

Abstract
In an effort to further probe metal binding to metallo-β-lactamase L1 (mβl L1), Cu- (Cu-L1) and Ni-substituted (Ni-L1) L1 were prepared and characterized by kinetic and spectroscopic studies. Cu-L1 bound 1.7 equivalents of Cu and small amounts of Zn(II) and Fe. The EPR spectrum of Cu-L1 exhibited two overlapping, axial signals, indicative of type 2 sites with distinct affinities for Cu(II). Both signals indicated multiple nitrogen ligands. Despite the expected proximity of the Cu(II) ions, however, only indirect evidence was found for spin-spin coupling. Cu-L1 exhibited higher kcat (96 s−1) and Km (224 μM) values, as compared to the values of dinuclear Zn(II)-containing L1, when nitrocefin was used as substrate. The Ni-L1 bound 1 equivalent of Ni and 0.3 equivalents of Zn(II). Ni-L1 was EPR-silent, suggesting that the oxidation state of nickel was +2; this suggestion was confirmed by 1H NMR spectra, which showed relatively sharp proton resonances. Stopped-flow kinetic studies showed that ZnNi-L1 stabilized significant amounts of the nitrocefin-derived intermediate and that the decay of intermediate is rate-limiting. 1H NMR spectra demonstrate that Ni(II) binds in the Zn2 site and that the ring-opened product coordinates Ni(II). Both Cu-L1 and ZnNi-L1 hydrolyze cephalosporins and carbapenems, but not penicillins, suggesting that the Zn2 site modulates substrate preference in mβ1 L1. These studies demonstrate that the Zn2 site in L1 is very flexible and can accommodate a number of different transition metal ions; this flexibility could possibly offer an organism that produces L1 an evolutionary advantage when challenged with β-lactam containing antibiotics.
β-Lactams are inexpensive and widely-used antibiotics against microbes since the 1940’s(1). There are three different major classes of β-lactams, penicillins, cephalosporins, and carbapenems, that have been used clinically. However, most microorganisms have obtained the ability to either pump the β-lactams out of the cell via transporter proteins (2) or to hydrolyze these compounds by secreting β-lactamases into the periplasm or milieu (3). Four distinct classes of β-lactamases have been identified (4). Unlike class A, C, and D β-lactamases, which utilize an active site serine as a nucleophile, class B β-lactamases, metallo-β-lactamases or Mβl’s, are a group of enzymes that require Zn(II) to hydrolyze β-lactams (5). There have been >50 Mβl’s identified and categorized into three subgroups, according to amino acid sequence homology, the requirement of Zn(II) (1 or 2) for maximal activity, the identity of the metal binding ligands, and substrate preference. Although the amino acid sequence homology is less than 30% between the different subgroups of Mβl’s, the Zn(II) binding motif, HXHXD, is highly-conserved (5). Most Mβl’s have a Zn1 site, consisting of three histidines and one bridging hydroxide, and a relatively more variable Zn2 site, consisting of two histidines (or one histidine and one cysteine in B1 and B2 Mβl’s), one aspartate, one terminally-bound water, and the bridging hydroxide (Figure 1).
Figure 1.
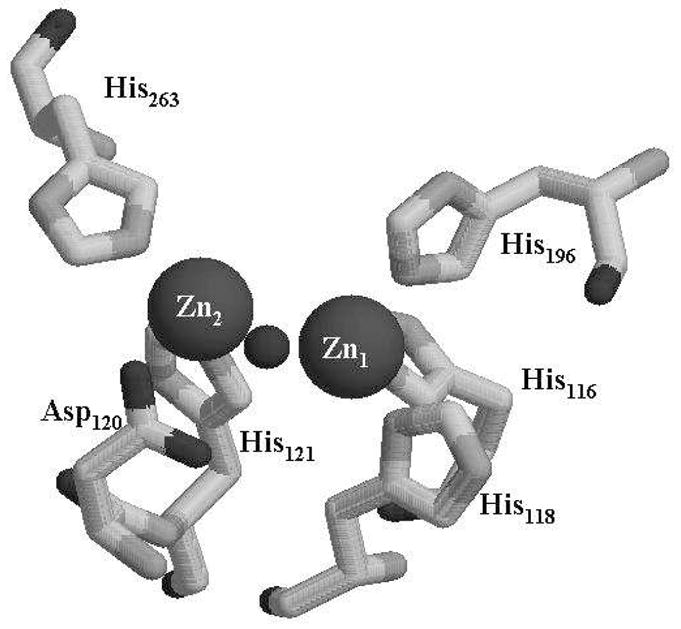
Active site of Mβl L1. The metal ion in the Zn1 site is coordinated by 3 histidines (His116, His118, His196), and one bridging hydroxide (sphere in between two metal ions). The metal ion in the Zn2 site is coordinated by two histidines (His121 and His263), one aspartate (Asp120), one bridging hydroxide, and one terminally bound H2O (not shown).
Since the electronic configuration of Zn(II) is Ar[d10], which makes the metal center in Mβl’s spectroscopically-silent with the most common techniques (6), Zn(II) has often been replaced by Co(II), resulting in catalytically-active analogs that can be characterized by a number of common spectroscopic techniques (7–10). The Zn(II) ions have also been substituted with Cd(II) in Mβl CcrA (11), and this analog was catalytically-active and was characterized with 119Cd NMR spectroscopy (12). While the Mβl’s demonstrate a preference for Zn(II) binding, Crowder and coworkers have recently reported that the metallation of Mβl L1 depends on the bioavailability of metal ions (13). In these studies, L1 was shown to bind Fe, Zn(II), and Mn. This result suggests that L1 contains a highly-flexible metal binding site like that previously reported for plant glyoxalase 2’s (14, 15), raising the question of whether other 1st row transition metal ions could bind to L1. In this work, we explored whether Ni- and Cu-containing analogs of L1 could be prepared, and we characterized the resulting analogs using spectroscopic and kinetic studies. These studies demonstrate that Cu- and Ni-containing analogs of L1 are active and suggest that the flexible metal binding site in L1 offers organisms an evolutionary advantage by being able to produce an enzyme that confers antibiotic resistance in environments containing transition metal ions other than Zn(II).
Experimental Procedures
Preparation of Ni- and Cu-containing analogs of L1
Mature L1 (M-L1) was over-expressed as previously described, and 100 μM NiSO4 or CuSO4 were added to the minimal medium during cell growth and protein production (16, 17). After protein over-expression the E. coli cells were centrifuged for 15 minutes (8,200 × g), and the cell pellet was resuspended in 300 mL of 50 mM Hepes (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid), pH 6.0. The suspension was centrifuged for 15 minutes (8,200 × g), and the resulting pellet was resuspended in 50 mM Hepes, pH 6.0. The cells were lysed by using a French press as previously described (18), and the mixture was centrifuged for 25 minutes (23,400 × g). The cleared, crude protein solution was loaded onto a 25 mL SP-Sepharose column that was equilibrated with 50 mM Hepes, pH 6.0, and bound proteins were eluted from the column using a linear 0 – 500 mM NaCl gradient in the same buffer. L1 typically eluted at 80–120 mM NaCl, and the fractions were analyzed for the presence of L1 by using SDS-PAGE, as previously described (18).
