

Abstract
Motivation: The study of complex biological relationships is aided by large and high-dimensional data sets whose analysis often involves dimension reduction to highlight representative or informative directions of variation. In principle, information theory provides a general framework for quantifying complex statistical relationships for dimension reduction. Unfortunately, direct estimation of high-dimensional information theoretic quantities, such as entropy and mutual information (MI), is often unreliable given the relatively small sample sizes available for biological problems. Here, we develop and evaluate a hierarchy of approximations for high-dimensional information theoretic statistics from associated low-order terms, which can be more reliably estimated from limited samples. Due to a relationship between this metric and the minimum spanning tree over a graph representation of the system, we refer to these approximations as MIST (Maximum Information Spanning Trees).
Results: The MIST approximations are examined in the context of synthetic networks with analytically computable entropies and using experimental gene expression data as a basis for the classification of multiple cancer types. The approximations result in significantly more accurate estimates of entropy and MI, and also correlate better with biological classification error than direct estimation and another low-order approximation, minimum-redundancy–maximum-relevance (mRMR).
Availability: Software to compute the entropy approximations described here is available as Supplementary Material.
Contact: tidor@mit.edu
Supplementary information: Supplementary data are available at Bioinformatics online.
1 INTRODUCTION
As the size and dimension of biological data sets have grown, a variety of data mining and machine learning techniques has been employed as analytical tools. Among these are techniques aimed at a class of problems generally known as dimension reduction problems (Golub et al., 1999; Janes et al., 2005 Slonim et al., 2005). Dimension reduction techniques can improve the interpretability of data, either by representing high-dimensional data in a reduced space for direct inspection, or by highlighting important features of data sets that warrant more detailed investigation. For many biological applications, notably the analysis of high-dimensional signaling data, principal component analysis (PCA) and partial least squares (PLS) decomposition are increasingly popular dimension reduction techniques (Janes et al., 2005; Kumar et al., 2007). Whereas these techniques reduce the number of variables in a system by including only statistically important linear combinations of the full set of variables, the related techniques of representative subset selection (RSS) and feature selection (FS) instead aim to identify subsets of variables that are statistically important. These techniques can be used as preprocessing steps prior to application of machine learning methods such as classification (Ding and Peng, 2005), and have also been applied in chemical library design (Landon and Schaus, 2006) and biomarker discovery (Liu et al., 2005).
While many tools reduce dimensionality to maintain variance (variance-based techniques), recent directions have led to information theoretic phrasings (Ding and Peng, 2005; Slonim et al., 2005). Compared with variance-based methods, information theory has notable advantages. Information theoretic statistics can capture all relationships among a set of variables, whereas variance-based methods may miss non-linear relationships. Additionally many information theoretic values are invariant to reversible transformations, limiting the need for such common (and somewhat ad hoc) methods as mean-centering, variance-scaling and log-transforming. Finally, information theory provides a framework for treating both continuous and categorical data, in contrast to variance-based methods, which are unsuitable for categorical data (Cover and Thomas, 2006; MacKay, 2003). This common framework can be especially important when incorporating categorical data, such as the classification of a type of cancer, into the analysis of a continuous data set, such as mRNA expression microarrays.
A variety of dimension reduction problems has already been phrased using high-dimensional information theoretic statistics (Landon and Schaus, 2006; Peng et al., 2005; Slonim et al., 2005). Notably, the maximum-dependency criterion [maximizing the mutual information (MI) between the feature set and the output] has been proposed for FS (Peng et al., 2005). While the high-dimensional phrasing is theoretically more correct, difficulties in estimating high-dimensional statistics with finite sample sizes have resulted in poor performance when compared with techniques using only lower order statistics (Peng et al., 2005). That is, methods that are better in principle perform worse in practice due to their need for larger sample sizes. While some low-order methods have been shown to be related to the high-dimensional phrasing (Peng et al., 2005), they have generally been developed for a specific application, and their utility in other problems is unclear. To our knowledge, there is no available method for systematically replacing high-order metrics with associated low-order ones. Such a method would enable utilization of the general high-dimensional phrasing but avoid the sampling issues that plague direct applications.
In this article, we present a general framework for approximating high-dimensional information theoretic statistics using associated statistics of arbitrarily low order. Due to a relationship to the minimum spanning tree over a graph representation of the system, we refer to these approximations as Maximum Information Spanning Trees (MIST). The framework is demonstrated on synthetic data and a series of microarray data sets relevant to cancer classification, and the performance is compared with other approaches.
