

Abstract
We report the development of a new method to induce glioblastoma multiforme in adult immunocompetent mice by injecting Cre-loxP–controlled lentiviral vectors expressing oncogenes. Cell type- or region-specific expression of activated forms of the oncoproteins Harvey-Ras and AKT in fewer than 60 glial fibrillary acidic protein–positive cells in the hippocampus, subventricular zone or cortex of mice heterozygous for the gene encoding the tumor suppressor Tp53 were tested. Mice developed glioblastoma multiforme when transduced either in the subventricular zone or the hippocampus. However, tumors were rarely detected when the mice were transduced in the cortex. Transplantation of brain tumor cells into naive recipient mouse brain resulted in the formation of glioblastoma multiforme–like tumors, which contained CD133+ cells, formed tumorspheres and could differentiate into neurons and astrocytes. We suggest that the use of Cre-loxP–controlled lentiviral vectors is a novel way to generate a mouse glioblastoma multiforme model in a region- and cell type-specific manner in adult mice.
Mouse models of human cancers have been instructional in understanding the basic principles of cancer biology. Currently, there are three major types of animal models: xenografts, human tumor tissues or cell lines transplanted in immunodeficient mice; transgenesis, transgenic mice containing oncogenes with tissue-specific expression; and genetic knockouts, transgenic mice in whom a gene, usually a suppressor gene, is in the heterozygous state or is fully deleted1. Additional modifications to these methods, such as conditional knock-ins and knockouts, have become useful tools to study initiation, maintenance and progression of a wide variety of neoplasias. Cancers arise from a single cell or a small number of cells in specific cell types, and the cellular origin of cancers is one of the major determinants of the characteristics of tumor cells. But in most animal models, either the oncogene is expressed in all of the cells in the tissue or a gene is genetically knocked out in the whole tissue. It is, therefore, desirable to develop animal models of human cancers in which one or multiple oncogenes are activated or tumor suppressor genes are inactivated in a single cell or a few cells in an immunocompetent adult mouse. We have established a new method of inducing a mouse cancer model by directly transducing oncogenes into a small number of cells.
To test the validity of our technology, we have established a new mouse model of glioblastoma multiforme, a tumor in adult central nervous system neoplasms that is highly malignant and is resistant to existing treatments2. Transduction of as few as 60 GFAP+ cells into the subventricular zone or hippocampus of adult immunocompetent mice with Cre-loxP–controlled lentiviral vectors expressing activated forms of the oncogenes Harvey-Ras (H-Ras) and AKT led to the formation of human high-grade glioma (World Health Organization grades III–IV)–like tumors. However, when transduction of activated H-Ras- and AKT-expressing lentiviral vectors is carried out in these regions of the brains of adult Tp53 heterozygotes, the frequency of formation of human glioblastoma multiforme (World Health Organization grade IV)–like tumors is substantially increased. The resulting tumors faithfully recapitulate the pathophysiology of human glioblastoma multiforme. The technology reported here might be useful to model various human cancers in animals.
Results
Oncogenic H-Ras and activated AKT induce glioma
Brain tumors in adults, including glioblastoma multiforme, are thought to arise from a series of somatic mutations leading to the activation of oncogenes or inactivation of tumor suppressors that occur in a few cells or even a single founder cell in adults3,4. Thus, to recapitulate the initiation of glioblastoma multiforme, it is necessary to induce these oncogenic events in a single cell or in a small population of a specific type of cells in an adult mouse brain. To achieve this goal, we performed region-specific injection of lentiviral vectors that can transduce both neural stem and progenitor cells and terminally differentiated astrocytes5. To target specific cell types, we chose the Cre-loxP system, which is a reliable method of specifically expressing or deleting a target gene in Cre-expressing cells6. We generated Cre-loxP–controlled lentiviral vectors, Tomo lentiviral vectors, capable of inducing the expression of one or more oncogenes in a cell type–specific manner in an adult mouse brain (Fig. 1).
Figure 1.
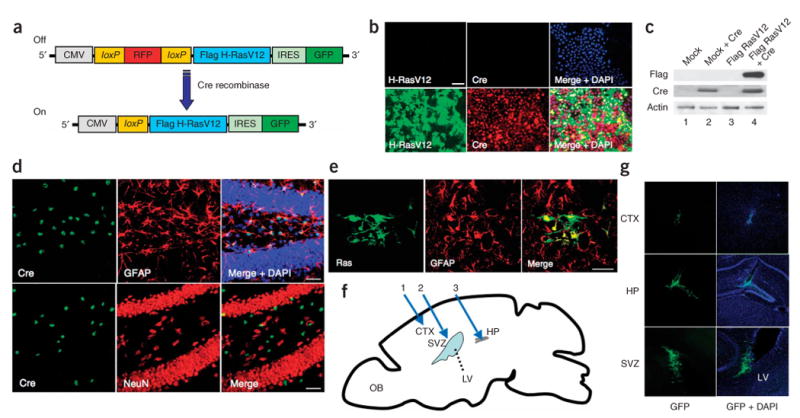
Cre recombinase–dependent expression of Flag H-RasV12 by pTomo H-RasV12 lentiviral vectors in vitro and in vivo. (a) Diagram of pTomo H-RasV12 lentiviral vector (LV). The stuffer fragment, RFP, keeps the translation of Flag H-RasV12 in an ‘off’ state. The excision of the stuffer sequence by Cre recombinase results in the expression of Flag H-RasV12. (b) Immunocytochemistry showing Flag H-RasV12 expression induced by Cre recombinase. HeLa cells were infected with Tomo H-RasV12 LVs with (bottom) or without (top) Cre-expressing LVs. After fixation, the cells were stained with the indicated antibodies. Scale bar, 50 μm. (c) Western blot showing Flag H-RasV12 expression induced by Cre recombinase. HeLa cells were infected with Tomo mock LVs (lanes 1 and 2) or Tomo H-RasV12 LVs (lanes 3 and 4) with (lanes 2 and 4) or without (lanes 1 and 3) Cre-expressing LVs, and the cell lysates were processed for western blotting. β-actin detection was used as a loading control. (d) Confocal images of Cre recombinase specifically expressed in GFAP+ cells in GFAP-Cre mice. Brain sections from 8-week-old GFAP-Cre mice were stained with the indicated antibodies. A merged image is shown in the right panel. Scale bars, 50 μm. (e) Confocal images showing H-RasV12 expression specifically in GFAP+ cells. Seven days after the injection of Tomo H-RasV12 LVs into cortex of GFAP-Cre mice, the mice were killed and fixed. Sections of the brain were stained with the indicated antibodies. A merged image is shown in the right panel. Scale bar, 40 μm. (f) A schematic diagram of the locations injected with LVs in a sagittal view of an adult mouse brain. Blue arrows show the locations (1: cortex (CTX), 2: subventricular zone (SVZ), 3: hippocampus(HP)) where the injections of LVs were performed. OB, olfactory bulb; LV, lateral ventricle. (g) Tomo H-RasV12 LVs were successfully injected into three different locations. Confocal images were taken 7 d after the injection of Tomo H-RasV12 LVs into cortex, hippocampus and subventricular zone in GFAP-Cre mice. GFP signals (green) indicate the cells infected with Tomo H-RasV12 LVs. Merged images with DAPI are shown in right panels.
