

Abstract
Thionein (T) has not been isolated previously from biological material. However, it is generated transiently in situ by removal of zinc from metallothionein under oxidoreductive conditions, particularly in the presence of selenium compounds. T very rapidly activates a group of enzymes in which zinc is bound at an inhibitory site. The reaction is selective, as is apparent from the fact that T does not remove zinc from the catalytic sites of zinc metalloenzymes. T instantaneously reverses the zinc inhibition with a stoichiometry commensurate with its known capacity to bind seven zinc atoms in the form of clusters in metallothionein. The zinc inhibition is much more pronounced than was previously reported, with dissociation constants in the low nanomolar range. Thus, T is an effective, endogenous chelating agent, suggesting the existence of a hitherto unknown and unrecognized biological regulatory system. T removes the metal from an inhibitory zinc-specific enzymatic site with a resultant marked increase of activity. The potential significance of this system is supported by the demonstration of its operations in enzymes involved in glycolysis and signal transduction.
Metallothionein (MT) has been studied very extensively since its discovery (1), but the biochemistry of thionein (T), its apoprotein, has received relatively little experimental attention thus far. Efforts to demonstrate its endogenous production have consistently failed, in some measure owing to its lack of any appropriate spectroscopic property that could be a guide to its isolation. A series of manuscripts from this laboratory (2–4) have indicated its transient existence and generation at high local concentrations at the instant of its formation, emphasizing its importance as an endogenous and potent zinc-chelating agent effective at exceedingly low cellular concentrations. We are unaware of any analogous biological substance with corresponding properties. T avidly binds metal ions and is highly susceptible to proteolytic digestion and oxidation. It suppresses the DNA-binding capacity of zinc-finger transcription factors in vitro by sequestering zinc and removing it from their structural sites (5–7). We have now established conditions to identify, isolate, and store T so that it completely retains its function including cluster formation through binding by its sulfhydryl groups (4). We have also shown that zinc is transferred from MT to the apoforms of zinc metalloenzymes and that T is indeed transiently formed in situ during this process (3, 4). For the reverse reaction, T itself does not remove significant amounts of zinc from zinc metalloenzymes. Instead, agents such as glutathione or citrate, which can bind zinc themselves but do not remove it from the active site of zinc metalloenzymes, can serve in the transfer process (4).
In addition to its catalytic role in more than 300 zinc metalloenzymes and its structural role in an even greater number of nonenzymatic proteins, zinc is also a known inhibitor of enzymes in general, including zinc metalloenzymes. In 1960, zinc binding experiments demonstrated a second, nonactive zinc site in carboxypeptidase A (8). Thirty years later this was established as an inhibitory site that is located in the vicinity (3.3 Å) of the catalytic zinc atom and to inhibit the enzyme with an apparent KI value of 24 μM (9). If it is taken into account that ZnOH+ is the inhibitory species, the actual KI value is 0.7 μM. Larsen and Auld (9, 10) proposed that the inhibitory zinc atom is bound to Glu-270 and binds both an anion and a water molecule and that a hydroxide bridges the inhibitory and catalytic zinc atoms, as has been confirmed by x-ray diffraction (11, 12). This particular inhibitory site has proven to be but one of many such inhibitory zinc sites found in nonmetalloenzymes. These have also been recognized recently in neurotransmitter receptors of the central nervous system (13).
We now report that a number of metabolically critical enzymes are also inhibited by nanomolar concentrations of zinc and that T reverses this inhibition. Moreover, under prevalent conditions, T proves to be a very effective agent for the removal of zinc from such inhibitory sites in zinc-inhibited enzymes. Thus, both T and zinc could jointly control enzymatic processes in metabolic and signal-transduction pathways through a mechanism that revolves about site-specific thionein-reversible inhibitory zinc binding to enzymes.
MATERIALS AND METHODS
Materials.