2 THEORY
Information theory is a framework for describing relationships of random variables (Shannon, 1948). The two most heavily used concepts from information theory with regard to dimension reduction are the concepts of information entropy and MI. The entropy of a random variable, H(x), quantifies the uncertainty or randomness of that variable and is a function of its probability distribution, p(x), also called the Probability Mass Function (PMF)
| (1) |
where the summation is over all b bins representing the states of x. To describe the relationship between two random variables x and y, one can consider the conditional entropy of x given that y is known, H(x|y). If x and y are related in some way, knowledge of y may reduce the uncertainty in x, thus reducing the entropy. Conditioning can never increase the entropy of a variable, so H(x)≥H(x|y). The difference between the entropy and the conditional entropy of a variable is a measure of the amount of information shared between the two variables. This difference is defined as the MI, I(x;y), and is symmetric
| (2) |
All of these concepts are similarly defined for vectors x and y, where they are functions of the associated higher order probability distributions (Cover and Thomas, 2006; MacKay, 2003).
MIST entropy approximation framework:the goal is to find an approximation Hnk to the joint entropy of n variables using entropies of order no greater than some k<n,
| (3) |
where Hi denotes a true entropy of order i and Hij denotes a j-th order approximation to an entropy of order i. To arrive at such an approximation, we begin with an exact expansion of the joint entropy of n variables (Cover and Thomas, 2006)
| (4) |
Note that Equation (4) produces the same LHS information entropy Hn for all permutations of the indices of the xi and that the RHS is a series of terms of increasingly higher order. We collect the first k terms on the RHS and identify this as the k-th order information entropy of the first k variables, giving
| (5) |
We replace each term in the summation by its k-th order approximation. Because conditioning cannot increase the entropy, each approximation term is an upper bound on the term it replaced,
| (6) |
All the terms in this sum are k-th order, providing an approximation, Hnk, which is formally an upper bound. Note that for k=n this expression returns to the exact expansion from Equation (4).
Because the indexing of the variables is arbitrary, there are a combinatorial number of approximations consistent with Equation (6), all of which are upper bounds to the true joint entropy. There are actually two levels of arbitrary indexing, one being which variables make up the first k and the second being the selection of k−1 variables used to bound each term beyond the first on the RHS of Equation (6). The best of these approximations is therefore the one that generates the minimum Hnk, as this will provide the tightest bound consistent with this framework. To complete the approximation, we therefore desire a method for choosing the indexing that produces the best of these bounds.
For low-dimensional problems one can enumerate the space of consistent approximations and use the smallest one. To provide a general solution, we first separate out elements that are independent of the indexing. Each conditional entropy term can be divided into an entropy and a MI component, as shown in Equation (2).
| (7) |
Because all individual self-entropy terms will ultimately be included in the summation, they are not affected by the indexing, whereas the MI terms do depend on the indexing. For k=2, we arrive at a compact expression of the best second-order approximation within this framework that depends only upon the indexing of the pairwise MI terms,
| (8) |
The goal is to select the ordering of the indices, i, and the conditioning terms, j, to minimize the expression. The selection of i and j has no effect on the left-hand sum, so it can be ignored during the optimization. We are then left with n−1 second-order terms to consider. To phrase the optimization of indices over these terms, consider a graph where the nodes are the variables and the edges are all possible pairwise MI terms. The result is a fully connected graph of n nodes from which we choose n−1 edges to maximize the sum of the edge weights. The choice of edges is constrained such that every node must have at least one edge. Because only n−1 edges are chosen, this also constrains the graph to be acyclic.
By negating the edge weights and adding a sufficiently large constant to ensure positivity, the problem is equivalent to the Minimum Spanning Tree (MST) from graph theory. A variety of algorithms has been developed to find the optimal solution, including Prim's algorithm (Cormen et al., 2001), a greedy scheme in which the smallest allowed edge is chosen during each iteration. Using this algorithm, we define a method for efficiently finding the best second-order approximation consistent with Equation (8). The computational complexity of Prim's algorithm for a fully connected graph, and thus of our method, is O(N2). For the higher order approximations, we apply the greedy algorithm to select the best k-th order approximation consistent with Equation (6). Although it is not guaranteed to be optimal, in small test systems where enumeration is possible, the greedy scheme resulted in bounds nearly as tight. Note that the MST phrasing, as used here, is merely an optimization method for finding the best approximation consistent with the mathematical framework, and is not necessarily an inherently meaningful representation.
Bias-estimation and propagation:the bias associated with computing the MIST approximation can be estimated by propagating the bias associated with estimating each of the low-order terms. For clarity we focus on the second-order approximation (MIST2) although the method can be easily extended for arbitrarily high approximation order. The error model we use takes advantage of two properties of entropy estimation: (i) higher entropy variables are more difficult to estimate (have higher errors), and (ii) entropy estimates are negatively biased (direct estimates are generally underestimates) (Paninski, 2003). While neither of these properties is guaranteed for any single estimate, they are true on average. We also assume that the estimation errors associated with the first-order entropies are negligible with respect to the errors in the higher order terms.
