on this modification. A recombinant peptide, enzymatically sulfonated using recombinant expressed TPST-1,20; 35 served as a model for the N-terminal extracellular domain of CXCR4. By comparing aromatic 1H/13C and amide 1H/15N chemical shifts of the modified and unmodified peptides, we confirmed that sulfation was limited to Y21 and neither Y7 nor Y12 were altered in the reaction. Since Y21 sulfation did not induce the formation of secondary or tertiary structures in the soluble CXCR4 domain that could enhance its interaction with SDF-1α, we investigated the sulfotyrosine binding site on SDF-1α by directly comparing complexes with CXCR4 peptide containing and devoid of sulfation.
Crystal structures of protein-protein complexes involving sulfotyrosine show consistent salt bridges between the sulfonate and the positively charged side chain of a His, Lys or Arg residue.29; 30 Of the 15 basic residues in SDF-1α, NMR titrations using CXCR4 peptide with and without Y21 sulfation identified R47 as the most likely salt bridge partner (Figure 5(c)). If the R47-sY21 pairing was due to a non-specific electrostatic interaction, similar ligands could be expected to bind the same location. For example, crystallization buffers for both SDF-1α X-ray structures (PDB 1QG7 and 1A15) contained high concentrations (1.5-2 M) of SO4-2. In each structure an inorganic sulfate ion is observed near residues 20-22 of the short 310 helix, but SO4-2 is absent from the putative sY21 binding site suggesting that the interaction detected by NMR represents a specific element of CXCR4-SDF-1α recognition. While we have focused on the role of sulfation at Y21 in this study, CXCR4 residues Y12 and Y7 display some of the largest chemical shift perturbations upon binding to SDF-1α (Figure 2(a) and (e)), and deletion studies showed that the first ∼10 residues of CXCR4 are essential for SDF-1α binding.17 Thus, modification of either Y7 or Y12 in vivo may further enhance chemokine binding through sulfotyrosine recognition at another site on the SDF-1α surface.
Like many chemokines, SDF-1α self-associates.25; 36 We attempted to explore the thermodynamics and stoichiometry of the SDF-1α/P38 binding equilibria by isothermal titration microcalorimetry, however, our preliminary analysis showed that simple binding models wherein one SDF-1α molecule combines with either one or two peptide ligands to form a complex were insufficient to fit the data. Furthermore, our FP results demonstrate that binding of an N-terminal peptide from CXCR4 shifts the SDF-1α monomer-dimer equilibrium towards dimer. These results indicate that a more complex model accounting for ligand induced dimer formation is required for accurate analysis of the microcalorimetry data.
The physiological relevance of the SDF-1α monomer-dimer equilibrium for CXCR4 signaling is not known. In particular, it has been shown for a number of chemokines that dimerization is not required for receptor interaction in vitro.37-40 However, dimerization of the CC chemokines RANTES, MIP-1β, and MCP-1 is crucial for their function in vivo.41 Furthermore, interactions between chemokines and extracellular matrix glycosaminoglycans (GAGs) have been shown to promote chemokine dimer formation and are essential for cell migration in vivo.41 For example, heparan sulfate binding shifts the monomer-dimer equilibria for both SDF-1α and MIP-1β toward dimer.25; 42 While others have interpreted similar chemical shift data strictly in the context of a CXCR4 peptide binding on the surface of a monomeric SDF-1α,43 our results demonstrate that extracellular CXCR4 domains also have the ability to alter the SDF-1α monomer-dimer equilibrium. Our observed in vitro equilibrium dimer dissociation constants are clearly too high for SDF-1α to exist as a dimer at physiological concentrations found in serum and extracellular fluids. However, the local SDF-1α concentrations on the cell surface are not known and it is also not clear how the interaction with GAGs and chemokine receptors might promote SDF-1α dimerization in vivo and under steady state flow conditions. Furthermore, the observation that CXCR4 exists as a constitutive dimer44 suggests that the stoichiometry of the functional chemokine-receptor complex may be other than 1:1.