Rabbit liver thionein was prepared from cadmium MT-1 (14), which was a gift from G. J. Xu (Shanghai Institute of Biochemistry), and stored under liquid nitrogen (4). To avoid metal contamination, deionized water (resistivity of ≥15 MΩ⋅cm) and metal-free pipet tips (Fisher) were used throughout. In addition, adventitious metals were removed by treatment of all buffer and substrate stock solutions with 5% (vol/wt) Chelex (Bio-Rad) for 2 h at room temperature and subsequent filtration through Millex-GS microfilters (Millipore). Buffers and stock solutions were purged with nitrogen gas for 30 min. Enzymes were brought into 20 mM Hepes-Na+, pH 7.5 (“buffer”) by passage through a PD-10 gel filtration column (Amersham Pharmacia). Ultrapure MgSO4, MgCl2, ZnCl2, ZnSO4, and KCl were obtained from Johnson Matthey, Valley Forge, PA. Ac-DEVD-AMC was purchased from PharMingen.
Enolase.
Enolase (rabbit muscle) from Sigma that contained no detectable zinc was diluted to a final concentration of 10 nM in buffer containing 2 mM MgCl2, and its activity was measured spectrophotometrically at 237 nm for 2 min after the reaction was started with d-2-phosphoglyceric acid at a final concentration of 1 mM. Analogous experiments were performed with enolase (yeast) from Sigma.
Fructose 1,6-Diphosphatase.
Fructose 1,6-diphosphatase (rabbit liver) from Sigma that contained no detectable zinc was diluted to a final concentration of 5 nM in buffer containing 1 mM MgCl2. Enzyme assays were initiated by addition of d-fructose 1,6-diphosphate to a final concentration of 100 μM, quenched after 15 min, and the phosphate released titrated with the molybdenum blue method (15). This time interval is in the linear range of phosphate production.
Glyceraldehyde 3-Phosphate Dehydrogenase.
Glyceraldehyde 3-phosphate dehydrogenase (rabbit muscle) from Sigma was diluted to final concentrations of 2.5 and 10 nM in buffer containing 30 mM Na2HAsO4. Glyceraldehyde 3-phosphate (30 mM stock solution) and NAD+ (15 mM stock solution) were added to final concentrations of 500 and 250 μM, respectively, and the reaction was monitored spectrophotometrically at 340 nm for 2 min.
Aldehyde Dehydrogenase.
Yeast aldehyde dehydrogenase from Sigma was diluted to a final concentration of 155 nM in buffer containing 100 mM KCl. Acetaldehyde (40 mM stock solution) and NAD+ (5 mM stock solution) were added to final concentrations of 2 and 0.5 mM, respectively, and the reaction was monitored spectrophotometrically at 340 nm for 2 min.
Protein Tyrosine Phosphatase.
The recombinant protein tyrosine phosphatase from Calbiochem used for our studies is a truncated form of the human T cell protein tyrosine phosphatase with an 11-kDa deletion at the C terminus (16). Phosphatase was diluted to a final concentration of 20 nM in buffer. An aliquot of 4-nitrophenyl phosphate was added (100 mM stock solution) to give a final concentration of 1 mM, and the formation of 4-nitrophenolate was monitored spectrophotometrically at 400 nm for 2 min.
Caspase-3.
Recombinant human caspase-3 from PharMingen was diluted to a final concentration of 1.7 nM in buffer. Ac-DEVD-AMC (1.12 mM stock solution) was added to a final concentration of 22.4 μM, and the formation of amino-4-methylcoumarin (AMC) was monitored fluorimetrically for 5 min (excitation wavelength, 380 nm; emission wavelength, 440 nm) with a FluoroMax-2 (Instruments SA, Edison, NJ) fluorimeter (17).
Alcohol Dehydrogenase.
Alcohol dehydrogenase (horse liver) from Boehringer contained 4.8 zinc atoms per dimer. Alcohol dehydrogenase was diluted to a final concentration of 100 nM in buffer. Enzymatic activity was measured by following the formation of NADH spectrophotometrically at 340 nm for 2 min after adding ethanol (6 M stock solution) and NAD+ (25 mM stock solution) to final concentrations of 330 and 1 mM, respectively.
