

Abstract
Damage response pathways triggered by mechanical stress might reasonably be expected to be conserved throughout evolution. However, using an NF-κB reporter mouse we show here that this phylogenetically recent transcription factor plays a major role in the response to mechanosensory stress in the mammalian inner ear. The protective action of NF-κB is exerted in neither sensory nor non-sensory epithelial cells, but rather in connective tissue cells within the spiral ligament and spiral limbus. In the spiral ligament, predominantly type I fibrocytes are activated following noise exposure, whereas type II fibrocytes are activated following systemic inflammatory stress. Immune-mediated and acoustic trauma-mediated hearing loss syndromes in humans may in part result from the vulnerability of type II and type I fibrocytes to systemic inflammatory stress and acoustic trauma, respectively. Unexpected cell-specific and stress-specific NF-κB activation found in the inner ear in this in vivo study suggest that this approach may have wide applications in demonstrating similar specializations of stress responses in other tissues, including the brain.
Keywords: fibrocytes, cochlear stress
Introduction
NF-κB is a polymorphic heterodimeric transcription factor that integrates a variety of signals arising from the activation of cell surface receptors or intracellular mediators, usually in the context of the detection of or response to inflammatory or pathogen stresses (Israel,2000; Hacker and Karin, 2006; Hayden and Ghosh, 2004). In unstimulated cells, NF-κB is sequestered in the cytoplasm through interaction with IκB, which masks nuclear localization signals. Phosphorylation of IκB results in ubiquitin conjugation and degradation followed by translocation of the NF-κB complex to the nucleus and activation of promoters that contain binding sites for the factor. NF-κB homologs are found in higher but not lower metazoans and are frequently activated as a component of the host immune response to inflammatory mediators. It is thought to be ubiquitous throughout the body. Downstream effectors influenced by NF-κB signaling, such as tumor necrosis factor alpha, are known to be elevated in a variety of settings in which the proximal elicitor is poorly understood, including such prevalent conditions as type 2 diabetes and congestive heart failure. In many cases, it is assumed that reactive oxygen species or other surrogate signals for pathogens or inflammation may be responsible for induction of transcriptional activity. Work on NF-κB signaling is widespread, with most assays of NF-κB activation being tissue destructive so that individually involved cells cannot be identified. In the present study an in vivo approach to identifying NF-κB activation in individual cells using a GFP reporter mouse is applied to the inner ear. Completely unexpected results regarding cell specificity and stress mode specificity were found, which casts stress responses of the cochlea in an entirely new light and suggests that the approach could well revise thinking about NF-κB activation in a host of other tissues.
The mammalian inner ear contains some two dozen cell types whose functions are not uniformly well understood. A large proportion of cells in the inner ear are connective tissue cells, located in the lateral wall of the cochlear duct, the spiral ligament. Genetic mutations that disrupt spiral ligament cells result in deafness (Minowa, 1999; Merchant et al., 2000; Abe et al., 2003; Delprat et al., 2005; Trowe et al., 2008). Moreover, pathologic remodeling of the bone enclosing the cochlea that occurs in otosclerosis and Paget’s disease, causes a sensorineural hearing loss if the remodeling front reaches the spiral ligament and disrupts its cells. This hearing loss appears to arise from malfunction of the spiral ligament, since sensorineural cells remain intact (Parahy and Linthicum, 1983; Kwok and Nadol, 1989; Teufert and Linthicum, 2005). Based upon characteristic anatomical and cytochemical properties, five cell classes have been distinguished among spiral ligament cells, and these are referred to as types I-V fibrocytes (Spicer and Schulte, 1991).
We report here the discovery that intense noise exposure is an effective proximal elicitor of NF-κB activation in cells of the mouse inner ear that are not mechanically stimulated by sound-induced motion of the organ and that different cochlear cells activate NF-κB following systemic inflammatory stimuli. We utilized NF-κB reporter mice to visualize individual cells within the inner ear that were activated by different forms of stress. Restricted populations of fibrocytes responded to different forms of stress by activating NF-κB, whereas epithelial and neural cells were remarkable for their lack of activation. The use of an in situ assay for NF-κB activation showed previously unrecognized specializations and vulnerabilities of specific cell classes. In contrast to acoustic stress, systemically administered inflammatory stress resulted in NF-κB activation predominantly in type II fibrocytes, indicating a previously unrecognized vulnerability of these cells to inflammatory stress. A better understanding of these novel cochlear stress responses should further our ability to predict and prevent acoustic and systemic-induced hearing loss.