In summary, we report the first heteronuclear NMR analysis of a sulfotyrosine containing protein and the recognition of this posttranslational modification by a specific binding partner, a member of the chemokine family. NMR chemical shift perturbations show that the CXCR4 peptide binds to the core domain of SDF-1α but does not interact with the chemokine N-terminus, consistent with the proposed “two-site / two-step” signaling mechanism.24 The NMR results also provide the first direct evidence that a sulfotyrosine at position 21 of CXCR4 interacts with SDF-1α at a specific site that includes R47.
METHODS
Cloning and Mutagenesis
The primers used to PCR amplify a fragment corresponding to CXCR4 residues 1-38 introduced 5′ BamHI and 3′ HindIII restriction sites for ligation into pQE30GB1.45 To avoid peptide dimerization through cysteine oxidation, C28 was replaced with alanine using the QuickChange™ kit (Stragene). Proteolytic removal of the N-terminal fusion protein yields a CXCR4 (1-38) C28A peptide (P38) that retains an N-terminal Gly-Ser dipeptide.
Protein expression and purification
E. coli strain SG13009[pRPEP4] (Qiagen) transformed with the P38 expression plasmid were grown at 37 °C in Luria-Bertani broth to a cell density of A600 = 0.6. The temperature was then reduced to 15 °C and overnight protein expression was induced by the addition of isopropyl-β-D-thiogalactopyranoside. Isotopically labeled proteins were produced using M9 medium containing 15N-NH4Cl and/or 13C-glucose as the sole nitrogen and carbon sources.
Cells were lysed using a French pressure cell and the GB1-P38 fusion protein was purified by metal affinity chromatography and cleaved according to published protocols.46 The GB1 fusion protein was removed by metal affinity chromatography and the column flow through/washes containing P38 were pooled, filtered, and purified to >98% homogeneity by reversed-phase HPLC.
A previously described method for producing SDF-1α with an additional N-terminal Gly-2-Ser-1 sequence25 was modified slightly in order to generate SDF-1α with a native N-terminus. Briefly, Ser-1 was mutated to methionine to facilitate cleavage of the resulting Gly-Met dipeptide by cyanogenbromide. Purification by reversed-phase HPLC was as previously described.25 Identity and purity of all proteins was confirmed by mass spectrometry.
Sulfation of P38 at Y21 using TPST-1
TPST-1 was prepared and incubated with 250 μM P38 as previously described for CCR5 peptides.20 Under the conditions of the sulfation reaction, sY21 P38 was the major sulfation product which enabled its purification to >98% homogeneity with reversed-phase HPLC on an analytical C18 column (Vydac). Protease digestion of sY21 P38 followed by mass spectrometry was used to confirm tyrosine 21 sulfation, as described for CCR5 sulfotyrosine peptides.20 The concentration of CXCR4 sulfo-peptide was determined by quantitative amino acid analysis at the Rockefeller University proteomics facility.