Metal anaylses
The metal content of the protein samples was determined by using a Varian Liberty 150 Inductively Coupled Plasma spectrometer with atomic emission spectroscopy detection (ICP-AES). All the proteins were diluted to 10 μM with 50 mM Hepes, pH 7.0. A calibration curve with 4 standards and a correlation coefficient of greater than 0.999 was generated using Zn(II), Cu, and Ni reference solutions from Fisher Scientific. The following emission wavelengths were chosen to ensure the lowest detection limits possible: Zn(II), 213.856 nm, Cu, 324.754 nm, and Ni, 221.647 nm.
1H NMR spectroscopy
1H NMR spectra were collected on a Bruker Avance 500 spectrometer operating at 500.13 MHz, 298 K, magnetic field of 11.7 T, recycle delay (AQ) of 41 ms, and sweep width of 400 ppm. Proton chemical shifts were calibrated by assigning the H2O signal the value of 4.7 ppm. A modified presaturation pulse sequence (zgpr) was used to suppress the proton signals originating from solvent. The presaturation pulse was as short as possible (500 ms) to avoid saturation of paramagnetically-shifted proton signals. The concentration of NMR samples was generally in the range of 1.0 – 1.2 mM. Samples in D2O were prepared by performing three or more dilution/concentration cycles in a Centricon-10.
EPR spectroscopy
Low temperature EPR spectroscopy was carried out using a Bruker EleXsys E600 spectrometer equipped with an Oxford Instruments ITC503 liquid helium flow system. EPR spectra were recorded at 9.63 GHz using an ER4116DM cavity, with 0.4 mT (4 G) magnetic field modulation at 100 kHz. Other EPR recording parameters are given in the legends to figures. Computer simulations were carried out using XSophe v.1.1.4 (Bruker Biospin).
Steady-state kinetics
All kinetic studies were conducted on an Agilent 8453 UV-Vis diode array spectrophotometer at 25 °C. Steady-state kinetic parameters, the Michaelis constant Km and the turnover number kcat, were determined by monitoring product formation at 485 nm using nitrocefin or substrate decay at 235 nm for pencillin G, 280 nm for cefaclor, or 295 nm for imipenem in 50 mM Chelex-treated, cacodylate, pH 7.0. Absorbance changes were converted to concentration changes using Beer’s law and the extinction coefficients (in M−1cm−1) of nitrocefin product (17,420), penicillin G (−926), cefaclor (−6,410), or imipenem (−9,000), respectively.
Stopped-flow kinetic studies
Stopped-flow kinetic experiments were performed on an Applied Photophysics SX18MV spectrophotometer equipped with a constant temperature circulating water bath as previously described (19). All experiments were performed in 50 mM Chelex-treated, cacodylate buffer, pH 7.0, at 10 °C. All the proteins were diluted with 50 mM Chelex-treated, cacodylate buffer to 100 μM and nitrocefin was prepared and diluted to 100 μM in the same buffer. The progress UV-Vis and fluorescence curves were fitted with single or double exponential equation.
Results
Preparation and characterization of Ni(II)- and Cu(II)-containing analogs of L1
L1 was over-expressed using an over-expression plasmid that contains the gene for mature L1 (M-L1), which lacks the N-terminal targeting sequence (20). This over-expression plasmid has been previously used to prepare Fe-containing analogs of L1 that are folded in the cytoplasm of E. coli. When M-L1 was over-expressed in minimal medium containing 100 μM Cu(II), 5 mg of purified protein could be obtained per liter of growth medium. The resulting protein was shown to bind 1.7 ± 0.1 (Table 1) equivalents of Cu, 0.10 ± 0.05 equivalents of Zn(II), and 0.20 ± 0.05 equivalents of Fe. When M-L1 was over-expressed in minimal medium containing 100 μM Ni(II), 4 mg of purified protein could be obtained per liter of growth medium. The resulting protein was shown to bind 1.0 ± 0.1 equivalents of Ni, 0.30 ± 0.05 equivalents of Zn(II), and no other metal ions at greater than 0.03 equivalents.
Table 1.
Steady-state kinetic studiesa and metal analyses on Cu- and Ni-containing analogs of L1.
| Analog | kcat (s−1) | Km (μM) | Metal content (eq) |
|---|---|---|---|
| ZnZn-L1b | 26 ± 1 | 4 ± 1 | 1.9 ± 0.1 Zn(II) |
| Cu-L1 | 96 ± 8 | 224 ± 20 | 1.7 ± 0.1 Cu 0.10 ± 0.05 Zn(II) 0.20 ± 0.05 Fe |
| Ni-L1c | 24 ± 2 | 18 ± 2 | 0.30 ± 0.05 Zn(II) 1.0 ± 0.1 Ni |
| NiZn-L1d | 36 ± 1 | 16 ± 1 | 1.0 Zn(II), 1.0 Ni(II) |
Steady state kinetic studies were conducted at 25 °C in 50 mM Chelex-treated, cacodylate, pH 7.0, using nitrocefin as the substrate
Data from reference (21).
As-isolated Ni-L1
Sample prepared by direct addition of Zn(II) to as-isolated Ni-L1.
The Cu- and Ni-containing analogs of L1 were characterized using steady-state kinetic studies. As-isolated Cu-containing L1 (called Cu-L1), which contained 1.7 eq. of Cu (Table 1), exhibited a kcat of 96 ± 8 s−1 and a Km of 224 ± 20 μM, when using nitrocefin as substrate. While exhibiting a larger kcat than wild-type L1, which binds 1.9 equivalents of Zn(II), Cu-L1 exhibits a Km that is 56 times larger than that of ZnZn-L1, resulting in a kcat/Km value that is >10-fold lower than that of ZnZn-L1. The as-isolated Ni-containing analog of L1 (called Ni-L1) exhibited a kcat of 24 ± 2 s−1 and a Km of 18 ± 2 μM, when using nitrocefin as substrate. Zn(II) (0.7 equivalents) was added to as-isolated Ni-containing L1 to generate ZnNi-L1, and this analog exhibited a kcat of 36 ± 1 s−1 and a Km of 18 ± 1 μM.