Materials and Methods
Generation of transgenic mice
The NF-κB GFP reporter construct contains eight copies of the NF-κB response element from the SV40 early promoter (a strong NF-κB element) inserted upstream of a synthetic consensus TATA box/basal promoter. The latter was designed by aligning the basal elements of the strongest reported mammalian promoters. A codon-optimized mutant GFP was placed downstream of the synthetic promoter and the performance of the promoter was verified in cell lines in culture. To create transgenic lines in which the reporter would be minimally influenced by adjacent chromatin, the NF-κB/GFP transcription unit was flanked by insulators derived from the chicken β-globin 3’ region.
Monitoring acoustic responses to Noise and LPS
Mice were placed in a small wire cage for two hours within a reverberant box in a uniform field of noise, which was filtered to pass 8-16 kHz. The sound pressure level of the noise varied between 90 and 112 dB. (See Wang et al., 2002), for complete descriptions of this apparatus and of the hearing tests. Following noise exposure, mice were returned to their home cages for 24 hours before their ears were assayed for NF-κB activation. Four mice were subjected to unilateral conductive hearing impairment before they were stimulated so that effects of the noise in the less stimulated ear could be compared with the ear that was exposed to more intense noise. To create a conductive hearing impairment the mice were sedated and the malleus was removed with forceps using an operating microscope. This results in approximately 30 dB conductive hearing loss. The following day the animals were exposed to noise.
To induce systemic stress, mice were injected (i.p.) with LPS from E. coli (Sigma-Aldrich, St. Louis, MO) dissolved in sterile saline. Control animals were either injected with saline or not injected with anything. LPS dosages varied from 50 ng to 200 μg per animal. Additional tests were performed with LPS from S. abortus, taxol (40-200 μg) and with anti-CD3. The effectiveness of anti-inflammatory steroids in blocking NF-κB activation was tested by injecting 4 reporter mice with 10 μg/gm dexamethasone i.p. 4 hours prior to an injection of 50 μg of LPS. These mice were allowed to survive for 24 hours before being processed. A total of eighty transgenic mice were utilized in these experiments. All experimental protocols were approved by the ICCUA of the Massachusetts Eye & Ear Infirmary.
Immunohistochemistry
Mice were administered a lethal dose of urethane (3 gm/kg) and perfused intracardially with phosphate buffered saline (PBS) followed by 10 ml 4% formaldehyde and 0.05% glutaraldehyde. The middle ear cavity was opened, the stapes removed, the round window membrane opened and fixative was slowly perfused through the cochlear scalae. The head was immersed in fixative for about 6 hours, decalcified in 120 mM EDTA, embedded in paraffin and 6 μm serial sections collected on slides. For visualization of GFP fluorescence, slides were dewaxed in xylene, hydrated through an alcohol series, mounted under coverslips in 50% glycerol in PBS and observed using a Zeiss fluorescence microscope equipped with a filter set optimized for GFP (Omega Optics, Austin, TX). Permanent slides, together with a more sensitive assay for GFP were produced by immunostaining for GFP. Hydrated slides were incubated in rabbit anti-GFP (Molecular Probes, Invitrogen, Carlsbad, CA) overnight at dilutions ranging from 1:1,000 to 1:50,000. To confirm that the antibody stained the same cells that showed GFP fluorescence, a red fluorescent marker was used to visualize the sites of antibody binding. A biotinylated secondary antibody (1:400 biotinylated donkey anti-rabbit, Jackson Immunoresearch, West Grove, PA) followed by streptavidin/Cy3 (Molecular Probes, Invitrogen, Carlsbad, CA) diluted 1:3000 was applied to the slides. This procedure was also used to show spatial relations of GFP and for Na+, K+-ATPase (CR4004C, Cortex Biochem, San Leandro, CA).
Permanent slides were produced by substituting ABC reagent (Vector Laboratories, Burlingame, CA) for the streptavidin/Cy3 and developing the peroxidase conjugation product of diaminobenzidine and hydrogen peroxide. In general, every tenth section was evaluated for GFP fluorescence and adjacent sections were stained for GFP. The permanent slide procedure was also used for immunodetection of the LPS receptor, TLR4 (Santa Cruz Biotechnology, Santa Cruz, CA and eBioscience, San Diego, CA), F4/80 (Serotec, Raleigh, NC), and for the NF-κB RelA protein (Santa Cruz Biotechnology, Santa Cruz, CA).
Brightfield micrographs were acquired with an Infinity X digital camera (Lumenera Corp.) on a Zeiss microscope with background subtraction applied for correction of illumination non-uniformities. Images were rendered monochromic, sharpened by unsharp masking, and gamma corrected using Photoshop. Fluorescence micrographs were acquired on a Zeiss fluorescence microscope. Exposures taken at a single focal plane with different filter sets were combined to show spatial relations of cells showing immunoreactivity to different antigens.