NMR spectroscopy
NMR experiments were performed on a Bruker DRX 600 equipped with a 1H/15N/13C Cryoprobe®. NMR samples contained 90% H2O, 10% D2O, and 0.02% NaN3 with various buffer, pH, and protein concentrations as specified in the text. 1H, 15N, and 13C resonance assignments for sY21 P38 and P38 (0.5 and 1mM, respectively, in 20mM MES, pH 6.8) were obtained using the following experiments:47 15N-1H HSQC, 3D HNCO, 3D HNCA, 3D HN(CO)CA, 3D 15N NOESY-HSQC, 3D 15N TOCSY-HSQC, 3D C(CO)NH, 3D HCCH TOCSY, 2D 13C constant time HSQC, and 3D 13C NOESY-HSQC (one each for aromatic and aliphatic regions). Heteronuclear 15N-1H NOE values were determined as previously described.46
NMR chemical shift assignments of SDF-1α have been described previously.24; 25; 43 [U-15N/13C]-SDF-1α (50 μM) in 20 mM MES pH 6.8 was titrated with incremental additions of sY21 P38 or P38 and monitored with 1D 1H, 2D 15N-1H HSQC, 2D aliphatic 13C-1H HSQC, and 2D aromatic 13C-1H HSQC spectra. Under these conditions SDF-1α peptide binding occurs in fast exchange on the chemical shift timescale, allowing most chemical shift assignments to be transferred by inspection. Final amide proton and nitrogen assignments were confirmed using a 3D HNCA spectrum. Amide 1H/15N chemical shift perturbations were computed as [(5ΔδNH)2 + (Δδ)2]1/2 N where ΔδNH and ΔδN are the changes in backbone amide 1H and 15N chemical shifts in ppm, respectively. Dose dependent changes in SDF-1α chemical shift perturbations upon titration with CXCR4 peptide were fit to the following equation that accounts for ligand depletion:
where Δδ is the chemical shift purturbation, Δδmax is the maximium chemical shift perturbation at 100% bound SDF-1α, Kd is the apparent SDF-1α peptide dissociation constant, and x is the peptide concentration. Non-linear fitting of chemical shifts for SDF1α residues 23, 25, 29, 31, 40-42, 47-51, 62, and 65-67 were used in apparent Kd determinations. Additionally, 1D 1H and 2D 15N-1H HSQC were used to monitor 15N/13C P38 and sY21 P38 in 20 mM MES pH 6.8 titrated with incremental additions of unlabeled SDF-1α.
The combined 1H/15N chemical shift difference values between both SDF-1α:peptide complexes (δ[P38:SDF-1α] and δ[sY21 P38: SDF-1α]) were calculated to determine the putative CXCR4 sY21 binding site.
Fluorescence Polarization
Fluorescence Polarization (FP) was performed as previously described.25 Briefly, intrinsic SDF-1α tryptophan FP values of serial dilutions of SDF-1α in 20 mM HEPES pH 7.4 with 0, 1, or 2 mM P38 were monitored. Non-linear fitting to an equation describing a monomer-dimer equilibrium was used to determine the apparent SDF-1α dimer dissociation constants in the presence of P38.25
ACKNOWLEDGMENTS
This work was supported by NIH grant AI058072 to BFV and a Northwestern Mutual Life fellowship to FCP from the MCW Cancer Center. The authors wish to thank Drs. Brian T. Chait and Martine Cadene for helpful discussions. The authors also thank The Rockefeller University proteomics facility for quantitative amino acid analysis, Bassam Wakim for performing CNBr digests and Jessie Mattmiller for assistance in protein production.
REFERENCES
- 1.Nagasawa T, Hirota S, Tachibana K, Takakura N, Nishikawa S, Kitamura Y, Yoshida N, Kikutani H, Kishimoto T. Defects of B-cell lymphopoiesis and bone-marrow myelopoiesis in mice lacking the CXC chemokine PBSF/SDF-1. Nature. 1996;382:635–638. doi: 10.1038/382635a0. [DOI] [PubMed] [Google Scholar]