As proof of principle, we first chose to express oncogenic H-RasV12 protein, which is known to be activated in glioblastoma multiforme in adults7. We constructed pTomo H-RasV12, which contains a human cytomegalovirus immediate-early promoter (CMV)-loxP-red fluorescent protein (RFP)-loxP-Flag-H-RasV12 followed by internal ribosomal entry site (IRES)-GFP (Fig. 1a). When Tomo H-RasV12 lentiviral vectors and Cre-expressing lentiviral vectors were used to infect HeLa cells, RFP signals were hardly detected (data not shown) and H-RasV12 was successfully expressed in vitro (Fig. 1b,c).
In addition to being expressed in mature astrocytes, glial fibrillary acidic protein (GFAP) has been reported to be expressed in multipotent neural stem cells in the hippocampus and subventricular zone in the postnatal and adult brain8. We next transduced oncogenes into GFAP+ cells in the cortex, subventricular zone and hippocampus by injecting Tomo lentiviral vectors into adult GFAP-Cre mice9,10 (Fig. 1f,g). Eight-week-old GFAP-Cre mice express Cre only in cells expressing GFAP in the central nervous system (Fig. 1d). Little or no Cre immunoreactivity was observed in neurons (detected by staining for the NeuN neuronal marker, Fig. 1d) and oligodendrocytes (detected by the marker O4, data not shown), which is consistent with a previous report10. Seven days after the injection, the brains were fixed and sectioned. Immunohistochemical analysis revealed that the injection of Tomo H-RasV12 lentiviral vectors into the brains of GFAP-Cre mice resulted in the expression of H-RasV12 specifically in GFAP+ cells (Fig. 1e).
About ten months after the injections into the three different locations of GFAP-Cre mouse brains, all of the mice were killed, and their brains were examined by histological analyses. Because none of the mice developed tumors (Supplementary Table 1 online), we concluded that activation of the Ras pathway alone in adult brain is not enough to form a brain tumor, which is consistent with a previous report11.
Loss of the tumor suppressor phosphatase and tensin homolog and the resulting activation of the AKT pathway is often associated with gliomas12,13. We next tried to activate AKT signaling by injecting Tomo AKT lentiviral vectors (Tomo lentiviral vectors harboring a hemagglutinin (HA)-tagged activated form of AKT) into brains of GFAP-Cre mice. However, there was no tumor formation induced by activated AKT alone, consistent with a previous report11 (Supplementary Table 1). Because both Ras and AKT pathways are activated downstream of growth factor receptors such as epidermal growth factor receptor (EGFR), which are frequently activated in glioblastoma multiforme14,15, we next transduced Tomo H-RasV12 lentiviral vectors and Tomo AKT lentiviral vectors into the cortex, subventricular zone and hippocampus of GFAP-Cre mice. We found that approximately 50–60 cells (48.4 ± 7 cells in the hippocampus, 57.0 ± 15 cells in the subventricular zone and 56.7 ± 13 cells in the cortex; Supplementary Fig. 1 online) were infected with both Tomo H-RasV12 lentiviral vectors and Tomo AKT lentiviral vectors, and almost all of the cells (>98%) showed the expression of both H-RasV12 and AKT proteins (Supplementary Fig. 1).
About 4–5 months (136 ± 32 d) after the injection, five out of twelve GFAP-Cre mice transduced with H-RasV12 and AKT lentiviral vectors into the right hippocampus showed an enlarged head and lethargy (Fig. 2a). In contrast, only one tumor was found from injections into subventricular zone, and no tumors were found in the cortex (Supplementary Table 1). Control GFAP-Cre mice injected with mock vectors showed no tumor formation (Supplementary Table 1). The gross appearance of the brain of a GFAP-Cre mouse with a massive tumor is shown in Figure 2b, and H&E staining shows the dark area of the tumor resulting from increased cellular density (Fig. 2c). Because Tomo lentiviral vectors also express GFP under the control of IRES (Fig. 1a), it is worth noting that a majority of the cells within the tumor area expressed GFP (Fig. 2d and Supplementary Fig. 2a,b online). Thus, it appears that a majority of the cells in the tumor were derived from a small number of cells (approximately 50 cells, Supplementary Fig. 1b) transduced with Tomo lentiviral vectors. A minority of the cells (less than 1%) was negative for GFP in the tumor and was probably taken up during the development of the tumor.
Figure 2.