Carbonic Anhydrase.
Carbonic anhydrase (bovine erythrocytes) from Sigma contained 0.93 zinc atoms per monomer. Carbonic anhydrase was diluted to a final concentration of 250 nM in buffer. Enzymatic activity was measured spectrophotometrically at 400 nm for 2 min after adding 4-nitrophenyl acetate (20 mM stock solution in acetonitrile) to a final concentration of 0.5 mM (18).
Glutathione Peroxidase.
Glutathione peroxidase (bovine erythrocytes) from Sigma was diluted to a final concentration of 2.4 nM in buffer. Enzymatic activity was measured spectrophotometrically at 260 nm for 1 min after adding t-butyl hydroperoxide (50 mM stock solution) and glutathione (0.2 M stock solution) to final concentrations of 5 and 2 mM, respectively.
Glutathione Reductase.
Glutathione reductase (yeast) from Sigma was diluted to a final concentration of 8 nM in buffer. Enzymatic activity was measured spectrophotometrically at 340 nm after adding NADPH (4.5 mM stock solution) and glutathione (5 mM stock solution) to final concentrations of 50 and 2 μM, respectively.
Zinc Inhibition.
Enzymes were incubated with ZnSO4 (from 10–100 μM stock solutions in water) and then assayed under initial velocity conditions. Inhibition was instantaneous in all cases. All kinetic experiments, including spectrophotometric measurements with a Cary 1E UV/VIS spectrophotometer, were performed at 25°C.
Effect of T on Enzyme Activities.
Enzymes were preincubated with ZnSO4 for 10 min, and their remaining activity was determined. Zinc concentrations sufficient to inhibit at least 50% of the enzyme’s original activity were chosen. The inhibited enzymes were then incubated with T for 30 min and the recovered activity determined. In a control, T lost <10% of its thiol content in 30 min as ascertained by titrations with 5,5′-dithio-bis(2-nitrobenzoic acid) (4). In another set of experiments, enzymes were incubated with T first, and enzyme activities were measured 10 min after the addition of ZnSO4. The effect of T on alcohol dehydrogenase and carbonic anhydrase was investigated by measuring enzymatic activity after preincubation of the enzymes with an excess of T for 30 min.
RESULTS
Zinc Inhibition of Enzymes.
A number of eukaryotic, cytosolic enzymes not known to require zinc for activity are inhibited by this metal. Among these are glycolytic enzymes, a protease involved in apoptosis, and a tyrosine phosphatase. Some of these enzymes were found to be inhibited by much lower concentrations of zinc than had been reported previously (Table 1). This is in part because of the fact that we have here avoided the use of buffers and reducing agents such as DTT or 2-mercaptoethanol that are also very effective zinc-binding agents. Furthermore, we have reduced the level of zinc contamination of buffers and substrates to below 1 ppb. Under these conditions, the IC50 values for zinc inhibition of the enzymes studied (save rabbit enolase) are below 250 nM. Because inhibition is in the nanomolar range, the IC50 values are a function of the enzyme concentration, which on occasion had to be lowered significantly, sometimes to the limit of sensitivity of the assay. Thus, these data only reflect upper limits, and the true inhibition constants are likely even lower. The inhibition of caspase-3 turns out to be most notable among those examined because it reflects a virtual 1:1 stoichiometric reaction: At equimolar concentrations of enzyme (1.7 nM) and zinc, only 50% of the initial enzymatic activity remains (Fig. 1). Again, the 5-fold higher enzyme concentration employed in a previous study clearly required a higher zinc concentration for inhibition (19).
Table 1.
Zinc inhibition of enzymes
| Enzyme | Concentration, nM | IC50 measured, nM | IC50 reported, nM | Reference |
|---|---|---|---|---|
| Caspase-3 | 1.7 | <10 | 100 | 19 |
| Fructose 1,6-diphosphatase | 5 | 100 | 300* | 20 |
| Glyceraldehyde 3-phosphate dehydrogenase | 2.5 | 150 | — | — |
| Aldehyde dehydrogenase | 155 | 154 | — | — |
| Tyrosine phosphatase | 20 | 200 | 100,000† | 21 |
| Enolase (yeast)‡ | 10 | 250 | 2,500 | 22 |
KI
15% activity remaining.