- 2.Tachibana K, Hirota S, Iizasa H, Yoshida H, Kawabata K, Kataoka Y, Kitamura Y, Matsushima K, Yoshida N, Nishikawa S, Kishimoto T, Nagasawa T. The chemokine receptor CXCR4 is essential for vascularization of the gastrointestinal tract. Nature. 1998;393:591–594. doi: 10.1038/31261. [DOI] [PubMed] [Google Scholar]
- 3.Zou YR, Kottmann AH, Kuroda M, Taniuchi I, Littman DR. Function of the chemokine receptor CXCR4 in haematopoiesis and in cerebellar development. Nature. 1998;393:595–599. doi: 10.1038/31269. [DOI] [PubMed] [Google Scholar]
- 4.Orlic D, Kajstura J, Chimenti S, Limana F, Jakoniuk I, Quaini F, Nadal-Ginard B, Bodine DM, Leri A, Anversa P. Mobilized bone marrow cells repair the infarcted heart, improving function and survival. Proc. Natl. Acad. Sci. U. S. A. 2001;98:10344–10349. doi: 10.1073/pnas.181177898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Abbott JD, Huang Y, Liu D, Hickey R, Krause DS, Giordano FJ. Stromal cell-derived factor-1alpha plays a critical role in stem cell recruitment to the heart after myocardial infarction but is not sufficient to induce homing in the absence of injury. Circulation. 2004;110:3300–3305. doi: 10.1161/01.CIR.0000147780.30124.CF. [DOI] [PubMed] [Google Scholar]
- 6.Imitola J, Raddassi K, Park KI, Mueller FJ, Nieto M, Teng YD, Frenkel D, Li J, Sidman RL, Walsh CA, Snyder EY, Khoury SJ. Directed migration of neural stem cells to sites of CNS injury by the stromal cell-derived factor 1alpha/CXC chemokine receptor 4 pathway. Proc. Natl. Acad. Sci. U. S. A. 2004;101:18117–18122. doi: 10.1073/pnas.0408258102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kucia M, Reca R, Miekus K, Wanzeck J, Wojakowski W, Janowska-Wieczorek A, Ratajczak J, Ratajczak MZ. Trafficking of normal stem cells and metastasis of cancer stem cells involve similar mechanisms: pivotal role of the SDF-1-CXCR4 axis. Stem Cells. 2005;23:879–894. doi: 10.1634/stemcells.2004-0342. [DOI] [PubMed] [Google Scholar]
- 8.Muller A, Homey B, Soto H, Ge N, Catron D, Buchanan ME, McClanahan T, Murphy E, Yuan W, Wagner SN, Barrera JL, Mohar A, Verastegui E, Zlotnik A. Involvement of chemokine receptors in breast cancer metastasis. Nature. 2001;410:50–56. doi: 10.1038/35065016. [DOI] [PubMed] [Google Scholar]
- 9.Liotta LA. An attractive force in metastasis. Nature. 2001;410:24–25. doi: 10.1038/35065180. [DOI] [PubMed] [Google Scholar]
- 10.Geminder H, Sagi-Assif O, Goldberg L, Meshel T, Rechavi G, Witz IP, Ben-Baruch A. A possible role for CXCR4 and its ligand, the CXC chemokine stromal cell-derived factor-1, in the development of bone marrow metastases in neuroblastoma. J. Immunol. 2001;167:4747–4757. doi: 10.4049/jimmunol.167.8.4747. [DOI] [PubMed] [Google Scholar]
- 11.Cooper CR, Chay CH, Gendernalik JD, Lee HL, Bhatia J, Taichman RS, McCauley LK, Keller ET, Pienta KJ. Stromal factors involved in prostate carcinoma metastasis to bone. Cancer. 2003;97:739–747. doi: 10.1002/cncr.11181. [DOI] [PubMed] [Google Scholar]
- 12.Libura J, Drukala J, Majka M, Tomescu O, Navenot JM, Kucia M, Marquez L, Peiper SC, Barr FG, Janowska-Wieczorek A, Ratajczak MZ. CXCR4-SDF-1 signaling is active in rhabdomyosarcoma cells and regulates locomotion, chemotaxis, and adhesion. Blood. 2002;100:2597–2606. doi: 10.1182/blood-2002-01-0031. [DOI] [PubMed] [Google Scholar]