We first consider the bias associated with estimating a single second-order entropy. For any pair of variables with fixed self entropies, non-zero MI between them will reduce the joint entropy of the pair. Because higher entropy variables have higher estimation bias, the highest possible bias comes when the variables are independent. By forcibly decoupling any pair of variables (by shuffling their order with respect to each other), we compute an estimate that is greater than or equal to the true bias,
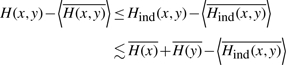 |
(9) |
where the angled brackets indicate averages over repeated samples and the overbars indicate entropy estimates. All quantities on the RHS are directly computable, and by repeating the shuffling procedure, the average estimation bias can be estimated or confidence limits can be established quantifying the likelihood of the true estimation error being greater than the computed value.
With a reasonable estimate of the bias associated with computing each second-order entropy, we need to propagate the bias through the MIST approximation. We start by rewriting Equation (8) assuming that the indexing i,j has been determined using the MST approach as described above, and by expanding the MI term into the corresponding difference of entropies
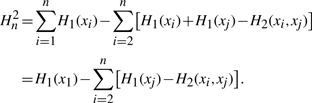 |
(10) |
Because we assume the bias in estimating first-order entropies to be small with respect to the bias in higher order terms, the propagated bias in this expression is dominated by the errors in approximating the n−1 second-order entropies. Because all of these terms are negatively biased, we expect the overall propagated error to be negatively biased as well; i.e. the computed Hn2 is expected to be an underestimate of the approximation assuming no estimation errors in the low-order terms. Consequently, by summing the second-order bias approximated by Equation (9), we arrive at an expected bias for the full approximation:
| (11) |
As with Equation (9), repeated shuffling allows one to estimate the expected bias and to compute confidence limits on the calculation.
3 METHODS
Direct entropy estimation:while the framework developed here is equally applicable to continuous phrasings of information theory, all variables in this work were treated as discrete. For continuous data, variables were discretized into three equiprobable bins unless otherwise stated. Similar results were achieved using different binning protocols and numbers of bins. For discrete data no preprocessing was performed. Entropies of arbitrary order were computed from data by approximating the PMF by the frequencies and using the resulting PMF estimate in Equation (1). The MIs were then computed from the estimated entropies according to Equation (2).
Bias estimation: Bias estimates were computed as described in Section 2. The bias of all pairs of variables was first estimated using Equation (9) by shuffling the ordering of samples for each pair and recomputing the entropy directly. This procedure was repeated until the bias estimate computed from two halves of the shuffling samples agreed within 0.01 nats. The pairs' biases were then used to approximate the bias of each high-order approximation according to Equation (11). The terms included in the summation were chosen according to the MIST method prior to any error analysis. Two cases were examined for computing the term in angled brackets. Either the converged mean value was used to compute the expected bias, or 100 samples were drawn and the maximum error from this set was used for each term in the sum, resulting in a P=0.01 confidence limit that the true value of the entropy approximation lies below this max-error value.
Validation framework: to evaluate the approximation, we developed a framework for generating relational models with analytically determinable entropies from which we could draw sample data. These networks consisted of 5–11 discrete nodes connected by randomly placed unidirectional influence edges. All nodes initially had an unnormalized uniform probability of 1 for each state. If node A influenced node B with weight w, then B was favored to adopt the same state as A by adding w to the unnormalized probability of that state in B. For higher dimensional influences, the states of all parents where summed and remapped to the support of the child, and the corresponding state in the child was favored by adding the influence weight to that state. Influences including 1–4 parents were included, with 4–19 influences of each order, depending on the number of nodes in the system. Influence weights ranged from 1–10 and all variables had three bins. For each system, the joint entropy of all combinations of nodes was computed analytically and 10 000 samples were drawn from each network.
FS and classification error: for the FS task, an incremental method was used in which features were added one at a time to the set of already chosen features either at random or in order to maximize the score of the new feature set according to: (i) maximum dependency using direct estimation, (ii) maximum dependency using MIST of order two (MIST2), or (iii) a second-order approximation proposed elsewhere specifically for feature selection known as minimum-redundancy–maximum-relevance (mRMR) (Ding and Peng, 2005). All FS methods were evaluated by training on 75% of the samples and testing on the remaining 25%. This procedure was repeated 200 times and the mean behavior is reported. The data were discretized and the features chosen using only the training data. The frequency of each gene across the 200 trials was also recorded, and the Bonferroni-adjusted P-value for each gene occurring this many times was computed compared to a null model in which features are chosen at random. The subset of features was then used to train support vector machine (SVM) using a linear kernel, linear discriminant analysis (LDA), 3-nearest neighbor (3NN) or 5-nearest neighbor (5NN) classifiers (Gokcen and Peng, 2002 and references therein). Additional SVM kernels (polynomials of order 2 and 3, Gaussian Radial Basis Function and Multilayer Perceptron) where also examined; while these kernels generally resulted in better fits to the training sets, they performed worse than the linear kernel in cross-validation. To compute the correlation between the metric scores and classification error, 100 subsets each of 1–15 features were chosen at random and the cross-validation classification error was computed. Additionally, the MI of each feature set was computed using all samples according to MIST2, mRMR and direct estimation.