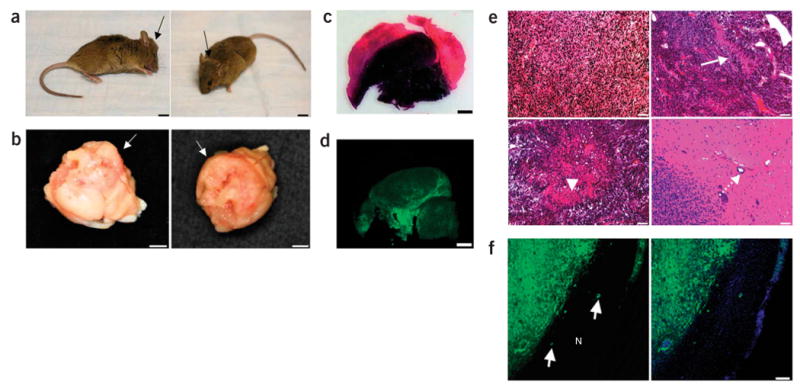
Brain tumors induced by combined activation of H-Ras and AKT in GFAP-Cre mice. (a) Representative images of GFAP-Cre mouse showing tumor formation. A GFAP-Cre mouse injected with Tomo H-RasV12 LVs into the right hippocampus showed an enlarged head (black arrows) 93 d after the injection. Scale bars, 5 mm. (b) Photograph showing gross appearance of the brain, suggesting a massive lesion in the cerebrum. White arrows indicate the irregular surface of the cerebrum. Scale bars, 3 mm. (c) Representative image of an H&E-stained section (40 μm). A darker area indicates increased cellular density of the tumor. Scale bars, 1.5 mm. (d) Representative confocal image of a section (40 μm). GFP+ cells are found throughout the tumor. Scale bars, 1.5 mm. (e) H&E staining showing the characteristics of glioma, including increased cell density (top left panel), pseudopalisading (top right panel, white arrow), necrosis within the dense cellular region (bottom left panel, arrowhead) and perivascular invasion (bottom right panel, dotted arrow). Scale bars, 50 μm. (f) Tumor section (40 μm) analyzed by confocal microscopy. White arrows in the left panel show invasion of the GFP+ tumor cells into normal tissues (indicated by N). A merged image with DAPI (blue) is shown in the right panel. Scale bar, 100 μm.
The brains of the five mice with tumors showed extensive parenchymal lesions that vary in size (Fig. 2b and data not shown). Microscopic characterization of the lesions showed abnormally increased cell density, microvascular proliferation and serpiginous zones of tumor necrosis bordered by dense palisades of viable tumor cells (necrosis with pseudopalisading, Fig. 2e). It is noteworthy that these tumor cells invaded normal tissues in a manner similar to glioma cells in humans (Fig. 2f). These histological characteristics are the hallmarks of human high grade gliomas (World Health Organization grades III–IV). However, nuclear pleomorphism, a phenotype generally found in human high-grade gliomas, was not obvious.
Impact of p53 loss
The Tp53 gene is known to be frequently mutated in human glioblastoma multiforme and is associated with malignant transformation of astrocytoma16,17. We therefore injected H-RasV12 and AKT lentiviral vectors into three locations of the brains of GFAP-Cre Tp53+/− mice. All mice (17 of 17 mice) injected with H-RasV12 and AKT lentiviral vectors into the hippocampus formed tumors (Fig. 3a–e), whereas 75% (12 of 16) of the subventricular zone–injected mice and about 7% (1 of 15) of the cortex-injected mice showed tumor formation (Supplementary Figs. 3 and 4 and Supplementary Table 1 online). The tumor latency in GFAP-Cre Tp53+/− mice was much shorter than that in GFAP-Cre mice injected with H-Ras- and AKT-expressing vectors into the hippocampus or subventricular zone (Fig. 3a). Histological analyses of the tumor from hippocampus revealed lesions showing high cellular density, intratumoral hemorrhage (Fig. 3b), necrosis within a dense cellular lesion (Fig. 3c), nuclear pleomorphism (giant cells, Fig. 3d) and high mitotic activity (Fig. 3f), all of which are the hallmarks of human glioblastoma multiforme. The tumors were mostly GFP positive (Fig. 3e). Expression of both H-Ras and AKT was readily detected in the tumor (Fig. 4a,b), and, furthermore, the tumor contained cells positive for the astrocyte marker GFAP, the oligodendrocyte marker myelin basic protein and the neuron-specific β III tubulin marker Tuj1 (Fig. 4), indicating the presence of different cell types, which is also a phenotype of human glioblastoma multiforme. It is noteworthy that many of the tumor cells expressed a neural progenitor cell marker, nestin (Fig. 4d), which is often found in human glioblastoma multiforme18. Microvascular proliferation was found by the detection of CD31- and von Willebrand factor–positive endothelial cells in the tumor (Fig. 4g and Supplementary Fig. 2f–h). Consistent with this result, the overexpression of vascular endothelial growth factor (VEGF) and VEGF receptor-2 (VEGFR2) in the tumor tissues were observed (Fig. 4h,i and Supplementary Fig. 5a online). Five tumors that arose from the hippocampus, five tumors that arose from the subventricular zone and one tumor that arose from the cortex were examined by histological analyses, and most of the histological characteristics of the tumors from the hippocampus were also found in the tumors from the subventricular zone and cortex (Supplementary Figs. 3 and 4 and Supplementary Table 2 online).
Figure 3.