- 13.Phillips RJ, Burdick MD, Lutz M, Belperio JA, Keane MP, Strieter RM. The stromal derived factor-1/CXCL12-CXC chemokine receptor 4 biological axis in non-small cell lung cancer metastases. Am. J. Respir. Crit. Care Med. 2003;167:1676–1686. doi: 10.1164/rccm.200301-071OC. [DOI] [PubMed] [Google Scholar]
- 14.Murakami T, Maki W, Cardones AR, Fang H, Tun Kyi A, Nestle FO, Hwang ST. Expression of CXC chemokine receptor-4 enhances the pulmonary metastatic potential of murine B16 melanoma cells. Cancer Res. 2002;62:7328–7334. [PubMed] [Google Scholar]
- 15.Mori T, Doi R, Koizumi M, Toyoda E, Ito D, Kami K, Masui T, Fujimoto K, Tamamura H, Hiramatsu K, Fujii N, Imamura M. CXCR4 antagonist inhibits stromal cell-derived factor 1-induced migration and invasion of human pancreatic cancer. Mol. Cancer Ther. 2004;3:29–37. [PubMed] [Google Scholar]
- 16.Zhou N, Luo Z, Luo J, Liu D, Hall JW, Pomerantz RJ, Huang Z. Structural and functional characterization of human CXCR4 as a chemokine receptor and HIV-1 co-receptor by mutagenesis and molecular modeling studies. J. Biol. Chem. 2001;276:42826–42833. doi: 10.1074/jbc.M106582200. [DOI] [PubMed] [Google Scholar]
- 17.Brelot A, Heveker N, Montes M, Alizon M. Identification of residues of CXCR4 critical for human immunodeficiency virus coreceptor and chemokine receptor activities. J. Biol. Chem. 2000;275:23736–23744. doi: 10.1074/jbc.M000776200. [DOI] [PubMed] [Google Scholar]
- 18.Farzan M, Babcock GJ, Vasilieva N, Wright PL, Kiprilov E, Mirzabekov T, Choe H. The role of post-translational modifications of the CXCR4 amino terminus in stromal-derived factor 1 alpha association and HIV-1 entry. J. Biol. Chem. 2002;277:29484–29489. doi: 10.1074/jbc.M203361200. [DOI] [PubMed] [Google Scholar]
- 19.Wang J, Babcock GJ, Choe H, Farzan M, Sodroski J, Gabuzda D. N-linked glycosylation in the CXCR4 N-terminus inhibits binding to HIV-1 envelope glycoproteins. Virology. 2004;324:140–150. doi: 10.1016/j.virol.2004.03.005. [DOI] [PubMed] [Google Scholar]
- 20.Seibert C, Cadene M, Sanfiz A, Chait BT, Sakmar TP. Tyrosine sulfation of CCR5 N-terminal peptide by tyrosylprotein sulfotransferases 1 and 2 follows a discrete pattern and temporal sequence. Proc. Natl. Acad. Sci. U. S. A. 2002;99:11031–11036. doi: 10.1073/pnas.172380899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Preobrazhensky AA, Dragan S, Kawano T, Gavrilin MA, Gulina IV, Chakravarty L, Kolattukudy PE. Monocyte chemotactic protein-1 receptor CCR2B is a glycoprotein that has tyrosine sulfation in a conserved extracellular N-terminal region. J. Immunol. 2000;165:5295–5303. doi: 10.4049/jimmunol.165.9.5295. [DOI] [PubMed] [Google Scholar]
- 22.Farzan M, Chung S, Li W, Vasilieva N, Wright PL, Schnitzler CE, Marchione RJ, Gerard C, Gerard NP, Sodroski J, Choe H. Tyrosine-sulfated peptides functionally reconstitute a CCR5 variant lacking a critical amino-terminal region. J. Biol. Chem. 2002;277:40397–40402. doi: 10.1074/jbc.M206784200. [DOI] [PubMed] [Google Scholar]
- 23.Fong AM, Alam SM, Imai T, Haribabu B, Patel DD. CX3CR1 tyrosine sulfation enhances fractalkine-induced cell adhesion. J. Biol. Chem. 2002;277:19418–19423. doi: 10.1074/jbc.M201396200. [DOI] [PubMed] [Google Scholar]