Data sets: gene expression data sets relating to the classification of four cancer types were used for the FS task. Samples from prostate (Singh et al., 2002), breast (van de Vijver et al., 2002), leukemia (Golub et al., 1999) and colon (Alon et al., 1999) were analzyed. Additional information on the data sets is available in Supplementary Table S1.
4 RESULTS
4.1 Direct validation
To validate the method, we examined the performance of the MIST approximation in systems with analytically computable entropies. For real-world applications the entropies of the true distribution are estimated from limited data sets, and the corresponding numerical experiments were performed here. To serve this function, we developed a framework to generate networks with a variable number of nodes, interactions, orders of interaction, discrete states and weights of influence between nodes. For each of these networks, all of the joint entropies were analytically determined for comparison to the approximations (see Section 2).
Using this framework we randomly generated 100 networks containing between 5 and 11 variables each with widely varied topologies, and we sampled 10 000 points from the joint distribution. For each network, we then computed the joint entropy of all variables in the network either (i) analytically, (ii) directly from the data, (iii) using the the second- through fifth-order MIST approximations with analytical low-order entropies up to and including k or (iv) using MIST after estimating the low-order entropies from the sampled data. Additionally, half of the nodes in each network were randomly chosen and the MI between the chosen set and the unchosen set was computed according to all the metrics. The results for entropy and MI approximation are shown in Figure 1 and Supplementary Figure S1, respectively.
Fig. 1.

Direct validation of MIST entropy approximation. To evaluate the MIST framework, we simulated 100 randomly generated networks with analytically computable joint entropies and applied the metrics using a range of sample sizes. When the analytical entropies are known exactly (A), the higher order approximations perform increasingly well. When the entropies are estimated from a finite sample, however (C–E), the approximations provide the best estimates, with the higher order approximations performing better as more data become available. This behavior is quantified by computing the sum-of-squared error of each metric as a function of the sampling regime (B). The best approximation to use depends upon the amount of data available, but for all cases examined with finite sample size, the approximations outperform direct estimation and the second-order approximation provides a good estimate.
The scatter-plots show the relationship between each of the MIST approximations and the analytical value. As guaranteed by the theory, when the exact low-order entropies are known (Figs 1A and S1A), all joint entropy approximations are greater than or equal to the true joint entropy, and the higher order approximations are increasingly accurate. Although there are no guarantees for the behavior of the MI approximation, all approximations tend to underestimate the true MI and the higher order approximations generally perform better. In some cases the lower order approximations are able to fully represent the network, resulting in perfect accuracy and in all cases the MIST approximations tend to be fairly accurate.
For biological applications, the exact low order terms are not available and must instead be estimated from a finite sample of the underlying distribution (Figs 1C–D and S1C-D). Because estimating high-order joint entropies requires larger sample sizes than estimating low-order entropies, the relative performance of the approximations is crucially tied to the number of samples available. In the least sampled case shown here (100 points, Figs 1C and S1C), the second-order approximation (MIST2) yielded more accurate results than any of the other methods for computing entropy, while the second- and third-order approximations performed about equally well for MI. As more samples were used to estimate the low-order terms, the higher order approximations began to outperform the lower order ones. This trend is quantified in the upper-right plots (Figs 1B and S1B), which show the sum-of-squared error (SSE) for each approximation tested. For all sample sizes tested here, direct estimation performed the worst, demonstrating the impracticality of estimating high-order information theoretic terms directly. Furthermore as can be seen in Figure 1C–E and Supplementary Figure S1C–E, the MIST2 approximation is quite accurate for all sample sizes. When more data are available, the higher order approximations can provide even better accuracy than MIST2, but MIST2 itself appears to be a good metric for all sample sizes tested.
We also examined the behavior of our bias approximation framework in the same systems for MIST2. For each pair of variables, we computed the converged bias and the maximum observed error over 100 shuffling iterations. For each MIST-approximated joint entropy we propagated both error sets through to determine a bias-adjusted entropy (BA-MIST2) and P=0.01 confidence limit. We then compared these values with the analytically determined ones in different sampling regimes (Fig. 2).
Fig. 2.

Bias-adjusting for the MIST entropy approximation. Networks were generated and simulated as in Figure 1. The joint entropy of each network was computed by the second-order MIST approximation with (BA-MIST2) or without (MIST2) bias adjusting. (A) The performance of both metrics as well as a P=0.01 confidence limit for MIST (CL-MIST2) approach the analytical MIST2 with increasing samples. (B) The SSE for estimating the analytical MIST2 is shown to decrease as a function of sample size. (C–D) MIST2 and BA-MIST2 were computed using 10, 50 or 100 samples and are plotted against the analytical MIST2.