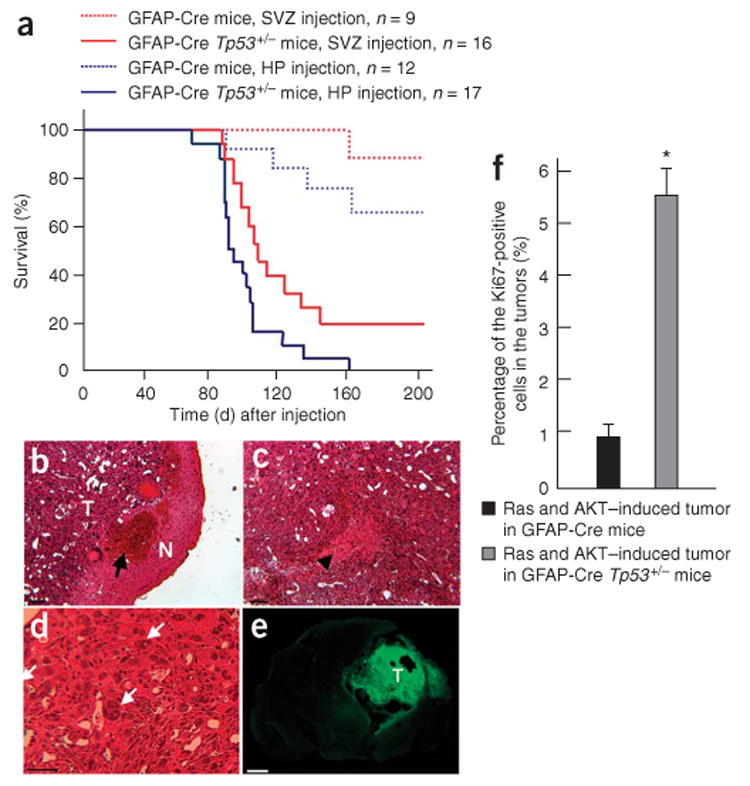
Effects of p53 loss on tumor formation induced by combined activation of H-Ras and AKT. (a) A Kaplan-Meier curve showing glioma-free survival. GFAP-Cre or GFAP-Cre Tp53+/− mice were injected with Tomo H-RasV12 LVs and Tomo AKT LVs into hippocampus or subventricular zone. (b–d) H&E staining of the tumor induced by combined activation of H-Ras and AKT in the right hippocampus in a GFAP-Cre Tp53+/− mouse. T and N in b indicate the tumor area and the normal tissue area, respectively. Black arrow in b indicates intratumoral hemorrhage. Black arrowhead in c indicates the area of necrosis. White arrows in d indicate giant cell formation in the tumor. Scale bars, 50 μm. (e) Confocal micrograph showing GFP+ cells throughout the tumor area. T indicates tumor area. Scale bar, 1.5 mm. (f) A graph showing the proportion of the cycling cells in tumors. Tumor tissues originated from the right hippocampus found in GFAP-Cre or GFAP-Cre Tp53+/− mice were stained with Ki67-specific antibody, at least 700 cells in three different locations were examined in each tumor, and the number of cells positive for Ki67 was calculated. *P < 0.05; data represent means ± s.d.
Figure 4.
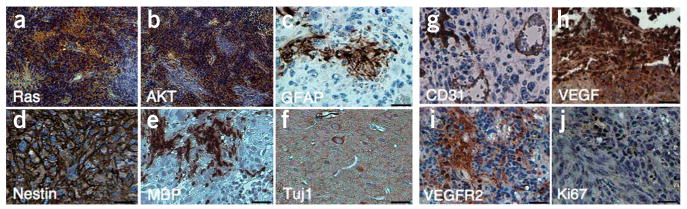
Tumors induced by combined activation of H-Ras and AKT in GFAP-Cre Tp53+/− mice contain the cells expressing various cell markers including astrocytes, oligodendrocytes and neurons. (a–j) Immunohistochemical analysis of sections from the tumor induced by combined activation of H-Ras and AKT in the right hippocampus of GFAP-Cre Tp53+/− mice stained with antibodies to Flag (a), HA (b), GFAP (c), nestin (d), myelin basic protein (MBP, e), Tuj1 (f), CD31 (g), VEGF (h), VEGFR2 (i) and Ki67 (j). Scale bars, 50 μm.
P53 has been shown to have a role in inducing senescence in response to oncogenic stimuli, resulting in resistance to cellular transformation, and, therefore, the loss of Tp53 is thought to allow the tumor cells to bypass the oncogene-induced senescence19. The p53 was more abundant in tumors from GFAP-Cre mice than in tumors from GFAP-Cre Tp53+/− mice (Supplementary Fig. 6a,b online). We next examined senescence-associated β-galactosidase (SA–β-gal) activity in the tumors derived from GFAP-Cre or GFAP-Cre Tp53+/− mice. SA–β-gal activity was more prominently found in the tumors from GFAP-Cre mice than in those from GFAP-Cre Tp53+/− mice (Supplementary Fig. 6c–f), indicating that loss of Tp53 promotes the cells' escape from the oncogene-induced senescence program activated by H-Ras and AKT, which may result in the shorter latency, higher incidence and higher mitotic activity of tumors found in GFAP-Cre Tp53+/− mice compared to those from GFAP-Cre mice (Fig. 3a,f and Supplementary Table 1). Consistent with this result, the degree of apoptosis in the tumors from GFAP-Cre mice was much lower than that in the tumors from GFAP-Cre Tp53+/− mice (Supplementary Fig. 6g,h).
The expression of EGFR, which is frequently amplified and overexpressed in human glioblastoma multiforme, was not prominent in this mouse model of glioblastoma multiforme (Supplementary Fig. 5a), probably because H-Ras and AKT, which are known to be downstream of EGFR, have sufficiently been activated in this model14,15. Also, the expression of MDM2, an oncoprotein that degrades p53, is often amplified or overexpressed in human glioblastoma multiforme but is low in this mouse model of glioblastoma multiforme (Supplementary Fig. 5a), probably because p53 expression is low in the tumor (Supplementary Fig. 6b) and MDM2 is known to be transcriptionally activated by p53 (ref. 14,15,20). Another major tumor suppressor gene, Cdkn2a (encoding p16), which is often deleted in human glioblastoma multiforme, was not deleted in this mouse model of the disease (Supplementary Fig. 5b). Finally, a magnetic resonance image (MRI) of the tumor-bearing mouse showed a gadolinium-enhanced mass lesion, which is commonly found in imaging of human glioblastoma multiforme (Supplementary Fig. 7 online).