- 24.Crump MP, Gong JH, Loetscher P, Rajarathnam K, Amara A, Arenzana-Seisdedos F, Virelizier JL, Baggiolini M, Sykes BD, Clark-Lewis I. Solution structure and basis for functional activity of stromal cell-derived factor-1; dissociation of CXCR4 activation from binding and inhibition of HIV-1. EMBO J. 1997;16:6996–7007. doi: 10.1093/emboj/16.23.6996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Veldkamp CT, Peterson FC, Pelzek AJ, Volkman BF. The monomer-dimer equilibrium of stromal cell-derived factor-1 (CXCL 12) is altered by pH, phosphate, sulfate, and heparin. Protein Sci. 2005;14:1071–1081. doi: 10.1110/ps.041219505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ohnishi Y, Senda T, Nandhagopal N, Sugimoto K, Shioda T, Nagal Y, Mitsui Y. Crystal structure of recombinant native SDF-1alpha with additional mutagenesis studies: an attempt at a more comprehensive interpretation of accumulated structure-activity relationship data. J. Interferon Cytokine Res. 2000;20:691–700. doi: 10.1089/10799900050116390. [DOI] [PubMed] [Google Scholar]
- 27.Dealwis C, Fernandez EJ, Thompson DA, Simon RJ, Siani MA, Lolis E. Crystal structure of chemically synthesized [N33A] stromal cell-derived factor 1alpha, a potent ligand for the HIV-1 “fusin” coreceptor. Proc. Natl. Acad. Sci. U. S. A. 1998;95:6941–6946. doi: 10.1073/pnas.95.12.6941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Horiike K, Shiga K, Isomoto A, Yamano T. Mathematical analysis of ligand-induced monomerization and dimerization in the monomer dimer equilibrium of proteins. Application to D-amino acid oxidase. J. Biochem. (Tokyo) 1977;81:179–186. doi: 10.1093/oxfordjournals.jbchem.a131433. [DOI] [PubMed] [Google Scholar]
- 29.Skrzypczak-Jankun E, Carperos VE, Ravichandran KG, Tulinsky A, Westbrook M, Maraganore JM. Structure of the hirugen and hirulog 1 complexes of alpha-thrombin. J. Mol. Biol. 1991;221:1379–1393. [PubMed] [Google Scholar]
- 30.Somers WS, Tang J, Shaw GD, Camphausen RT. Insights into the molecular basis of leukocyte tethering and rolling revealed by structures of P- and E-selectin bound to SLe(X) and PSGL-1. Cell. 2000;103:467–479. doi: 10.1016/s0092-8674(00)00138-0. [DOI] [PubMed] [Google Scholar]
- 31.Moore KL. The biology and enzymology of protein tyrosine O-sulfation. J. Biol. Chem. 2003;278:24243–24246. doi: 10.1074/jbc.R300008200. [DOI] [PubMed] [Google Scholar]
- 32.Baeuerle PA, Huttner WB. Tyrosine sulfation of yolk proteins 1, 2, and 3 in Drosophila melanogaster. J. Biol. Chem. 1985;260:6434–6439. [PubMed] [Google Scholar]
- 33.Farzan M, Mirzabekov T, Kolchinsky P, Wyatt R, Cayabyab M, Gerard NP, Gerard C, Sodroski J, Choe H. Tyrosine sulfation of the amino terminus of CCR5 facilitates HIV-1 entry. Cell. 1999;96:667–676. doi: 10.1016/s0092-8674(00)80577-2. [DOI] [PubMed] [Google Scholar]
- 34.Farzan M, Vasilieva N, Schnitzler CE, Chung S, Robinson J, Gerard NP, Gerard C, Choe H, Sodroski J. A tyrosine-sulfated peptide based on the N terminus of CCR5 interacts with a CD4-enhanced epitope of the HIV-1 gp120 envelope glycoprotein and inhibits HIV-1 entry. J. Biol. Chem. 2000;275:33516–33521. doi: 10.1074/jbc.M007228200. [DOI] [PubMed] [Google Scholar]