In these systems, the bias-adjusted entropy proved to be a significantly better estimator of the MIST approximation than the unadjusted estimator. This result is not necessarily expected, as the bias was computed using the different, but related, system in which all variables were forcibly decoupled. That the bias-adjusted values are not strictly greater than the approximation using analytically determined values is likely a result of the approximations made in the analysis: namely, neglecting the errors in first-order terms and adjusting from a single observed value, rather than a mean from repeated samplings. As expected, the bias decreases as more samples are used, resulting in the bias-adjusted and unadjusted approximations converging for higher sampling regimes. Because the BA-MIST is always greater than MIST without bias-adjusting, and the MIST approximation itself is an upper bound to the true entropy, for higher sampling regimes, bias-adjusting actually results in poorer performance with respect to the analytical answer. While the bias is likely to be small in these cases, this result suggests that while BA-MIST is likely more accurate for low-sampling regimes, when more data are available, MIST without bias-adjusting may have lower error with respect to the true joint entropy.
The confidence limit also shows the expected behavior. While it is not as good an estimator as the bias-adjusted metric, it does provide an upper bound to the approximation computed with analytical entropies within the resolution of the estimation techniques. As such, this metric can provide a guide towards the convergence of the MIST approximation techniques and may lend some insight into the selection of the appropriate order of approximation.
4.2 Biological application
To further characterize the MIST approximation and to evaluate performance in tasks relevant to the interpretation of biological data, we employed MIST in the task of FS, which has been previously phrased using information theory (Peng et al., 2005). FS is the task of choosing a subset of available features for use in some learning task, such as classification; the information theoretic phrasing seeks the feature subset with maximal MI with the classification. A well-studied example is that of selecting a subset of gene expression levels to use when building classifiers to discriminate among cancer types (Ding and Peng, 2005; Draminski et al., 2008; Goh and Kasabov, 2005). To explore the performance of the MIST approximation in this task, we analyzed four gene expression data sets (which varied both in the number of samples and the number of genes) that had previously been used to classify cancer type in prostate (Singh et al., 2002), breast (van de Vijver et al., 2002), leukemia (Golub et al., 1999) and colon (Alon et al., 1999).
The rationale behind using MI to choose gene subsets comes from the relationship between MI and classification error (Ney, 2003). To evaluate the relationship between MIST2 and the true relationships in these biological data sets, we therefore computed the cross-validated classification error using 100 randomly chosen subsets including 1–15 genes and a range of classifiers. We also computed the MI of the same feature sets with the class variable according to MIST2 and direct estimation, as well as an existing incremental FS metric that has been shown to be an approximation of high-dimensional MI known as mRMR (Peng et al., 2005). The Pearson correlation coefficient between the SVM cross-validation classification error and the MI metrics for each set size is shown in Figure 3. Results using 3NN, 5NN or LDA classification error showed similar trends, as did those using the fit error rather than the cross-validation error (data not shown). The SVM classifier was chosen due to its superior performance across the four data sets.
Fig. 3.

Correlation of MI metrics with classification error. The classification error of randomly chosen subsets of 1–15 genes was computed through cross-validation with an SVM-based classifier. The same sets were then scored by MIST2, MI computed with direct estimation, and mRMR. The Pearson correlation between each metric and the error was computed for gene expression data sets collected in (A) breast, (B) leukemia, (C) colon and (D) prostate tissue. For all cases, MIST2 shows strong negative correlation with cross-validation error, meaning high MI is associated with low error. While correlated in some cases, both mRMR and direct estimation show poor correlation for some set sizes and data sets.
For all four systems, all three metrics have a strong negative correlation coefficient for the feature sets of size one, indicating that high MI corresponds to low classification error, as expected. For larger numbers of features, however, while the MIST2 approximation maintains reasonable negative correlation for all sizes and data sets, the direct estimation has virtually no correlation with classification error for sets larger than five. For breast (A) and leukemia (B), MIST2 and mRMR are relatively close though MIST2 generally exhibits slightly better correlation. For colon (C) and prostate (D), however, MIST2 exhibits significantly better correlation for larger feature sets. The correlation across sets of different size was also computed and is shown in Supplementary Figure S4. While correlation between different sizes is not necessary for standard FS phrasings, the strong negative correlation of MIST2, even across sets of varied size is further evidence that the approximation reflects the underlying relationships of the system.