Brain tumor–initiating stem cells
Normal neural stem cells can be isolated in culture as clonally derived colonies termed neurospheres21. We next established primary cultures of the brain tumor induced by the combined activation of H-Ras and AKT in GFAP-Cre Tp53+/− mice and determined that the cultures contained brain tumor–initiating stem cells (BTICs). The tumor cells (005 tumor cells) were taken from a mouse tumor (originating from the hippocampus in a GFAP-Cre Tp53+/− mouse) and cultured in the normal medium often used for cancer cell lines or in the medium prepared for neural stem cells. 005 tumor cells in the normal medium survived for 1 week but eventually died within 2 weeks, whereas 005 tumor cells in the medium prepared for culturing neural stem cells could survive and continue to proliferate for more than 30 passages (data not shown). Furthermore, GFP+ tumor cells cultured in the medium prepared for neural stem cells formed neurosphere-like structures (often called a ‘tumorsphere’, Fig. 5a). We next examined the self-renewal capacity of 005 tumor cells by dissociating primary tumorspheres and replating single-cell suspensions of these cells at serial dilutions down to one cell per well. Primary tumorsphere–derived GFP+ cells formed secondary spheres with a range of 76–88 secondary spheres per 100 primary sphere cells (mean 81.3 ± 5.1) over a period of 1–2 weeks. The secondary sphere-forming cells possessed the potential to grow infinitely, and the proportion of the cells positive for nestin, a marker for immature neural progenitors, remained stable throughout the course of cell culture (at least 2 months), indicating that 005 tumor cells have the potential to self renew.
Figure 5.
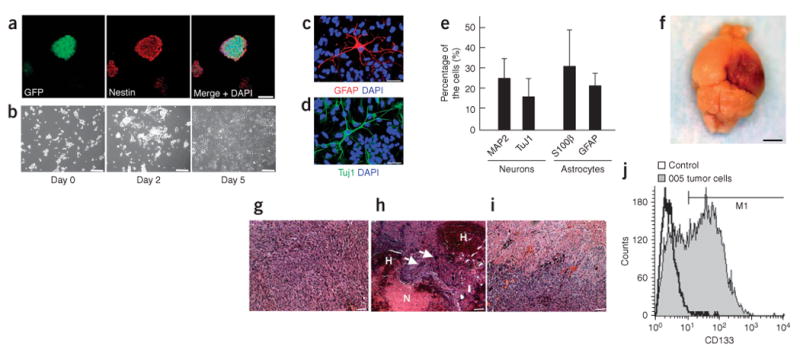
005 tumor cells show the characteristics of BTICs. (a) Confocal microscopic analysis of neurosphere-like structures formed by 005 tumor cells. Cells stained with nestin-specific antibody are shown in the middle panel. A merged image with DAPI staining is shown in the right panel. Scale bar, 50 μm. (b) Phase-contrast images of 005 tumor cells showing elongated processes in response to serum addition in the medium. Images were taken on days 0, 2 and 5 after the serum addition. Scale bars, 50 μm. (c–e) 005 tumor cells expressing neuronal or astrocyte markers. Five days after the addition of FBS to the medium (final 10% FBS), cells were fixed and stained with GFAP-specific antibody (c) or Tuj1-specific antibody (d) and observed by confocal microscopy. Merged mages with DAPI are shown. Scale bars, 30 μm. The proportions of cells positive for neuronal or astrocyte marker are shown in e. Data represent means ± s.d. (f) The gross appearance of tumor formation in the brain of a NOD-SCID mouse after injection of 100 of 005 tumor cells. Scale bar, 3 mm. (g–i) Histological characteristics of the tumor caused by the injection of 005 tumor cells in NOD-SCID mice, including increased cellular density (g), intratumoral hemorrhage (indicated by H in h), necrosis (indicated by N in h), giant cell formation (indicated by white arrows in h) and infiltrative characteristics (i). Scale bars, 50 μm. (j) FACS analysis of 005 tumor cells in culture incubated with CD133-specific antibody. Unstained cells were used as a control.
A vast majority of tumorsphere-forming cells (>99% of 005 tumor cells) were positive for nestin (Fig. 5a) and negative for neuronal markers (microtubule-associated protein 2, and Tuj1, data not shown), astrocyte markers (s100β and GFAP, data not shown) and oligodendrocyte markers (RIP and O4, data not shown). We next tested whether 005 tumor cells can differentiate in response to serum addition to the medium. We found that 005 tumor cells showed elongated processes (Fig. 5b) and expressed markers for astrocytes and neurons (Fig. 5c–e) upon addition of 10% FBS, although they did not express oligodendrocyte markers under these conditions (data not shown).
It has been shown that most xenograft models for brain tumors using tumor cell lines require at least 1 × 105 cells to form gliomas22,23, whereas only 100 BTICs are enough to form tumors in the immunodeficient mouse brain24. Thus, we next examined whether 005 tumor cells can form brain tumors when injected into the brains of nonobese diabetic–severe combined immunodeficient (NOD-SCID) mice. Injection of only 100 005 tumor cells into the hippocampus of NOD-SCID mice resulted in the formation of tumors (seven of nine mice, average latency 40 ± 6 d, Fig. 5f–i), whereas no control mice (zero of eight) injected with 1 × 105 wild-type mouse neural stem cells formed any tumors (data not shown). Moreover, injection of only ten 005 cells into the hippocampus in NOD-SCID mice resulted in tumor formation (four of eight mice, average latency: 63.2 ± 6.8 d), indicating the strong tumor-forming activity of 005 tumor cells. The gross appearance of the brains of the injected mice showed a darker area, which suggests a massive lesion with hemorrhage (Fig. 5f). The tumor showed glioma-like histopathology, including increased cellular density, necrosis within the dense cellular lesion and intratumoral hemorrhage (Fig. 5g–i). Notably, the tumor also showed infiltrative characteristics (Fig. 5i), which is not observed in xenograft models using glioblastoma multiforme cell lines25. The characteristics of the 005 tumor cells described here were also observed in experiments using 006 tumor cells, which were independently isolated from a tumor induced by combined activation of H-Ras and AKT in the hippocampus of a GFAP-Cre Tp53+/− mouse (Supplementary Fig. 8 online).
In addition to being a marker hematopoietic stem and progenitor cells, CD133 has also been found to be a marker for BTICs. FACS analysis revealed that CD133 was found in 40–50% of 005 tumor cells (Fig. 5j). We observed that the proportion of the cells that express CD133 gradually decreased during the course of the culture (especially after 3 months of culture), indicating that 005 tumor cells differentiate upon culturing in the dish, even in the medium for neural stem cells. We conclude that tumorsphere-forming cells derived from our mouse model of glioblastoma multiforme, self renew, differentiate in vitro and have strong tumor-initiating activity similar to previously reported BTICs4,24. These results indicate that this mouse model of glioblastoma multiforme is useful not only for the understanding of the initiation of glioblastoma multiforme but also for the investigation of BTICs.