Enolase (rabbit muscle), 10 nM: IC50 = 1.3 μM.
Figure 1.

Inhibition of caspase-3 by zinc. Caspase-3 (1.7 nM) was incubated with various amounts of zinc. Activity was assayed after 5 min.
Because a catalytic cysteinyl residue is known to be characteristic of glyceraldehyde 3-phosphate dehydrogenase, aldehyde dehydrogenase, protein tyrosine phosphatase, and caspase-3, we have searched for other enzymes that might have potential cysteine (or selenocysteine) ligands at their active site. Unlike the above enzymes, nanomolar concentrations of zinc do not inhibit glutathione peroxidase from bovine erythrocytes (catalytic selenocysteine) or glutathione reductase from yeast (catalytic vicinal cysteines). In the presence of 1 μM zinc, glutathione peroxidase (2.4 nM) loses only 15% of its activity. The IC50 value for glutathione reductase (8 nM) is about 10 μM. Apparently, the presence of a cysteine residue at the catalytic site is not sufficient to provide an inhibitory site: the active site of enzymes with potential metal-binding ligands may be designed in such a way that they are protected from inhibition by zinc or other metal ions.
T-Reversible Inhibition of Enzymatic Zinc Sites.
Incubation with T reactivates zinc-inhibited enzymes, as illustrated for glyceraldehyde 3-phosphate dehydrogenase (Fig. 2). Reactivation is a function of the concentration of zinc relative to that of T but apparently is not a function of the nature of the enzyme. The concentration of T required to overcome the inhibition (Fig. 2) shows that, on average, T sequesters about six to seven zinc atoms, corresponding to the capacity of T to bind seven zinc atoms. T rapidly reactivates these zinc-inhibited enzymes, e.g., during the mixing time, as illustrated for glyceraldehyde 3-phosphate dehydrogenase (Fig. 3).
Figure 2.
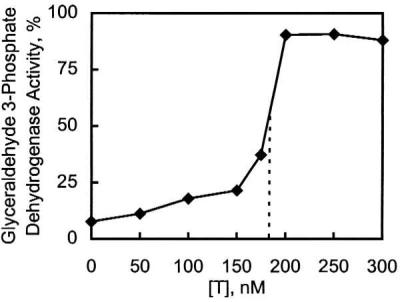
Reactivation of glyceraldehyde 3-phosphate dehydrogenase with T in the presence of zinc. Glyceraldehyde 3-phosphate dehydrogenase (10 nM) was incubated in buffer containing 30 mM Na2HAsO4 with various amounts of T introduced 5 min after ZnSO4 was added to a final concentration of 1 μM. Activities were assayed after 30 min.
Figure 3.

Reactivation of zinc-inhibited glyceraldehyde 3-phosphate dehydrogenase with T. Glyceraldehyde 3-phosphate dehydrogenase (10 nM) was incubated with ZnSO4 (1 μM) for 10 min in buffer containing 30 mM Na2HAsO4. Enzyme activity was measured for 0.5 min; T (250 nM) was then injected into the cuvet, and the measurement continued.
This remarkably efficient removal of zinc from these inhibitory sites prompted us to investigate the effect of T on two zinc enzymes in addition to carboxypeptidase A and alkaline phosphatase, which were studied previously (4). Up to a 20-fold excess of T over alcohol dehydrogenase (100 nM) results in only a marginal loss of its enzymatic activity (6%). Similarly, in the presence of a 12-fold excess of T, carbonic anhydrase (250 nM) retains 85% of its original activity, as has been described (23). Thus, low concentrations of T rapidly react with zinc bound to inhibitory sites of enzymes, but, under these conditions, it cannot extract significant amounts of zinc from the active sites of zinc metalloenzymes.