In practice, for FS the MI metric would be used to select a single subset of features that is expected to have low classification error. In this task, correlation across all sets is not necessary as long as the top ranked set is a good one. To evaluate the utility of MIST in this application, we included it, as well as direct estimation and mRMR, in an incremental FS task to choose subsets of genes with which to build a classifier for each of the four tissue types. For each data set, 75% of the samples were used to select the best set of size 1–15 (or 1–10 for direct estimation) according to each metric in an incremental fashion. SVM classifiers were then trained on the same 75% and used to predict the class of the remaining 25% of the samples. This procedure was repeated 200 times to determine the average cross-validation error of the FS/classification methods. The performance of randomly chosen feature sets was also computed and in all cases was significantly worse than all tested methods (Supplementary Fig. S2). Parallel studies were performed using 3NN, 5NN and LDA classifiers (Supplementary Fig. S3), as well as ones in which features were preselected using the full data set rather than only 75% (data not shown). Leave-one-out cross-validation schemes were also examined (data not shown). While the results in all cases showed similar trends, the SVM classifier consistently outperformed the other classifiers and the 75% cross-validation scheme seemed to be the most stringent test. The mean SVM classification errors are shown in Figure 4.
Fig. 4.

Gene subset selection for cancer classification. Subsets of gene expression levels were chosen incrementally to maximize the information with the cancer class according to MIST2, direct estimation of MI or mRMR and scored by the cross-validation error of an SVM classifier. For all data sets, 75% of the data were separated and used to select features and train the model; the classifier was then used to classify the remaining 25%. The mean classification error and standard error of the mean for 200 training/testing partitionings are reported. Genes were selected for data sets relating to (A) breast, (B) leukemia, (C) colon and (D) prostate cancer.
For all cases, the MIST2 feature sets showed lower classification errors relative to direct estimation and mRMR when choosing a small number of features (2–5). This is consistent with the better correlation with the classification error for MIST2 shown in Figure 3. For the breast data, this improvement was maintained for feature sets of all sizes. For the other three systems, however, both direct estimation and mRMR generated sets with lower classification errors for sets including more than 5–7 genes. This result is particularly surprising given that this is the regime in which MIST showed improved correlation with classification error relative to the other metrics. Regardless, while MIST appears to select superior subsets of size 2–5, this behavior does not generally appear to extend to large set sizes and deserves further study.
In the above validation scheme, many different feature sets were chosen using different subsets of sample data so as to characterize the expected performance of the metric for predictive tasks. In application however, the features would be selected using all the samples available for training. We therefore incrementally selected the set of 10 most informative genes according to MIST2 for each of the data sets. An ordered list of these genes along with references demonstrating the relevance to cancer biology or cancer diagnosis for a subset of the genes can be found in Supplementary Table S2. All of the selected feature sets contained genes that have been either statistically or functionally related to cancer. Many of the genes have also been identified in other computational studies. The most informative gene for all four data sets had previously been identified in multiple studies. For the highly studied leukemia and colon data sets, nearly all of the genes have been identified in some study, though not always in the top 10 ranked genes. Notably, three of the genes identified in the breast data set (NM_003981, AI918032 and AF055033) consistently appeared in the globally optimal feature sets of sizes 2–7 in (Choudhary et al., 2006).
We also evaluated the robustness of the chosen genes by observing how often they were chosen in the 200 cross-validation trials. The P-value for having at least this frequency for each of the chosen genes is shown in Supplementary Table S2. While some of the globally chosen genes are not robustly reselected, the majority (32/40) of the genes appear in the 200 trials more often than expected at random (Bonferroni-corrected P-value < 0.01), particularly for the breast (8/10) and prostate (10/10) data sets which have larger sample sizes.
5 DISCUSSION
Here, a novel framework for approximating high-order information theoretic statistics with associated statistics of arbitrarily low order has been developed and validated. Due to the generality of information theory, the MIST approximation should allow the use of high-dimensional information theoretic phrasings for a variety of problems, even in cases when data quantities are limited. Information theoretic phrasings exist for such tasks as FS (shown here), RSS (Landon and Schaus, 2006), clustering (Slonim et al., 2005), network inference (Liang et al., 1998) and other applications where relationships of multiple variables are important. Though high-dimensional phrasings are theoretically correct, difficulties in estimating these terms has led to low-order approximations having better performance. While these approximations have been applied to many problems, task-specific metrics were usually developed that are not generally usable across multiple applications. Instead, by developing a principled approximation to joint entropy and MI, we propose a general method for application to many problems.
In regards to the FS task shown here, while MIST2 correlates well with the classification error and generates low-error sets when picking a small number of genes, the overall behavior for choosing larger sets could still likely be improved. For incremental FS, MIST and mRMR are similar with the primary difference being that MIST selects a subset of MI terms to consider, whereas mRMR averages all gene–gene terms to compute the redundancy. While both have been shown to relate to the maximum dependency criterion, MIST represents a more general framework for extension to different problem phrasings. In contrast, mRMR has been well calibrated for FS, and some features of mRMR may be useful in improving the performance of MIST in FS. In particular, preliminary work on incorporating weighting factors to influence the relative importance of the relevance and redundancy suggests that such a scheme may result in a better FS method. Additionally, while the current work has focussed on incremental FS, the generality of MIST and the good correlation with classification error suggest that global search methods using MIST could be feasible. In its current form, MIST provides a well-principled framework without any ad hoc parameterization that performs comparably to current FS methods. Furthermore, MIST can be generalized and ported to other problem phrasings and takes advantage of larger data quantities when they become available.