Discussion
The use of somatic cell gene transfer using retroviral vectors in to the neonatal mouse brain to model gliomas has been previously described26,27. In one such case, avian leukosis virus (ALV)-based replication-competent ALV-LTR (long terminal repeat) splice acceptor (RCAS) vectors were injected into the brain of neonatal mice expressing the RCAS receptor, tv-a (a receptor for the ALV envelope glycoprotein) from GFAP or the nestin promoter11. The researchers showed that combined activation of K-Ras and AKT in nestin+ neural progenitors caused the formation of glioblastoma multiforme–like tumor11. They did not observer tumor formation when K-Ras and AKT were induced in GFAP+ astrocytes11; thus, nestin+ neural progenitors may be the cellular origin of glioblastoma multiforme. In our model, combined activation of H-Ras and AKT and the loss of p53 in GFAP+ cells in the hippocampus or subventricular zone caused the formation of glioblastoma multiforme–like disease. Because GFAP+ cells in the hippocampus or subventricular zone are likely to contain both differentiated astrocytes and neural progenitor cells8,28, these cell types could be the cellular origin of the glioblastoma multiforme–like tumors in our model. To induce glioblastoma multiforme–like disease in mice, the other group used mice with a wild-type Tp53 genetic background, whereas we needed to use Tp53 heterozygotes to obtain all of the neoplastic features associated with glioblastoma multiforme (Fig. 3). Some of the discrepancies between the two models might be attributed to the differences between K-Ras and H-Ras in vivo. The other researchers also used neonatal mice, whereas we used adult mice, and thus the discrepancies may have originated from the differences between the cells used in their proliferative and differentiation characteristics.
One of the advantages of our strategy is that this method can be used to clarify the differences between tumors caused by the same oncogenic events occurring in different regions of the brain. There was little tumor formation from the cortex, whereas more than 75% of mice showed tumor formation from the neurogenic areas such as the subventricular zone and hippocampus (Supplementary Table 1). This result is consistent with a previous report showing that neurogenic areas such as the subventricular zone and hippocampus are more sensitive to oncogenic stimuli, such as chemicals, than other areas29. One explanation for this is that there may be unknown growth factors in neurogenic areas that promote the transformation of GFAP+ cells. Another explanation is that GFAP+ neural progenitor cells, which are abundant in neurogenic areas, may be more susceptible to transformation than GFAP+ differentiated astrocytes, which are abundant in the cortex.
Because there are growing numbers of transgenic mice that express Cre in a specific cell type, our proposed strategy of introducing oncogenes could become a versatile and useful method to model various tumors in any organ.
Methods
Plasmids
To generate pTomo, we inserted a loxP-mRFP-loxP fragment amplified from the pSETB mRFP1 vector by PCR between the XbaI and BamHI sites of the p156RRLsin PPTCMVIRESPRE vector (a third-generation lentiviral vector)30. To construct pTomo H-RasV12, we amplified N-terminus Flag-tagged H-RasV12 from pGEM H-RasV12 by PCR and inserted it into the BamHI site of the pTomo vector. To construct pTomo AKT, we amplified the HA-tagged constitutively active AKT (we added the myristoylation signal of the c-Src oncoprotein, MGSSKSKPKDPSQR, to the N terminus of human AKT1) from p156RRLsin PPTCMVIRESPRE myr-AKT by PCR and inserted it into the BamHI site of the pTomo vector. We confirmed all PCR fragments by sequencing.
Viral vector production
We produced all lentiviral vectors were by the method described previously31. We determined the biological titer of lentiviral vectors by infecting HeLa cells (American Type Culture Collection) with various amounts of the viruses.
Mice
We obtained GFAP-Cre transgenic mice9 from The Jackson Laboratories. We maintained the colonies of this strain by crossing to wild -C57BL/6 mice. We crossed Tp53−/− mice with the GFAP-Cre line. All mice in this study were cared for according to the guidelines that were approved by the Animal Care and Use Committee of the Salk Institute.
In vivo vector injection
We stereotaxically injected a small amount of viruses (0.8 μl, 1 × 108 international units) into mice at the age of 8–16 weeks old who were under anesthesia with ketamine-xylazine solution. To inject a mixture of Tomo H-RasV12 lentiviral vectors and Tomo AKT lentiviral vectors, we mixed the two viral preparations (1:1) and injected 0.8 μl.
Western blotting
We lysed cells or tissues in NP-40 lysis buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1 mM EDTA and 0.5% NP-40), and we subjected equal amounts of the lysates to SDS-PAGE. We transferred the proteins to a nitrocellulose membrane and probed the membrane with the antibodies listed in Supplementary Table 3 online.
Southern blotting
We digested genomic DNA (20 μg) with BamHI, separated the fragments through an 0.8% agarose gel and blotted them on a nylon membrane before hybridization with a 32P-random-prime–labeled, 737–base pair SalI-BstXI GFP fragment from the pTomo vectors or a 501–base pair genomic probe (corresponding to the DNA sequence from exon 1 to exon 2) for Cdkn2a, which we PCR-amplified from genomic DNA of normal mouse endothelial cells.
β-galactosidase staining and TUNEL staining
We subjected 40-μm coronal tumor sections fixed with 4% paraformaldehyde in PBS to SA–β–gal staining and TUNEL staining as previously described19.
Cell culture in vitro
We cultured tumor cells obtained from mouse glioblastoma multiforme–like tumors (005 and 006) in N2-supplemented (Invitrogen) DME:Ham's F12 (Omega Scientific) medium containing 20 ng ml−1 fibroblast growth factor-2 (PeproTech), 20 ng ml−1 epidermal growth factor (Promega) and 50 μg ml−1 heparin (Sigma), which is prepared for mouse neural stem cells32. To induce differentiation, we allowed cells to proliferate in DME:Ham's F12 medium supplemented with 10% FBS for 5 d.
Immunocytochemistry
We performed immunocytochemistry as previously described33. The antibodies used are listed in Supplementary Table 3.