In order to test the substrate specificity of the Cu- and Ni-containing analogs of L1, three different substrates, penicillin G, cefaclor, and imipenem, were used in steady-state kinetic studies with the metal-substituted forms of L1, and the results from these studies were compared to results with ZnZn-L1 (Table 2). When cefaclor, a cephalosporin like nitrocefin, was used as substrate, Cu- and Ni-containing L1 exhibited kcat values similar to those exhibited by ZnZn-L1; however, the Km values exhibited by both metal-substituted analogs were significantly higher. With the carbapenem imipenem as substrate, Cu- and ZnNi-L1 exhibited much higher kcat and Km values than ZnZn-L1. The largest difference in kinetic behavior, however, was observed when penicillins were used as substrates. Neither Cu- nor ZnNi-L1 hydrolyzed penicillin G (Table 2) or ampicillin, while the ZnZn-analog hydrolyzed penicillin G with a high kcat value.
Table 2.
Steady state kineticsa of different L1 analogs with different substrates.
| Penicillin G | Cefaclor | Imipenem | ||
|---|---|---|---|---|
| ZnZn-L1 | kcat (s−1) | 761 | 38 | 13 |
| Km (μM) | 278 | 8 | 2 | |
| kcat/Km (μMs)−1 | 2.7 | 4.8 | 6.5 | |
| ZnCo-L1 | kcat (s−1) | 692 | 26 | 12 |
| Km (μM) | 218 | 40 | 23 | |
| kcat/Km (μMs)−1 | 3.2 | 0.65 | 0.52 | |
| CoCo-L1 | kcat (s−1) | 118 | 14 | 43 |
| Km (μM) | 36 | 43 | 13 | |
| kcat/Km (μMs)−1 | 3.3 | 0.33 | 3.3 | |
| ZnFe-L1 | kcat (s−1) | < 1 | 16 | 59 |
| Km (μM) | ND | 35 | 27 | |
| kcat/Km (μMs)−1 | ND | 0.46 | 2.2 | |
| Cu-L1 | kcat (s−1) | < 1 | 29 | 205 |
| Km (μM) | ND | 58 | 42 | |
| kcat/Km (μMs)−1 | ND | 0.50 | 4.9 | |
| ZnNi-L1 | kcat (s−1) | < 1 | 27 | 166 |
| Km (μM) | ND | 91 | 61 | |
| kcat/Km (μMs)−1 | ND | 0.30 | 2.7 |
Standard deviation < 10%. ND = not determined.
Steady-state kinetic studies were conducted at 25 °C in 50 mM Chelex-treated cacodylate, pH 7.0.
To test whether other metal-substituted analogs of L1 exhibited similar substrate specificity profiles, the ZnCo-, CoCo-, and ZnFe-analogs were prepared as previously-reported (13, 20, 21) and used in steady-state kinetic studies with the same substrates (Table 2). The ZnCo analog exhibited steady-state kinetic constants most similar to those of ZnZn-L1; however, the ZnCo analog did exhibit Km values for cephalosporins and carbapenems much higher than those of ZnZn-L1. The CoCo analog of L1 exhibited activity with all substrates tested; however, this analog exhibit a much lower Km value for penicillins and higher Km values for cephalosporins and penicillins as compared to those of ZnZn-L1. The ZnFe analog of L1 behaved similarly as Cu-L1 and ZnNi-L1 with respect to the values of Km; however, this analog did not exhibit the higher kcat values that were exhibited by Cu- and ZnNi-L1. Like Cu- and ZnNi-L1, the ZnFe analog of L1 exhibited no hydrolysis activity towards pencillin G.
Spectroscopic studies on Cu- and Ni-containing analogs of L1
As-isolated Cu-L1 was pale blue, and the UV-Vis spectrum of the enzyme revealed a broad peak between 500–950 nm (molar absorptivity at 780 nm of 120 M−1cm−1) (data not shown). In contrast, the as-isolated Ni-containing analog of L1 was pale green; however, there were no resolved UV-Vis peaks between 330 and 1000 nm, although there was a broad, low intensity absorbance starting at 500 nm and extending to almost 1000 nm. The EPR spectrum of L1 (Figure 2) was complex but some information was available from computer simulations. Comparison of the spectra recorded at 0.1 mW and at 2 mW indicated that the spectrum was comprised of a component that exhibited a resolved 63/65Cu hyperfine pattern in the g|| region, and an additional component that was most evident as an broad feature at 2900 G - 3250 G (290 mT - 325 mT) and that exhibited no resolved hyperfine lines. The component with resolved Cu hyperfine appeared to account for an estimated 65 % of the total Cu(II), and was itself a mixture of two species, as is evident from the number and intensity of hyperfine lines (Figure 2B). A very good simulation of the resolved features in the 63/65Cu hyperfine region of the spectrum was obtained assuming two species, both indicative of Type 2 Cu(II) with nitrogen ligation. The major component accounted for 70 % of the resolved signal (45 % of Cu(II)), and exhibited g|| = 2.284 and A||(Cu) = 15.5 × 103 cm−1 (full and detailed parameters for the simulations are given in the caption to Figure 2). Peisach-Blumberg analysis (Figure 3A) (22) suggests three nitrogen and one oxygen ligands in the equatorial coordination plane for this copper ion. The minor component accounted for 30 % of the resolved signal (20 % of Cu(II)), and exhibited g|| = 2.222 and A||(Cu) = 16.0 × 10−3 cm−1. These values do not lie in a well-defined region of the Peisach-Blumberg diagram (Figure 3B), but are consistent with nitrogen ligands in the equatorial plane and additional ligation by some moiety with electron-withdrawing character, though perhaps not so much as a thiolate ligand.
Figure 2.

EPR spectrum from Cu(II)-containing L1. (A) Experimental ∂χ/∂B EPR spectrum recorded at 10 K, 0.1 mW microwave power (thick line) and 10 K, 2 mW power (thin line). (B) Derivative (∂χ/∂B2) of the 63/65Cu hyperfine region of the spectrum. (C) Simulation of (B) assuming two species; the spin Hamiltonian parameters for the major species (accounting for 70 % of the spins) were g|| = 2.284, A||(Cu) = 15.5 × 10−3 cm−1, g⊥ = 2.055, A⊥ (Cu) = 1.10 × 10−3 cm−1, A⊥ (3 × 14N) = 1.35 × 10−3 cm−1; the parameters for the minor species (30 %) were g|| = 2.222, A||(Cu) = 16.0 ×10−3 cm−1, g⊥ = 2.032 (D) Derivative (∂χ/∂B2 of the g⊥ region of the spectrum. (E) – (G) Calculated, (∂χ/∂B2) spectra, assuming g|| = 2.284 A||(Cu) = 15.5 × 10−3 cm−1, g⊥2.055 ± 0.005, and; (E) A⊥ (Cu) = 1.10 × 10−3 cm−1, A⊥ (3 × 14N) = 1.35 × 10−3 cm−1; (F) A⊥ (3 × 14N) = 1.35 × 10−3 cm−1; (G) A⊥ (Cu) = 1.10 × 10−3 cm−1, A⊥ (2 × 14N) = 1.35 × 10−3 cm−1; (H) A⊥ (Cu) = 1.60 × 10−3 cm−1, A⊥ (2 × 14N) = 1.35 × 10−3 cm−1.