One natural extension of the MIST approximation is FS with multiple outputs. Typical FS phrasings focus on a single output variable, resulting in most FS methods not being directly applicable to multiple-output scenarios. Instead, separate subsets may be chosen for each output and combined subsequently, or multiple outputs can be combined into a single variable. With high-dimensional statistics, rephrasing the maximum dependency criterion for multiple outputs is trivial, by replacing the single output variable with the set of all outputs of interest (i.e. find the set that maximizes MI between the gene set and the output set). In cases where different feature sets can be used for each output, such as preprocessing before machine learning, multiple output FS may not be appropriate, as a single consensus set will not represent each output as well as the individually chosen sets. In other cases, however, a fixed number of features may be needed to describe multiple outputs and a single optimization for this task could be valuable. Considering the relationships between multiple outputs could be particularly important if the outputs are closely related. For example, in the case of FS for cancer classification, one might consider tumor progression measurements at multiple time points. Alternatively, defining a compact set of features that can classify multiple disease states could be valuable in more efficient diagnostic tools. Designing experiments that are richly informative of a particular set of output variables might also benefit from such methods. In general, having metrics that support multiple outputs allows phrasing FS problems that better reflect questions of interest.
The ability to maintain the general information theoretic phrasing also allows the results between different tasks and experiments to be compared. Information theory is able to treat data from different experimental modalities within the same framework, enabling one to quantitatively compare the information content of different data types without significant preprocessing. Information theory also allows the treatment of categorical and continuous data, and can consider non-linear relationships, unlike variance-based techniques. While these benefits of information theory have long been understood, the inability to estimate information theoretic terms has often precluded their use in biological systems. By reducing the data requirements for computing high-order entropies, MIST enables the use of information theoretic statistics even when few samples are available, as is often true in biological systems.
Although we have used only the second-order MIST approximation here, the framework provides a range of approximations of higher order, allowing increased accuracy when sufficient quantities of data are available. As high-throughput data collection continues to improve, the framework extends to incorporate third- and fourth-order relationships. Even as larger quantities of data become available, MIST is likely to be useful, as in our synthetic system, even with 104 samples, all orders of approximation tested outperformed direct estimation. In Figure 1, we have shown how one might select an approximation order based on the sample size. For applications where the analytical solutions are unknown, however, it is unclear how to choose the best approximation order. Additional work is required to fully enable such a method. Despite this, it is encouraging that the second-order approximation performs well both on synthetic and microarray data, even though high-order relationships are known to exist.
While the MIST framework arises from a mathematical approximation, it can alternatively be thought of as a method to infer a relational model of low-order interactions. This model is then used to estimate the high-order statistics of interest. Currently, this model is used only for the approximation, however, the good agreement between the approximation and the analytical entropies suggests that the inferred model captures many of the relevant relationships. The generation of relational models for biomarker discover has been previously proposed (van der Greef et al., 2007), and network inference tools have been proposed that use pairwise MI as the primary metric (Liang et al., 1998; Meyer et al., 2007). There is reason to believe, therefore, that the relational models inferred may be meaningful, as they reasonably represent the system's statistical relationships.
6 CONCLUSIONS
Here, we have presented a novel method for approximating high-dimensional information theoretic statistics with significantly improved performance when data quantities are limited, as is often true when dealing with biological data. While we have demonstrated the utility of this approximation in FS, the generality of information theory should enable application in a number of different learning tasks, including RSS, clustering and network inference. While previous low-dimensional information theoretic phrasings exist for these problems, they have generally been developed on a problem-by-problem basis, and are thus not directly portable between tasks. Instead, by focusing on ways to approximate the information theoretic statistics directly, we can take advantage of general information theoretic phrasings in a variety of problems. In addition, our MIST approximation naturally allows for incorporating arbitrarily high-order information as sample sizes increase, providing a consistent framework as the collection of biological data continues to increase in scale.
Supplementary Material
ACKNOWLEDGEMENTS
The authors gratefully acknowledge the MIT Computational and Systems Biology community, particularly Doug Lauffenburger, K. Dane Wittrup, Jared Toettcher, Ben Cosgrove, Paul Kopesky and Ericka Noonan, for stimulating and thoughtful discussion.
Funding: DuPont MIT Alliance (partial); National Institutes of Health (U54 CA112967 and T32 GM008334, partial).
Conflict of Interest: none declared.