Immunohistochemistry and hematoxylin and eosin staining
We perfused mice with 0.9% NaCl followed by 4% paraformaldehyde in PBS. We then excised the brains, stored them in fixative overnight and transferred them to 30% sucrose in PBS. For detection with fluorescence, we cut 40-μm coronal sections on a sliding microtome and processed the sections for standard immunohistochemical staining as previously described34. We obtained the images by confocal laser-scanning microscopy (Leica TCS SP2 ABS). For H&E staining, we embedded the fixed brains in paraffin, cut them into 6-μm sections and stained with H&E. For immunohistochemical analysis of paraffinized tissues, we deparaffinized the tissues, rehydrated them and stained them as previously described35. We used diaminobenzidine (Vector) as a substrate for a peroxidase. After counterstaining with Mayer's hematoxylin (Sigma), we mounted the sections with Vectamount AQ (Vector). We obtained the images with a Retiga digital camera on a Nikon optical microscope and analyzed them with OpenLab software (Improvision).
Flow cytometric analysis
We stained 005 tumor cells with CD133-specific antibody and analyzed them on a BD LSRI flow cytometer (Becton Dickinson) with CELLQuest software.
Magnetic resonance imaging
We imaged the mice in vivo both before and after contrast. We acquired images with a 1.5-cm custom-built surface MRI coil, which was manually tuned and placed in a horizontal-bore 7T MR scanner (General Electric). We acquired images both before and after administration of the magnetic resonance contrast agent gadolinium dimeglumine (Magnevist) with a three-dimensional fast spoiled gradient sequence: TR (repitition time) / TE (echo time) = 7.5/3.2, FA (flip angle) 20, FOV (field of view) 1.4 mm, matrix 160 × 160 × 300, BW (bandwidth) 15.63; ten averages providing three-dimensional datasets of the brain at 87 μm in plane resolution. We also acquired T2-weighted anatomical images before with a two-dimensional fast spin-echo sequence TR / TE = 2900/35.5, FA 90, BW 15.63. The after-contrast dataset was semi-manually segmented and volume-rendered with Amira software (Template Graphic Software) to produce quantitative three-dimensional models of the area of contrast enhancement in the brain tissue.
Supplementary Material
Acknowledgments
We thank R. Shaw for discussions and critical reading of the manuscript; N. Varki and H. Powell for pathological analyses; S. Ylä-Herttuala, K. Suzuki, N. Tanaka, G. Pao, A. Parker and H. Suh for useful discussions; T. Sawai, G. Estepa and M. Lawrence for technical assistance; M. Schmitt and B. Coyne for administrative assistance; and S. Withoff (St. Jude Hospital), V. Tergaonkar (Bioprocessing Technology Institute), O. Singer (Salk Institute) and G. Wahl (Salk Institute) for providing pSETB mRFP1, pGEM H-RasV12, p156RRLsin PPTCMVIRESPRE myr-AKT vectors and Tp53−/− mice, respectively. T.M. is supported by the American Brain Tumor Association. I.M.V. is an American Cancer Society Professor of Molecular Biology and is supported in part by grants from the US National Institutes of Health and the H.N. and Frances C. Berger Foundation. The project described was supported in part by the National Institutes of Health. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Author Contributions: T.M. conducted most of the experiments, prepared figures and wrote the manuscript. A.T., D.F.-M. and Y.S. participated in in vivo studies and histological analysis. M.S. performed MRI imaging. F.H.G. supervised the experiments and the project. I.M.V. is the principle investigator, supervised the experiments and the project and wrote the manuscript.
Note: Supplementary information is available on the Nature Medicine website.
References
- 1.Hesselager G, Holland EC. Using mice to decipher the molecular genetics of brain tumors. Neurosurgery. 2003;53:685–694. doi: 10.1227/01.neu.0000081304.57547.b5. discussion 695. [DOI] [PubMed] [Google Scholar]
- 2.Holland EC. Glioblastoma multiforme: the terminator. Proc Natl Acad Sci USA. 2000;97:6242–6244. doi: 10.1073/pnas.97.12.6242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Holland EC. Gliomagenesis: genetic alterations and mouse models. Nat Rev Genet. 2001;2:120–129. doi: 10.1038/35052535. [DOI] [PubMed] [Google Scholar]
- 4.Vescovi AL, Galli R, Reynolds BA. Brain tumour stem cells. Nat Rev Cancer. 2006;6:425–436. doi: 10.1038/nrc1889. [DOI] [PubMed] [Google Scholar]
- 5.Kafri T, Blomer U, Peterson DA, Gage FH, Verma IM. Sustained expression of genes delivered directly into liver and muscle by lentiviral vectors. Nat Genet. 1997;17:314–317. doi: 10.1038/ng1197-314. [DOI] [PubMed] [Google Scholar]
- 6.Abremski K, Hoess R, Sternberg N. Studies on the properties of P1 site-specific recombination: evidence for topologically unlinked products following recombination. Cell. 1983;32:1301–1311. doi: 10.1016/0092-8674(83)90311-2. [DOI] [PubMed] [Google Scholar]
- 7.Guha A, Feldkamp MM, Lau N, Boss G, Pawson A. Proliferation of human malignant astrocytomas is dependent on Ras activation. Oncogene. 1997;15:2755–2765. doi: 10.1038/sj.onc.1201455. [DOI] [PubMed] [Google Scholar]
- 8.Doetsch F, Caille I, Lim DA, Garcia-Verdugo JM, Alvarez-Buylla A. Subventricular zone astrocytes are neural stem cells in the adult mammalian brain. Cell. 1999;97:703–716. doi: 10.1016/s0092-8674(00)80783-7. [DOI] [PubMed] [Google Scholar]
- 9.Zhuo L, et al. hGFAP-Cre transgenic mice for manipulation of glial and neuronal function in vivo. Genesis. 2001;31:85–94. doi: 10.1002/gene.10008. [DOI] [PubMed] [Google Scholar]