Figure 3.

Peisach Blumberg plot showing the suggested coordination for the major (A) and minor (B) species of Cu(II) in L1.
Further information from the spectra was obtained from an analysis of the g⊥ region of the spectrum (Figure 2D). A well-resolved superhyperfine pattern was exhibited in this region and simulations indicated that this pattern was associated with the major component of the spectrum. The g⊥ region of Cu(II) spectra, particularly at X-band, is difficult to interpret due to contributions from 63/65Cu, 14N, and the angular anomaly, or “overshoot” line. However, comparison of matrix diagonalization simulations was instructive. A simulation (Figure 2E) assuming a single, relatively small A⊥ due to copper, and a splitting due to three nitrogen ligands with typical superhyperfine coupling constants, reproduced all of the features in the experimental spectrum with faithful reproduction of their intensities. Other models with only three nitrogens and no copper (Figure 2F), or with two nitrogens and a range of copper coupling constants (e.g., Figure 2G, H), either did not return the correct relative intensities or did not reproduce all of the features in the experimental spectrum. Thus, the higher affinity Cu(II) species in L1 is characterized by at least three equatorial nitrogen ligands.
In addition to the spectral features that were characterized by simulation, some broad absorption between 290 and 320 mT (2900 – 3200 G) was observed that resisted saturation more than the Type 2 signals (Figure 2A). Slight changes to the shape of the high-field side of the g⊥ line were also observed upon increasing the microwave power. The expected inter-Cu(II) distance of 3.5 Å (0.35 nm) would, in turn, be expected to yield features in the EPR spectrum due to dipolar coupling. In addition, any bridging ligand might be expected to lead to either further splitting of the spectrum, or loss of the spectrum completely, due to exchange coupling. Further analysis of the spectra did indeed yield some evidence for spin-coupling and is presented in Figure S1 of the Supplementary Material. A difference spectrum obtained by subtraction of the 0.1 mW spectrum from the 2 mW spectrum (Figure S1 A) exhibited intensity at 3150 G (315 mT) and 3520 G (352 mT) that would not be expected for mononuclear Cu(II). The difference spectrum could not be comprehensively reproduced by computer simulation, but a reasonable model was obtained that invoked a Cu-Cu distance of 5 Å (Figure S1 B). Close examination of the high field region of the spectrum recorded at 2 mW also revealed a broad feature centered at 5000 G (500 mT) that was suggestive of a weak exchange interaction of about 0.2 cm−1. This feature is similar to that observed from a μ-hydroxy-bridged di-Cu center with a Cu-Cu distance of about 3.5 Å (23). However, for such a system, an easily observed half-field line would be expected but is not seen in the spectra of Cu-L1. For a longer distance of 5 Å, simulations indicated that the much less intense half-field line would have been obscured by even the small signal due to traces of Fe-L1 in the sample. Careful observation and simulation of the signals do suggest, then, that there is a subpopulation (30%) of the Cu(II) that experiences Cu-Cu spin coupling. However, while the precise magnitude of the Cu-Cu distance cannot be stated with any certainty, given the lack of an complete solution by simulation, it certainly appears to be significantly greater than 3.5 Å. Alternatively, if a Cu-Cu center with an inter-Cu distance of 3.5 Å is indeed present, there must be considerable structural inhomogeneity in the sample, such that a wide distribution of Euler angles and/or distances renders the expected dipolar features too broad to be observed. Either way, it appears that L1 does not form a well-defined dinuclear Cu center that structurally corresponds to the di-Zn center in the native enzyme. Up to 65 % of the Cu(II) in L1 containing 1.7 eq Cu can be accounted for by apparently magnetically-isolated Cu(II), although this figure should be treated as an upper limit due to the extensive overlap of multiple signals, including those with a putative dipolar component. Regarding the remainder, analogy can be drawn with the aminopeptidase from Vibrio proteolyticus (Aeromonas proteolytica), in which two Zn(II) ions at 3.5 Å apart can be substituted by two Cu(II) ions that form a center with an inter-Cu distance of 5 Å (30).
The Cu-containing analog of L1 did not yield a 1H NMR spectrum, and this result is not surprising given the very slow electron relaxation time exhibited by magnetically-isolated Cu(II) (24). EPR suggested that only maybe 30 % of Cu(II) may exhibit relaxation-enhancing magnetic interactions that would facilitate observation of an NMR signal, and these interactions are likely to be of a number of types or a distribution, rather than a discrete interaction typical of a well-defined dinuclear site. While the Ni-containing analog of L1 did not yield an EPR spectrum, Ni(II) exhibits an electronic relaxation time of 10−13–10−11s (24), which makes Ni(II) an excellent metal ion for 1H NMR studies. The 1H NMR spectrum of the Ni-containing analog of L1 showed 4 resolved, paramagnetically-shifted peaks: one peak at 18 ppm and three peaks between 55–80 ppm (Figure 4A). Peaks a, c, and d integrated to 1 proton each, and peak b integrated to 2 protons. Peak b was assigned to a non-exchangeable proton, possibly an ortho proton on a Ni(II)-bound histidine (24). Peaks c and d disappear completely when the buffer in the Ni-L1 samples is exchanged with buffer in 90% D2O, suggesting that these two protons can be assigned to NH protons on metal-bound histidines (Figure 4B). This result strongly suggests that Ni(II) binds to the Zn2 site in L1, since the Zn2 site has 2 metal binding histidines (25). We cannot unambiguously rule out the possibility that Ni(II) binds in a position that uses metal binding histidines from both the Zn1 and Zn2 site. While this behavior has been reported for Mβl BcII (26), the use of ligands from both sites to bind a single equivalent of metal has not been reported in any of the structural studies on L1.
Figure 4.

1H NMR spectra of Ni-L1. (A) Spectrum of Ni-L1 in buffer containing 10% D2O, (B) spectrum of Ni-L1 in buffer containing 90% D2O, and (C) spectrum of Ni-L1with nitrocefin in buffer containing 90% D2O. The enzyme concentration was 1.0 mM, and the buffer in these samples was 50 mM Hepes, pH 7.0. The spectra were collected at a temperature of 300 K on a 500 MHz NMR spectrometer.
In order to probe the function of Ni(II) in the Zn2 site of Ni-L1, one milligram of nitrocefin powder (to avoid dilution of the protein) was added to the Ni-L1 sample in 90% D2O. The 1H NMR spectrum was immediately obtained (Figure 4C), and a new peak at 65 ppm was observed. The intensity of this peak increased and stabilized over 15 minutes. We assign this peak to a proton on product (Figure 4C).