- 35.Ouyang Y, Lane WS, Moore KL. Tyrosylprotein sulfotransferase: purification and molecular cloning of an enzyme that catalyzes tyrosine O-sulfation, a common posttranslational modification of eukaryotic proteins. Proc. Natl. Acad. Sci. U. S. A. 1998;95:2896–2901. doi: 10.1073/pnas.95.6.2896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Holmes WD, Consler TG, Dallas WS, Rocque WJ, Willard DH. Solution studies of recombinant human stromal-cell-derived factor-1. Protein Expr. Purif. 2001;21:367–377. doi: 10.1006/prep.2001.1402. [DOI] [PubMed] [Google Scholar]
- 37.Laurence JS, LiWang AC, LiWang PJ. Effect of N-terminal truncation and solution conditions on chemokine dimer stability: nuclear magnetic resonance structural analysis of macrophage inflammatory protein 1 beta mutants. Biochemistry. 1998;37:9346–9354. doi: 10.1021/bi980329l. [DOI] [PubMed] [Google Scholar]
- 38.Paavola CD, Hemmerich S, Grunberger D, Polsky I, Bloom A, Freedman R, Mulkins M, Bhakta S, McCarley D, Wiesent L, Wong B, Jarnagin K, Handel TM. Monomeric monocyte chemoattractant protein-1 (MCP-1) binds and activates the MCP-1 receptor CCR2B. J. Biol. Chem. 1998;273:33157–33165. doi: 10.1074/jbc.273.50.33157. [DOI] [PubMed] [Google Scholar]
- 39.Rajarathnam K, Sykes BD, Kay CM, Dewald B, Geiser T, Baggiolini M, Clark-Lewis I. Neutrophil activation by monomeric interleukin-8. Science. 1994;264:90–92. doi: 10.1126/science.8140420. [DOI] [PubMed] [Google Scholar]
- 40.Rajarathnam K, Clark-Lewis I, Sykes BD. 1H NMR solution structure of an active monomeric interleukin-8. Biochemistry. 1995;34:12983–12990. doi: 10.1021/bi00040a008. [DOI] [PubMed] [Google Scholar]
- 41.Proudfoot AE, Handel TM, Johnson Z, Lau EK, LiWang P, Clark-Lewis I, Borlat F, Wells TN, Kosco-Vilbois MH. Glycosaminoglycan binding and oligomerization are essential for the in vivo activity of certain chemokines. Proc. Natl. Acad. Sci. U. S. A. 2003;100:1885–1890. doi: 10.1073/pnas.0334864100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McCornack MA, Boren DM, LiWang PJ. Glycosaminoglycan Disaccharide Alters the Dimer Dissociation Constant of the Chemokine MIP-1beta. Biochemistry. 2004;43:10090–10101. doi: 10.1021/bi049751u. [DOI] [PubMed] [Google Scholar]
- 43.Gozansky EK, Louis JM, Caffrey M, Clore GM. Mapping the binding of the N-terminal extracellular tail of the CXCR4 receptor to stromal cell-derived factor-1alpha. J. Mol. Biol. 2005;345:651–658. doi: 10.1016/j.jmb.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 44.Babcock GJ, Farzan M, Sodroski J. Ligand-independent dimerization of CXCR4, a principal HIV-1 coreceptor. J. Biol. Chem. 2003;278:3378–3385. doi: 10.1074/jbc.M210140200. [DOI] [PubMed] [Google Scholar]
- 45.Waltner JK, Peterson FC, Lytle BL, Volkman BF. Structure of the B3 domain from Arabidopsis thaliana protein At1g16640. Protein Sci. 2005;14:2478–2483. doi: 10.1110/ps.051606305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lytle BL, Peterson FC, Qiu SH, Luo M, Zhao Q, Markley JL, Volkman BF. Solution structure of a ubiquitin-like domain from tubulin-binding cofactor B. J. Biol. Chem. 2004;279:46787–46793. doi: 10.1074/jbc.M409422200. [DOI] [PubMed] [Google Scholar]
- 47.Markley JL, Ulrich EL, Westler WM, Volkman BF. Macromolecular structure determination by NMR spectroscopy. Methods Biochem. Anal. 2003;44:89–113. [PubMed] [Google Scholar]