- 10.Malatesta P, et al. Neuronal or glial progeny: regional differences in radial glia fate. Neuron. 2003;37:751–764. doi: 10.1016/s0896-6273(03)00116-8. [DOI] [PubMed] [Google Scholar]
- 11.Holland EC, et al. Combined activation of Ras and Akt in neural progenitors induces glioblastoma formation in mice. Nat Genet. 2000;25:55–57. doi: 10.1038/75596. [DOI] [PubMed] [Google Scholar]
- 12.Steck PA, et al. Identification of a candidate tumour suppressor gene, MMAC1, at chromosome 10q23.3 that is mutated in multiple advanced cancers. Nat Genet. 1997;15:356–362. doi: 10.1038/ng0497-356. [DOI] [PubMed] [Google Scholar]
- 13.Li J, et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science. 1997;275:1943–1947. doi: 10.1126/science.275.5308.1943. [DOI] [PubMed] [Google Scholar]
- 14.Ekstrand AJ, et al. Functional characterization of an EGF receptor with a truncated extracellular domain expressed in glioblastomas with EGFR gene amplification. Oncogene. 1994;9:2313–2320. [PubMed] [Google Scholar]
- 15.Maher EA, et al. Malignant glioma: genetics and biology of a grave matter. Genes Dev. 2001;15:1311–1333. doi: 10.1101/gad.891601. [DOI] [PubMed] [Google Scholar]
- 16.Lang FF, Miller DC, Koslow M, Newcomb EW. Pathways leading to glioblastoma multiforme: a molecular analysis of genetic alterations in 65 astrocytic tumors. J Neurosurg. 1994;81:427–436. doi: 10.3171/jns.1994.81.3.0427. [DOI] [PubMed] [Google Scholar]
- 17.Watanabe K, et al. Overexpression of the EGF receptor and p53 mutations are mutually exclusive in the evolution of primary and secondary glioblastomas. Brain Pathol. 1996;6:217–223. doi: 10.1111/j.1750-3639.1996.tb00848.x. discussion 223–214. [DOI] [PubMed] [Google Scholar]
- 18.Dahlstrand J, Collins VP, Lendahl U. Expression of the class VI intermediate filament nestin in human central nervous system tumors. Cancer Res. 1992;52:5334–5341. [PubMed] [Google Scholar]
- 19.Xue W, et al. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature. 2007;445:656–660. doi: 10.1038/nature05529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Barak Y, Juven T, Haffner R, Oren M. Mdm2 expression is induced by wild type p53 activity. EMBO J. 1993;12:461–468. doi: 10.1002/j.1460-2075.1993.tb05678.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Reynolds BA, Weiss S. Generation of neurons and astrocytes from isolated cells of the adult mammalian central nervous system. Science. 1992;255:1707–1710. doi: 10.1126/science.1553558. [DOI] [PubMed] [Google Scholar]
- 22.Houchens DP, Ovejera AA, Riblet SM, Slagel DE. Human brain tumor xenografts in nude mice as a chemotherapy model. Eur J Cancer Clin Oncol. 1983;19:799–805. doi: 10.1016/0277-5379(83)90012-3. [DOI] [PubMed] [Google Scholar]
- 23.Hu B, et al. Angiopoietin-2 induces human glioma invasion through the activation of matrix metalloprotease-2. Proc Natl Acad Sci USA. 2003;100:8904–8909. doi: 10.1073/pnas.1533394100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Singh SK, et al. Identification of human brain tumour initiating cells. Nature. 2004;432:396–401. doi: 10.1038/nature03128. [DOI] [PubMed] [Google Scholar]
- 25.Finkelstein SD, et al. Histological characteristics and expression of acidic and basic fibroblast growth factor genes in intracerebral xenogeneic transplants of human glioma cells. Neurosurgery. 1994;34:136–143. [PubMed] [Google Scholar]
- 26.Uhrbom L, Hesselager G, Nister M, Westermark B. Induction of brain tumors in mice using a recombinant platelet-derived growth factor B-chain retrovirus. Cancer Res. 1998;58:5275–5279. [PubMed] [Google Scholar]
- 27.Holland EC, Varmus HE. Basic fibroblast growth factor induces cell migration and proliferation after glia-specific gene transfer in mice. Proc Natl Acad Sci USA. 1998;95:1218–1223. doi: 10.1073/pnas.95.3.1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Garcia AD, Doan NB, Imura T, Bush TG, Sofroniew MV. GFAP-expressing progenitors are the principal source of constitutive neurogenesis in adult mouse forebrain. Nat Neurosci. 2004;7:1233–1241. doi: 10.1038/nn1340. [DOI] [PubMed] [Google Scholar]
- 29.Lantos PL, Cox DJ. The origin of experimental brain tumours: a sequential study. Experientia. 1976;32:1467–1468. doi: 10.1007/BF01937439. [DOI] [PubMed] [Google Scholar]
- 30.Dull T, et al. A third-generation lentivirus vector with a conditional packaging system. J Virol. 1998;72:8463–8471. doi: 10.1128/jvi.72.11.8463-8471.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ikawa M, Tanaka N, Kao WW, Verma IM. Generation of transgenic mice using lentiviral vectors: a novel preclinical assessment of lentiviral vectors for gene therapy. Mol Ther. 2003;8:666–673. doi: 10.1016/s1525-0016(03)00240-5. [DOI] [PubMed] [Google Scholar]
- 32.Ray J, Gage FH. Differential properties of adult rat and mouse brain-derived neural stem/progenitor cells. Mol Cell Neurosci. 2006;31:560–573. doi: 10.1016/j.mcn.2005.11.010. [DOI] [PubMed] [Google Scholar]
- 33.Marumoto T, et al. Aurora-A kinase maintains the fidelity of early and late mitotic events in HeLa cells. J Biol Chem. 2003;278:51786–51795. doi: 10.1074/jbc.M306275200. [DOI] [PubMed] [Google Scholar]
- 34.Gage FH, et al. Survival and differentiation of adult neuronal progenitor cells transplanted to the adult brain. Proc Natl Acad Sci USA. 1995;92:11879–11883. doi: 10.1073/pnas.92.25.11879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gagneux P, et al. Human-specific regulation of α 2–6-linked sialic acids. J Biol Chem. 2003;278:48245–48250. doi: 10.1074/jbc.M309813200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.