Pre-steady state kinetic studies on Cu and Ni-containing analogs of L1
In an effort to probe whether the different metal ions in Cu-L1 and Ni-containing L1 affected the kinetic mechanism of the analogs, pre-steady state kinetic studies were carried out on the reaction of 50 μM Cu-L1 or Ni-containing L1 with 50 μM nitrocefin (Figures 5 and 6). Stopped-flow fluorescence studies, in which changes in intrinsic tryptophan fluorescence were monitored over time, showed a rapid decrease in fluorescence over the first 0.05 seconds of the reaction and a relatively slower regain of fluorescence over the subsequent 1 second (Figure 5A) when using as-isolated Cu-L1. A similar stopped-flow fluorescence progress curve was observed with ZnZn-L1, and the fluorescence changes were attributed to changes in the fluorescence properties of Trp38 (19, 27), which is close to the metal ion in the Zn2 site (25), upon substrate binding. The stopped-flow fluorescence curve strongly indicates that Cu(II) in Cu-L1 plays a similar role as Zn(II) in the native enzyme. Stopped-flow UV-Vis studies on Cu-L1 showed a rapid decrease in substrate concentration (rate = 3.6 ± 0.1 s−1), and a rapid formation of product (rate = 2.6 ± 0.1 s−1), which matched the fluorescence recovery (2.8 ± 0.1 s−1). The rate of intermediate decay (rate = 1.0 ± 0.1 s−1) was slower than the rate of product formation. The rapid kinetic studies also revealed the presence of significant amounts of reaction intermediate, which absorbs at 665 nm. However, the intermediate in the reaction of Cu-L1 with nitrocefin was only 15% of that observed for ZnZn-L1 (21).
Figure 5.

Stopped-flow kinetic studies on Cu-L1. (A) Stopped-flow fluorescence trace of the reaction of 50 μM Cu-L1 and 50 μM nitrocefin at 10 °C. (B) Stopped-flow UV-Vis traces of the reaction of 50 μM Cu-L1 with 50 μM nitrocefin at 10 °C. The absorbance at 485 nm is due to the product, the absorbance at 390 nm is due to the substrate, and the absorbance at 665 nm is due to the intermediate.
Figure 6.

Stopped-flow kinetic studies on Ni-containing L1. (A) Stopped-flow UV-Vis traces of the reaction of 50 μM Ni-L1 with 50 μM nitrocefin 10 °C. The absorbance at 485 nm is due to the product, the absorbance at 390 nm is due to the substrate, and the absorbance at 665 nm is due to the intermediate. (B) Stopped-flow UV-Vis traces of the reaction of 50 μM ZnNi-L1 with 50 μM nitrocefin 10 °C. The absorbance at 485 nm is due to the product, the absorbance at 390 nm is due to the substrate, and the absorbance at 665 nm is due to the intermediate. (C) Stopped-flow fluorescence trace of the reaction of 50 μM ZnNi-L1 with 50 μM nitrocefin at 10 °C.
The pre-steady state kinetic behavior of Ni-L1, which contains 1 equivalent of Ni(II) and 0.3 equivalents of Zn(II), was unusual. The substrate depletion progress curve showed a very rapid decrease in absorbance over the first 20 milliseconds of the reaction, followed by a linear decrease in absorbance until substrate was depleted (Figure 6). The intermediate progress curve showed a rapid increase, a relatively-long presence of intermediate over 600 milliseconds, and a decrease in intermediate concentration. The product formation curve appears to be linear over the first 600 milliseconds of the reaction with no apparent rapid phase like that seen in the substrate decay or intermediate formation plots (Figure 6A). The stopped-flow UV-Vis plots of ZnNi-L1 (Figure 6B), which contained 1 eq. of Zn(II) and 1 eq. of Ni(II), were very similar to those of ZnZn-L1 (21). There was a rapid decrease (269 ± 20 s−1) in substrate concentration and increase (188 ± 1 s−1) in intermediate concentration, and a decrease (15.0 ± 0.1 s−1) in intermediate concentration that was similar to that of product formation (15.8 ± 0.1 s−1). The stopped-flow fluorescence curve of this analog showed the typical decrease (140 ± 3 s−1) in intrinsic tryptophan fluorescence (27), followed by a slower regain (12.3 ± 0.2 s−1) of fluorescence (Figure 6C).
Discussion
Recently, Hu et al. showed that the metallation of Mβl L1 is dependent on the bioavailability of metal ions (13). This study showed that mature L1, which is produced from an over-expression plasmid containing the gene for L1 lacking the targeting sequence, could be isolated containing iron, zinc, or cobalt when the enzyme was over-expressed in minimal medium containing Fe(II), Zn(II), or Co(II), respectively. In an effort to extend this study, we used the same method to demonstrate that L1 containing 1.7 equivalents of Cu(II) or 1.0 equivalents of Ni(II) can be isolated. Our data show that L1 can bind ~ 2 equivalents of Zn(II), Co(II), Cu(II), or Fe, and that the first three dinuclear metal ion-containing analogs are catalytically-active, while the last analog is inactive (13). Since the EPR spectrum of Fe-containing L1 shows evidence for an antiferromagnetically-coupled Fe(III)Fe(II) center (28), it is possible that Asp120 bridges the two Fe ions and is not available for orienting the bridging solvent molecule for nucleophilic attack or proton transfer (29).
L1 can also bind 1 equivalent of Ni(II) in the Zn2 site, and the Ni(II)-containing analog utilizes the same reaction mechanism as the wild-type enzyme, at least when nitrocefin is used as the substrate. This flexibility of the metal binding sites is unusual; however, there are examples of other metalloenzymes with similar properties. For example, AAP (aminopeptidase from Aeromonas proteolytica), which is an unrelated, dinuclear Zn(II)-containing enzyme, is also active containing dinuclear copper, nickel, and cobalt centers (30–33). In addition, several heterobimetallic analogs (CoZn, etc.) of AAP are active (30, 31). These heterobimetallic forms are possible because of the sequential metal binding properties of the enzyme. In L1, Zn(II) preferentially binds to the Zn1 site, which is made up of 3 histidines and 1 bridging hydroxide, and Co(II) and Ni(II) preferentially bind to the Zn2 site ((21) and data herein). The EPR data in the present study indicate two copper binding sites, one of high affinity (fully-occupied) and the other of lower affinity (~ 45 % occupied). The high affinity site provides at least three nitrogen ligands in the equatorial plane and is thus assignable to the Zn1 site. The lower affinity site is consistent with Type 2 Cu(II), with nitrogen ligation, and with a mildly electron-withdrawing moiety ligated to Cu(II). These characteristics are all consistent with the Zn2 site being the low affinity Cu(II) binding site. Contrarily, inconsistent with this proposal is the observation that the two Cu(II) signals are S = ½. The lack of EPR signals characteristic of dinuclear copper is not definitive evidence for the lack of a dinuclear center. However, a substantial amount of an EPR-silent dinuclear copper center would require that much of the copper in L1 is EPR-silent, and this is not the case. In addition, even strongly antiferromagnetically-coupled Cu(II)-Cu(II), e.g. 50 cm−1 (33), would have substantially populated excited states at ambient temperatures, and NMR signals would be expected. The available data suggest, then, that the low-affinity Cu site is not the Zn2 site. Cu(II) has a propensity to bind to amide nitrogen, even in the presence of available histidine, in a manner that is not generally observed for Zn(II) (34). This may account for binding of the second Cu(II) in an alternative site or sites. Cu(II) is similarly distributed in AAP between magnetically-isolated sites and a dinuclear site with an inter-Cu distance of 5 Å, in contrast to the inter-Zn distance of 3.5 Å in the native enzyme. As mentioned above, we believe that the spin-coupling in 2Fe-L1 is due to the recruitment of Asp120 as a bridging ligand, and EXAFS studies on this analog are currently in progress to confirm this hypothesis. Importantly, 2Fe-L1 is catalytically inactive. A recent crystal structure of L1 showed 3 equivalents of copper bound to the enzyme (Figure 7) (35). Two of the coppers were bound in the active site, and the M—M internuclear bond distance was 3.5 Å, which is probably too long for a dinuclear metal center with a bridging hydroxide and a bridging carboxylate. A third copper, in complex with phenanthroline, was bound at a remote site, utilizing a single His (His51) to bind to the enzyme. In the present case, with only 1.7 eq Cu(II) bound and with both EPR and NMR evidence against a 3.5 Å Cu-Cu center, it seems likely that only the Zn1 site of the dinuclear center is occupied, with additional copper bound at one or more other sites, one possibly within 5 Å of Zn1.