DISCUSSION
T and MT have persistently been described as unique because of their metal content, amino acid composition, and physicochemical characteristics (24). T binds zinc very tightly to form zinc/sulfur clusters that do not exist in the inanimate world. Thus far, T has not proven to be a particularly effective chelating agent for zinc metalloenzymes and does not inhibit or remove significant amounts of zinc from the catalytic sites of zinc metalloenzymes (4). However, it rapidly activates enzymes that are inhibited by zinc at what we here refer to as specific “zinc-inhibitory sites;” these may be contrasted with zinc-active sites in zinc metalloenzymes. The high specificity of both nanomolar zinc inhibition and removal of zinc from these zinc-inhibitory sites is commensurate with the capacity of T to bind seven zinc atoms and suggests the possible existence of a biological regulatory system that has not been recognized thus far.
The identity and characteristic properties of zinc metalloenzymes (24) are well established. Enzymes that are inhibited by zinc have also been known for a long time. However, reversal of inhibition by T had not been and could not have been described previously, because T was not available in sufficient quantities for such experimentation. We now recognized that T reverses zinc inhibition by competing for the metal at zinc inhibitory sites of enzymes (Figs. 2 and 3). In point of fact, zinc metalloenzymes must be protected from loss of zinc when exposed to T. T seems to distinguish between these active and inhibitory zinc sites likely on the basis of differences in their stability constants and/or their accessibility. The stability constants for zinc inhibitory sites are nanomolar, whereas those of catalytic or structural zinc sites are in the picomolar range. For a relatively large molecule, access to the inner coordination sphere of active site zinc atoms is limited, perhaps because such zinc atoms usually are not on the protein surface. Structural zinc atoms, although sometimes located at the surface of proteins, are fully coordinated and, hence, not readily accessible to chelating agents. Thus, although enzymes with zinc-inhibitory sites do bind zinc tightly, they are not recognized readily to contain zinc, because their isolation may require conditions where zinc dissociates, and then yield an enzyme which is partially active, because a commensurate mole fraction of zinc remains bound. Hence, such zinc-inhibited enzymes cannot be identified by the criteria established to recognize zinc metalloenzymes with catalytic or structural zinc atoms whose biological activity is a direct function of their total zinc content.
The signatures of zinc atoms at catalytic and structural sites are characteristic, e.g., in terms of the spacings of their ligands in the polypeptide chain and the number and types of ligands involved. Zinc metalloenzymes and proteins have been classified and generalized as families (25). The recognition of coactive zinc sites has extended this to binuclear and trinuclear sites (26). At present, the signatures, if any, of zinc inhibitory sites are still unknown and cannot be predicted as yet. Hence, their definition is entirely operational, i.e., based on their zinc inhibition and its reversibility by T. Recent descriptions of the action of d-penicillamine as a “catalytic chelator” (27) or of other agents (4) that further potentiate removal of intrinsic zinc atoms attest to the unknown nature of such mechanisms and point to the possible existence of yet other analogous systems. They seem to represent new inroads into the coordination dynamics and traffic of metal ions in cells and provide mechanisms and pathways for mobilizing, transferring, and distributing metal ions bound tightly to enzymes. Certainly, they would not be recognized easily for want of agents that remove metals with concomitant modulation of biological activity. They do raise the possibility that chelating agents might be effective as catalytic chelators in conjunction with T in a manner proposed by Chong and Auld (27).
Zinc is known to inhibit multiple biological processes, demonstrating that its cellular concentration must be controlled carefully and kept well below the micromolar range to preserve the activity of zinc metalloenzymes and other enzymes critical to metabolism. This is accomplished in part by the cellular MT/T system. If free zinc concentrations were to rise into the micromolar range, numerous enzymatic processes could be compromised. The binding constants for zinc in T-reversible inhibitory sites of enzymes on the one hand and in catalytically active zinc metalloenzyme sites on the other seem to establish the conditions for the availability of cellular zinc while demonstrating the need for its efficient regulation. Dissociation of zinc from zinc metalloenzymes occurs below 10−10 M, whereas zinc inhibition becomes significant above 10−8 M. The definition of this upper setpoint in the nanomolar range depends on the exact magnitudes of inhibition constants.