REFERENCES
- Alon U, et al. Broad patterns of gene expression revealed by clustering analysis of tumor and normal colon tissues probed by oligonucleotide arrays. Proc. Natl Acad. Sci. USA. 1999;96:6745–6750. doi: 10.1073/pnas.96.12.6745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhary A, et al. Genetic test bed for feature selection. Bioinformatics. 2006;22:837–842. doi: 10.1093/bioinformatics/btl008. [DOI] [PubMed] [Google Scholar]
- Cormen TH, et al. Introduction to Algorithms. 2. Cambridge, MA: MIT Press; 2001. [Google Scholar]
- Cover TM, Thomas JA. Elements of Information Theory. 2. Hoboken, NJ: Wiley-Interscience; 2006. [Google Scholar]
- Ding C, Peng H. Minimum redundancy feature selection from microarray gene expression data. J. Bioinform. Comput. Biol. 2005;3:185–205. doi: 10.1142/s0219720005001004. [DOI] [PubMed] [Google Scholar]
- Draminski M, et al. Monte Carlo feature selection for supervised classification. Bioinformatics. 2008;24:110–117. doi: 10.1093/bioinformatics/btm486. [DOI] [PubMed] [Google Scholar]
- Goh L, Kasabov N. An integrated feature selection and classification method to select minimum number of variables on the case study of gene expression data. J. Bioinform. Comput. Biol. 2005;3:1107–1136. doi: 10.1142/s0219720005001533. [DOI] [PubMed] [Google Scholar]
- Gokcen I, Peng J. Lecture Notes in Computer Science. Vol. 2457. Berlin: Springer; 2002. Advances in Information Systems; pp. 104–113. [Google Scholar]
- Golub TR, et al. Molecular classification of cancer: class discovery and class prediction by gene expression monitoring. Science. 1999;286:531–537. doi: 10.1126/science.286.5439.531. [DOI] [PubMed] [Google Scholar]
- Janes KA, et al. A systems model of signaling identifies a molecular basis set for cytokine-induced apoptosis. Science. 2005;310:1646–1653. doi: 10.1126/science.1116598. [DOI] [PubMed] [Google Scholar]
- Kumar N, et al. Modeling HER2 effects on cell behavior from mass spectrometry phosphotyrosine data. PLoS Comput. Biol. 2007;3:e4. doi: 10.1371/journal.pcbi.0030004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landon MR, Schaus SE. JEDA: joint entropy diversity analysis. Aninformation-theoretic method for choosing diverse and representative subsets from combinatorial libraries. Mol. Divers. 2006;10:333–339. doi: 10.1007/s11030-006-9042-4. [DOI] [PubMed] [Google Scholar]
- Liang S, et al. REVEAL, a general reverse engineering algorithm for inference of genetic network architectures. Pac. Symp. Biocomput. 1998;3:18–29. [PubMed] [Google Scholar]
- Liu JJ, et al. Multiclass cancer classification and biomarker discovery using GA-based algorithms. Bioinformatics. 2005;21:2691–2697. doi: 10.1093/bioinformatics/bti419. [DOI] [PubMed] [Google Scholar]
- MacKay DJC. Information Theory, Inference, and Learning Algorithms. Cambridge, UK: Cambridge University Press; 2003. [Google Scholar]
- Meyer PE, et al. Information-theoretic inference of large transcriptional regulatory networks. EURASIP J. Bioinform. Syst. Biol. 2007;2007:79879. doi: 10.1155/2007/79879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ney H. On the relationship between classification error bounds and training criteria in statistical pattern recognition. In: Perales López FJ, et al., editors. Pattern Recognition and Image Analysis. Berlin/Heidelberg: Springer; 2003. pp. 636–645. [Google Scholar]
- Paninski L. Estimation of entropy and mutual information. Neural Comput. 2003;15:1191–1253. [Google Scholar]
- Peng H, et al. Feature selection based on mutual information: criteria of max-dependency, max-relevance, and min-redundancy. IEEE Trans. Pattern Anal. Mach. Intell. 2005;27:1226–1238. doi: 10.1109/TPAMI.2005.159. [DOI] [PubMed] [Google Scholar]
- Shannon C. A mathematical theory of communication. Bell Syst. Tech. J. 1948;27:379–423. 623–656. [Google Scholar]
- Singh D, et al. Gene expression correlates of clinical prostate cancer behavior. Cancer Cell. 2002;1:203–209. doi: 10.1016/s1535-6108(02)00030-2. [DOI] [PubMed] [Google Scholar]
- Slonim N, et al. Information-based clustering. Proc. Natl Acad. Sci. USA. 2005;102:18297–18302. doi: 10.1073/pnas.0507432102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Greef,J, et al. The art and practice of systems biology in medicine: mapping patterns of relationships. J. Proteome Res. 2007;6:1540–1559. doi: 10.1021/pr0606530. [DOI] [PubMed] [Google Scholar]
- van de Vijver,MJ, et al. A gene-expression signature as a predictor of survival in breast cancer. N. Engl. J. Med. 2002;347:1999–2009. doi: 10.1056/NEJMoa021967. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.