Figure 7.

Left: Structure of the Cu-containing analog of L1 showing the third Cu binding site. Right: Metal binding site of the Cu-containing analog of L1.
1H NMR studies on ZnNi-L1 yielded novel information about product binding to the metal center. The 1H NMR spectra (Figure 4) of Ni-L1, which binds 1 equivalent of Ni(II) and 0.3 equivalents of Zn(II), showed that only two histidines were bound to the paramagnetic metal ion. We cannot unambiguously rule out the possibility that Ni(II) coordinates one histidine from the Zn1 site and one histidine from the Zn2 site; however, this type of binding mode is unprecedented in studies on L1. In addition, the NMR spectrum of the NiZn-sample showed that the non-solvent-exchangeable peak (peak b in Figure 4) did not shift, suggesting that Ni(II) did not change binding ligands upon addition of Zn(II) (data not shown). The fact that Ni-L1 contains some Zn(II), that Zn(II) can be added to Ni-L1 to generate a more active analog, and that there are only 2 histidines bound to Ni(II) strongly suggests that Ni(II) is bound in the consensus Zn2 site in L1. Peak c in Figure 4A and peak c* in Figure 4C are shifted downfield to similar positions, suggesting similar chemical environments for both protons. Given that the proton corresponding to peak c (Figure 4A) is assigned to the remote, solvent-exchangeable proton on a Ni(II)-bound histidine (proton is 2 bonds or 3 atoms from Ni(II)), it is possible that the proton corresponding to peak c* in Figure 4C is similarly positioned away from the Ni(II) ion. If Ni(II) binds to the nitrocefin product via the ring-opened nitrogen (5), the only proton that is 2 bonds (3 atoms) away from Ni(II) is the proton on the 7-position of the product. This proton is not solvent-exchangeable, like peak c*, and we tentatively assign peak c* to this proton. To our knowledge, this is the first NMR spectroscopic evidence of an enzyme-product species with the product coordinated to the metal center in the Mβl’s. The observation of the EP complex in NMR studies is surprising, given the prediction that the KD for the EP complex was predicted to be > 350 μM (36). Nonetheless, this observation validates the assumption, made in several kinetic mechanisms (7, 36), that a distinct EP can form.
Pre-steady state kinetic studies showed that both Cu-L1 and ZnNi-L1 utilize a similar kinetic mechanism (tryptophan fluorescence behavior, reaction intermediate) to hydrolyze nitrocefin as ZnZn-L1 and CcrA (Figures 5, 6) (19, 37); however, the kinetic behavior of the Cu-L1 analog is different. The rate of product formation in Cu-L1 is similar to the rate of fluorescence regain (2.6 and 2.8 s−1, respectively); these rates are higher than the rate of intermediate decay (1 s−1), and suggest that Cu-L1 may utilize two different pathways to hydrolyze nitrocefin. One pathway, which accounts for <30% of the turnover, utilizes an intermediate, as shown by the species that absorbs at 665 nm in Figure 5B. It is tempting to speculate that the catalytic species responsible for this is the subpopulation of L1 in which a dinuclear center is formed, albeit one with a longer inter-metal distance than in native enzyme. This, in turn, suggests a role for the second metal ion in stabilizing the ‘665 nm intermediate’. The remaining turnover, 70%, is accomplished through a mechanism in which no intermediate can be detected. Given that up to 65% of the Cu(II) in L1 is present as magnetically-isolated Cu(II), it appears that most of the turnover is accomplished by a mononuclear Cu(II) ion that resides in the Zn1 site. The lack of an adjacent, additional metal ion may force the adoption of an alternative mechanism, in which the 665 nm intermediate is not formed. This seems unlikely, given the comparable catalytic competencies of the two mechanisms. Alternatively, in the absence of a second metal ion, there is nothing to stabilize the 665 nm intermediate; the rate of its decay is much faster than the rate limiting step of the reaction and it is not detected in the pre-steady state kinetic experiments.
The hydrolytic activity of ZnNi-L1 was much higher compared than that of Cu-L1. The rates of substrate depletion and intermediate formation were very high, while the rates of production formation and intermediate decay were >10-fold lower (Figure 6). The unusual shape of the progress curve of intermediate is most likely due to the sample containing mixtures of ZnNi- and Ni-containing analogs (Figure 6A). When this sample is made to be predominantly ZnNi (Figure 6B), the shape of the progress curve for intermediate is identical to those of ZnZn-L1 (21). The presence of large amounts of reaction intermediate strongly suggests that the rate-limiting step of nitrocefin hydrolysis by Ni-L1 is protonation of the reaction of intermediate (36).