Zinc is bound tightly to MT (KA(Zn) (Zn7MT) = 3.2 × 1013 M−1), and the zinc content of MT accounts for approximately 50 μM zinc in the cell. The relative abundance of the cellular MT/T couple and its unique features make it an element central in the regulation of zinc. T expression is regulated by a metal response element binding factor (MTF-1), which in turn is itself activated by zinc (28). Because of its high affinity for zinc, T sequesters zinc to low nanomolar concentrations where zinc no longer inhibits enzymes. Although this mechanism leads to down-regulation of zinc, up-regulation mechanisms include oxidative zinc release (2). One may wonder whether such a release of zinc from MT and directed to an inhibitory site could also reflect a physiological control mechanism for the regulation of enzymatic processes.
Clearly, the list of zinc-inhibited enzymes that we have studied is far from exhaustive. Thus, the KI values of bacterial adenylosuccinate synthase and 6-phosphogluconate dehydrogenase are 29 and 21 nM, respectively (29, 30). Their eukaryotic counterparts have not been examined as yet in this regard. Hence, all consequences of zinc inhibition cannot be evaluated at present. Moreover, minimally, studies of additional cellular processes and a more precise definition of both the magnitude of inhibition and T reactivation need to be defined. The roles of T discussed herein may very readily become an important aspect of a mechanism by which metabolic and mitogenic pathways are regulated.
Acknowledgments
This work is the result of E.H.F.’s Visiting Professorship of the Bert L. and N. Kuggie Vallee Foundation, which also supported these studies in the Center for Biochemical and Biophysical Sciences and Medicine at Harvard Medical School. C.J. was supported in part by a BASF fellowship from the Studienstiftung des Deutschen Volkes. Sibylla Braedel distinguished herself by her constructive participation in this work while serving as a trainee in biochemistry from the University of Tübingen, Germany.
ABBREVIATIONS
- T
thionein
- MT
metallothionein
References
- 1.Margoshes M, Vallee B L. J Am Chem Soc. 1957;79:4813. [Google Scholar]
- 2.Maret W, Vallee B L. Proc Natl Acad Sci USA. 1998;95:3478–3482. doi: 10.1073/pnas.95.7.3478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jiang L-J, Maret W, Vallee B L. Proc Natl Acad Sci USA. 1998;95:3483–3488. doi: 10.1073/pnas.95.7.3483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jacob C, Maret W, Vallee B L. Proc Natl Acad Sci USA. 1998;95:3489–3494. doi: 10.1073/pnas.95.7.3489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zeng J, Heuchel R, Schaffner W, Kägi J H R. FEBS Lett. 1991;279:310–312. doi: 10.1016/0014-5793(91)80175-3. [DOI] [PubMed] [Google Scholar]
- 6.Zeng J, Vallee B L, Kägi J H R. Proc Natl Acad Sci USA. 1991;88:9984–9988. doi: 10.1073/pnas.88.22.9984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Roesijadi G, Bogumil R, Vašák M, Kägi J H R. J Biol Chem. 1998;273:17425–17432. doi: 10.1074/jbc.273.28.17425. [DOI] [PubMed] [Google Scholar]
- 8.Coleman J E, Vallee B L. J Biol Chem. 1960;235:390–399. [PubMed] [Google Scholar]