Steady-state kinetic studies on metal-substituted analogs of L1 yielded surprising results. The most efficient enzyme, in terms of kcat/Km, is the ZnZn-analog (Table 2). There are three analogs that presumably have Zn(II) in the Zn1 site: ZnCo-, ZnFe-, and ZnNi-L1; however, these analogs exhibit significantly different steady-state kinetic properties with the substrates tested. ZnCo-L1 (21) exhibits steady-state kinetics constants similar to those of ZnZn-L1 for penicillin G, but the Km values for studies with cefaclor, imipenem, and nitrocefin are much larger. On the other hand, the ZnFe- and ZnNi-containing analogs of L1 do not hydrolyze penicillin G at all, exhibit decreased kcat values when using cefaclor as substrate, exhibit increased kcat values when using imipenem and nitrocefin as substrate, and exhibit increased values of Km for all substrates tested. The metal-substituted analogs with metal ions other than Zn(II) in the Zn1 site (CoCo- and CuCu-L1) exhibit kinetic properties that do not follow any trend. For example, CoCo-L1 hydrolyzes penicillin G with significant kcat and Km values, while CuCu-L1 does not hydrolyze the same substrate. CoCo- and CuCu-L1 exhibit very large values for kcat for some substrates; however, the Km values for all tested substrates are also significantly higher. It is not clear why the metal-substituted analogs exhibit different steady state kinetic behavior, but the data suggest that substrate preference is somewhat modulated by the identity of the metal ion(s) in the active site of L1. However, we cannot rule out that some of the enzyme samples contain mixtures of metal centers, since the concentration of adventitious Zn(II) in the assay buffers is ca. 100 nM, while the enzyme concentration in the same assays is 10 nM. Studies to probe the metal centers in each of the analogs are on-going in an effort to probe whether M-M bond distances affect the kinetic behavior of the analogs.
The results of this work demonstrate further that the metal binding sites, particularly the Zn2 site, in L1 are very flexible and can accommodate a number of different divalent metal ions. This flexibility can possibly offer a competitive advantage to organisms that produce L1 when the organisms are threatened with β-lactam containing antibiotics. The ability of L1to be active containing a number of different metal ions would allow bacteria to survive in environments with limited Zn(II) concentrations. The different substrate preferences with different metal centers may be evolutionarily-exploited in the future to resistant inhibitors that resemble substrate.
Supplementary Material
Abbreviations
- AAP
aminopeptidase from Aeromonas proteolytica
- CcrA
metallo-β-lactamase from Bacteroides fragilis
- EPR
electron paramagnetic resonance
- EXAFS
extended X-ray absorption fine structure
- Hepes
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- ICP-AES
inductively coupled plasma – atomic emission spectroscopy
- L1
metallo-β-lactamase from Stenotrophomonas maltophilia
- mT
millitesla
Footnotes
This work was supported by the National Institutes of Health (AI056231 to BB, and EB001980 to the Medical College of Wisconsin) and Miami University/Volwiler Professorship (to MWC).
Additional supplemental material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Perez F, Endimiani A, Hujer KM, Bonomo RA. The continuing challenge of ESBLs. Curr Opin Pharmacol. 2007;7:459–469. doi: 10.1016/j.coph.2007.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dong HK, Dore MP, Kim JJ, Kato M, Lee M, Wu JY, Graham DY. High-level b-lactam resistance associated with acquired multidrug resistance in Helicobacter pylori. Antimicro Agents Chemo. 2003;47:2169–2178. doi: 10.1128/AAC.47.7.2169-2178.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Walsh TR, Toleman MA, Poirel L, Nordmann P. Metallo-β-lactamases: the quiet before the storm? Clin Microbiol Rev. 2005;18:306–325. doi: 10.1128/CMR.18.2.306-325.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bush K, Jacoby GA, Medeiros AA. A functional classification scheme for β-lactmases and its correlation with molecular structure. Antimicro Agents Chemo. 1995;39:1211–1233. doi: 10.1128/aac.39.6.1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Crowder MW, Spencer J, Vila AJ. Metallo-β-lactamases: Novel weaponry for antibiotic resistance in bacteria. Acc Chem Res. 2006;39:721–728. doi: 10.1021/ar0400241. [DOI] [PubMed] [Google Scholar]
- 6.Bennett B. EPR of Co(II) as a structural and mechanistic probe of metalloprotein active sites: Characterization of an aminopeptidase. Curr Topics Biophys. 2002;26:49–57. [Google Scholar]
- 7.Garrity JD, Bennett B, Crowder MW. Direct evidence that reaction intermediate in metallo-β-lactamase is metal bound. Biochemistry. 2005;44:1078–1087. doi: 10.1021/bi048385b. [DOI] [PubMed] [Google Scholar]
- 8.Periyannan G, Costello AL, Tierney DL, Yang KW, Bennett B, Crowder MW. Sequential binding of cobalt(II) to metallo-β-lactamase CcrA. Biochemistry. 2006;45:1313–1320. doi: 10.1021/bi051105n. [DOI] [PubMed] [Google Scholar]
- 9.Llarrull LI, Tioni MF, Kowalski J, Bennett B, Vila AJ. Evidence for a dinuclear active site in the metallo-β-lactamase BcII with substoichiometric Co(II): A new model for metal uptake. J Biol Chem. 2007;282:30586–30595. doi: 10.1074/jbc.M704613200. [DOI] [PubMed] [Google Scholar]
- 10.Crawford PA, Yang KW, Sharma N, Bennett B, Crowder MW. Spectroscopic studies on cobalt(II)-substituted metallo-β-lactamase ImiS from Aeromonas veronii bv. sobria. Biochemistry. 2005;44:5168–5176. doi: 10.1021/bi047463s. [DOI] [PubMed] [Google Scholar]
- 11.Concha NO, Rasmussen BA, Bush K, Herzberg O. Crystal structures of the cadmium- and mercury-substituted metallo-β-lactamase from Bacteroides fragilis. Prot Sci. 1997;6:2671–2676. doi: 10.1002/pro.5560061225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hemmingsen L, Damblon C, Antony J, Jensen N, Adolph HW, Wommer S, Roberts GCK, Bauer R. Dynamics of mononuclear cadmium β-lactamase revealed by the combination of NMR and PAC spectroscopy. J Am Chem Soc. 2001;123:10329–10335. doi: 10.1021/ja0112240. [DOI] [PubMed] [Google Scholar]
- 13.Hu Z, Gunasekera TS, Spadafora L, Bennett B, Crowder MW. Metal content of metallo-β-lactamase L1 is determined by the bioavailability of metal ions. Biochemistry. 2008;47:7947–7953. doi: 10.1021/bi8004768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wenzel NF, Carenbauer AL, Pfiester MP, Schilling O, Meyer-Klaucke W, Makaroff CA, Crowder MW. The binding of iron and zinc to glyoxalase II occurs exclusively as di-metal centers and is unique within the metallo-β-lactamase family. J Biol Inorg Chem. 2004;9:429–438. doi: 10.1007/s00775-004-0535-2. [DOI] [PubMed] [Google Scholar]
- 15.Schilling O, Wenzel N, Naylor M, Vogel A, Crowder M, Makaroff C, Meyer-Klaucke W. Flexible metal binding of the metallo-β-lactamase domain: Glyoxalase II incorporates iron, manganese, and zinc in vivo. Biochemistry. 2003;42:11777–11786. doi: 10.1021/bi034672o. [DOI] [PubMed] [Google Scholar]
- 16.Rajagopalan PTR, Grimme S, Pei D. Characterization of cobalt(II)-substituted peptide deformylase: Function of the metal ion and the catalytic residue Glu-133. Biochemistry. 2000;39:779–790. doi: 10.1021/bi9919899. [DOI] [PubMed] [Google Scholar]
- 17.Periyannan G, Shaw PJ, Sigdel T, Crowder MW. In vivo folding of recombinant metallo-β-lactamase L1 requires the presence of Zn(II) Prot Sci. 2004;13:2236–2243. doi: 10.1110/ps.04742704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Crowder MW, Walsh TR, Banovic L, Pettit M, Spencer J. Overexpression, purification, and characterization of the cloned metallo-β-lactamase L1 from Stenotrophomonas maltophilia. Antimicro Agents Chemo. 1998;42:921–926. doi: 10.1128/aac.42.4.921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Carenbauer AL, Garrity JA, Periyannan G, Yates RB, Crowder MW. Probing substrate binding to metallo-β-lactamase L1 from Stenotrophomonas maltophilia by using site-directed mutagenesis. BMC Biochemistry. 2002;3:4–10. doi: 10.1186/1471-2091-3-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hu Z, Periyannan GR, Crowder MW. Folding strategy to prepare Co(II)-substituted metallo-β-lactamase L1. Anal Biochem. 2008;378:177–183. doi: 10.1016/j.ab.2008.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hu Z, Periyannan G, Bennett B, Crowder MW. Role of the Zn1 and Zn2 sites in metallo-β-lactamase L1. J Am Chem Soc. 2008;130:14207–14216. doi: 10.1021/ja8035916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Peisach J, Blumberg WE. Structural implications derived from the analysis of electron paramagnetic resonance spectra of natural and artificial copper proteins. Arch Biochem Biophys. 1974;165:691–708. doi: 10.1016/0003-9861(74)90298-7. [DOI] [PubMed] [Google Scholar]
- 23.Holz RC, Bradshaw JM, Bennett B. Synthesis, molecular structure, and reactivity of dinuclear copper II complexes with carboxylate-rich coordination environments. Inorg Chem. 1998;37:1219–1225. doi: 10.1021/ic9707873. [DOI] [PubMed] [Google Scholar]
- 24.Bertini I, Banci L, Luchinat C. Meth Enzymol. Academic Press; New York: 1989. pp. 246–263. [DOI] [PubMed] [Google Scholar]
- 25.Ullah JH, Walsh TR, Taylor IA, Emery DC, Verma CS, Gamblin SJ, Spencer J. The crystal structure of the L1 metallo-β-lactamase from Stenotrophomonas maltophilia at 1.7 angstrom resolution. J Mol Biol. 1998;284:125–136. doi: 10.1006/jmbi.1998.2148. [DOI] [PubMed] [Google Scholar]
- 26.de Seny D, Heinz U, Wommer S, Kiefer M, Meyer-Klaucke W, Galleni M, Frere JM, Bauer R, Adolph HW. Metal ion binding and coordination geometry for wild type and mutants of metallo-β-lactamase from Bacillus cereus 569/H/9 (BcII) - A combined thermodynamic, kinetic, and spectroscopic approach. J Biol Chem. 2001;276:45065–45078. doi: 10.1074/jbc.M106447200. [DOI] [PubMed] [Google Scholar]
- 27.Garrity JD, Pauff JM, Crowder MW. Probing the dynamics of a mobile loop above the active site of L1, a metallo-β-lactamase from Stenotrophomonas maltophilia, via site-directed mutagenesis and stopped-flow fluorescence spectroscopy. J Biol Chem. 2004;279:39663–39670. doi: 10.1074/jbc.M406826200. [DOI] [PubMed] [Google Scholar]
- 28.Marasinghe GPK, Sander IM, Bennett B, Periyannan G, Yang KW, Makaroff CA, Crowder MW. Structural studies on a mitochondrial glyoxalase II. J Biol Chem. 2005;280:40668–40675. doi: 10.1074/jbc.M509748200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Garrity JD, Carenbauer AL, Herron LR, Crowder MW. Metal binding Asp-120 in metallo-β-lactamase L1 from Stenotrophomonas maltophilia plays a crucial role in catalysis. J Biol Chem. 2004;279:920–927. doi: 10.1074/jbc.M309852200. [DOI] [PubMed] [Google Scholar]
- 30.Bennett B, Holz RC. Spectroscopically distinct cobalt(II) sites in heterodimetallic forms of the aminopeptidase from Aeromonas proteolytica: Characterization of substrate binding. Biochemistry. 1997;36:9837–9846. doi: 10.1021/bi970735p. [DOI] [PubMed] [Google Scholar]
- 31.Bennett B, Antholine WE, D’Souza VM, Chen GJ, Ustinyuk L, Holz RC. Structurally distinct active sites in the copper(II)-substituted aminopeptidases from Aeromonas proteolytica and Escherichia coli. J Am Chem Soc. 2002;124:13025–13034. doi: 10.1021/ja026341p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bennett B, Holz RC. EPR studies on the mono- and dicobalt(II)-substituted forms of the aminopeptidase from Aeromonas proteolytica. Insight into the catalytic mechanism of dinuclear hydrolases. J Am Chem Soc. 1997;119:1923–1933. [Google Scholar]
- 33.Holz RC, Bennett B, Chen G, Ming LJ. Proton NMR spectroscopy as a probe of dinuclear copper(II) active sites in metalloproteins. Characterization of the hyperactive copper(II)-substituted aminopeptidase from Aeromonas proteolytica. J Am Chem Soc. 1998;120:6329–6335. [Google Scholar]
- 34.Chattopadhyay M, Walter ED, Newell DJ, Jackson PJ, Aronoff-Spencer E, Peisach J, Gerfen GJ, Bennett B, Antholine WE, Millhauser GL. The octarepeat domain of the prion protein binds Cu(II) with three distinct coordination modes at pH 7.4. J Am Chem Soc. 2005;127:12647–12656. doi: 10.1021/ja053254z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nauton L, Kahn R, Garau G, Hernandez JF, Dideberg O. Structural insights into the design of inhibitors for the L1 metallo-β-lactamase from Stenotrophomonas maltophilia. J Mol Biol. 2008;375:257–269. doi: 10.1016/j.jmb.2007.10.036. [DOI] [PubMed] [Google Scholar]
- 36.McMannus-Munoz S, Crowder MW. Kinetic mechanism of metallo-β-lactamase L1 from Stenotrophomonas maltophilia. Biochemistry. 1999;38:1547–1553. doi: 10.1021/bi9826512. [DOI] [PubMed] [Google Scholar]
- 37.Wang Z, Fast W, Benkovic SJ. Direct observation of an enzyme-bound intermediate in the catalytic cycle of the metallo-β-lactamase from Bacteroides fragilis. J Am Chem Soc. 1998;120:10788. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.