- 9.Larsen K L, Auld D S. Biochemistry. 1991;30:2613–2618. doi: 10.1021/bi00224a007. [DOI] [PubMed] [Google Scholar]
- 10.Larsen K L, Auld D S. Biochemistry. 1989;28:9620–9625. doi: 10.1021/bi00451a012. [DOI] [PubMed] [Google Scholar]
- 11.Gomez-Ortiz M, Gomis-Rüth F X, Huber R, Avilés F X. FEBS Lett. 1997;400:336–340. doi: 10.1016/s0014-5793(96)01412-3. [DOI] [PubMed] [Google Scholar]
- 12.Bukrinski J T, Bjerrum M J, Kadziola A. Biochemistry. 1998;37:16555–16564. doi: 10.1021/bi981678i. [DOI] [PubMed] [Google Scholar]
- 13.Chen N, Moshaver A, Raymond L A. Mol Pharmacol. 1997;51:1015–1023. doi: 10.1124/mol.51.6.1015. [DOI] [PubMed] [Google Scholar]
- 14.Vašák M. Methods Enzymol. 1991;205:41–44. doi: 10.1016/0076-6879(91)05082-7. [DOI] [PubMed] [Google Scholar]
- 15.Tashima Y, Yoshimura N. J Biochem (Tokyo) 1975;78:1161–1169. doi: 10.1093/oxfordjournals.jbchem.a131012. [DOI] [PubMed] [Google Scholar]
- 16.Cool D E, Tonks N K, Charbonneau H, Walsh K A, Fischer E H, Krebs E G. Proc Natl Acad Sci USA. 1989;86:5257–5261. doi: 10.1073/pnas.86.14.5257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nicholson D W, Ali A, Thornberry N A, Vaillancourt J P, Ding C K, Gallant M, Gareau Y, Griffin P R, Labelle M, Lazebnik Y A, et al. Nature (London) 1995;376:37–43. doi: 10.1038/376037a0. [DOI] [PubMed] [Google Scholar]
- 18.Henkens R W, Sturtevant J M. J Am Chem Soc. 1968;90:2669–2676. [Google Scholar]
- 19.Perry D K, Smyth M J, Stennicke H R, Salvesen G S, Duriez P, Poirier G G, Hannun Y A. J Biol Chem. 1997;272:18530–18533. doi: 10.1074/jbc.272.30.18530. [DOI] [PubMed] [Google Scholar]
- 20.Tejwani G A, Pedrosa F O, Pontremoli S, Horecker B L. Proc Natl Acad Sci USA. 1976;73:2692–2695. doi: 10.1073/pnas.73.8.2692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zander N F, Lorenzen J A, Cool D E, Tonks N K, Daum G, Krebs E G, Fischer E H. Biochemistry. 1991;30:6964–6970. doi: 10.1021/bi00242a022. [DOI] [PubMed] [Google Scholar]
- 22.Elliot J I, Brewer J M. J Inorg Biochem. 1980;12:323–334. doi: 10.1016/s0162-0134(00)80273-1. [DOI] [PubMed] [Google Scholar]
- 23.Li T-Y, Kraker A J, Shaw C F, III, Petering D H. Proc Natl Acad Sci USA. 1980;77:6334–6338. doi: 10.1073/pnas.77.11.6334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vallee B L, Falchuk K H. Physiol Rev. 1993;73:79–118. doi: 10.1152/physrev.1993.73.1.79. [DOI] [PubMed] [Google Scholar]
- 25.Vallee B L, Auld D S. Biochemistry. 1990;29:5647–5659. doi: 10.1021/bi00476a001. [DOI] [PubMed] [Google Scholar]
- 26.Vallee B L, Auld D S. Biochemistry. 1993;32:6493–6500. doi: 10.1021/bi00077a001. [DOI] [PubMed] [Google Scholar]
- 27.Chong C R, Auld D S. J Trace Elem Expl Med. 1998;11:335–336. [Google Scholar]
- 28.Radtke F, Heuchel R, Georgiev O, Hergersberg M, Gariglio M, Dembric Z, Schaffner W. EMBO J. 1993;12:1355–1362. doi: 10.1002/j.1460-2075.1993.tb05780.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kang C, Fromm H J. J Biol Chem. 1995;270:15539–15544. doi: 10.1074/jbc.270.26.15539. [DOI] [PubMed] [Google Scholar]
- 30.Niehaus W G, Richardson S B, Wolz R L. Arch Biochem Biophys. 1996;333:333–337. doi: 10.1006/abbi.1996.0399. [DOI] [PubMed] [Google Scholar]