Results
Noise treatment induces NF-κB activation in type I fibrocytes
To identify inner ear cells that respond to stress-specific NF-κB activation, we examined NF-κB GFP reporter mice. Exposure to intense noise or systemic injection with lipopolysaccharide (LPS) both resulted in induction of GFP in cochlear cells. Brightly fluorescent cells were present in the cochleas of animals that were exposed to noise and those injected with inflammatory agents (Figure 1). The identity of the green fluorescence as GFP was confirmed by immunostaining for GFP using a red fluorochrome so that individual cells could be checked for the presence of both red and green fluorescence. These experiments revealed that all fluorescent green cells were also immunopositive for GFP, indicating that there was no background of green fluorescing cells that could be mistaken for GFP-positive cells. On the other hand, some cells that were weakly immunopositive for GFP were not also clearly green, indicating that the immunoassay was more sensitive than the fluorescence of the GFP alone (not shown). Interestingly, photomicrographs of sequential exposure of both chromophores showed that, in some cases, the intracellular locations of the two were not identical, which suggested that the GFP may have been cleaved by lysosomes and the epitope recognized by the antibody was no longer associated with the fluorescent portion of the GFP molecule (Fig.1C, 1D).
Figure 1.
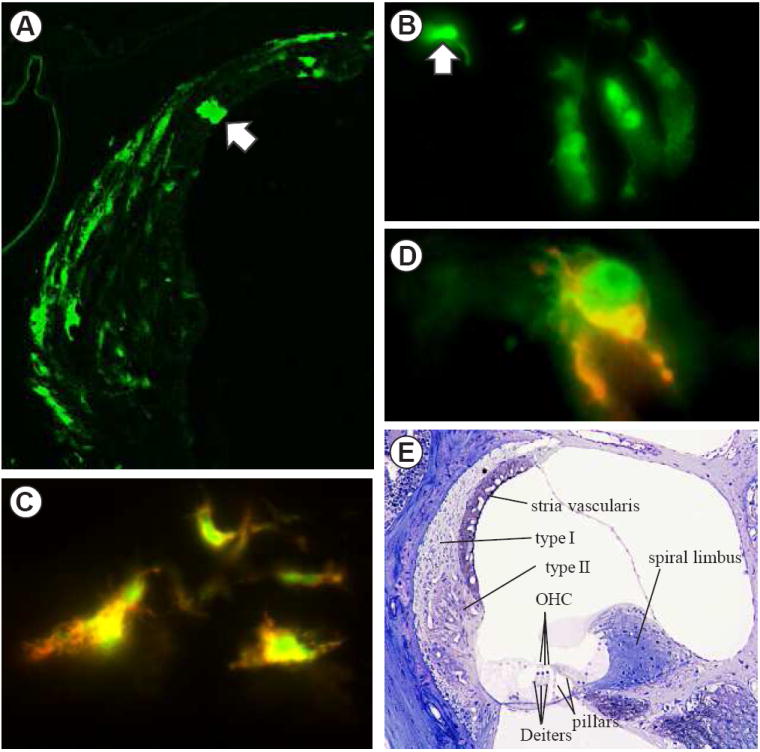
Fluorescent micrographs of GFP in NF-κB reporter mice (A-D). A. Spiral ligament in the lower basal turn of a mouse exposed to 100 dB noise for 2 hours and sacrificed 24 hours post-exposure. Most of the fluorescent cells are type I fibrocytes. The arrow indicates a rare fluorescent marginal cell of the stria vascularis. For orientation, see panel E. B. GFP fluorescent Deiters cells in a control mouse that was not exposed to noise or inflammatory stress. This is a rare case in which 3 adjacent Deiters cells were fluorescent. The arrow indicates the head of an outer pillar cell. C. Co-localization of green GFP fluorescence and red fluorescence reporting anti-GFP. D. A type II fibrocyte in a reporter mouse that was injected with LPS 24 hours prior to sacrifice showing green GFP fluorescence that was largely in different cellular compartments from the red immunostaining for GFP. E. A semithin section stained with osmium and toluidine blue showing the major structures of the cochlear duct. Outer hair cells (OHC), which are sensory cells, rest upon Dieters cells. The lateral wall of the duct is formed by the spiral ligament. Within the ligament the characteristic locations of type I and type II fibrocytes are indicated. The spiral limbus is situated toward the center of the cochlea.
In control mice that were placed in the noise chamber but not exposed to noise and those given saline injections in place of active compounds, there were a few GFP positive cells within the cochleas, which were taken to represent the background level of NF-κB activation. Most often these were non-sensory cells within the organ of Corti, specifically, pillar cells and Deiters cells (Figure 1B). In a cross section through the center of the mouse cochlea there are four half turns present, each of which contains two pillar cells and three Deiters cells (Figure 1E). When these cells were GFP positive, they usually occurred as single cells and were present in any of the four half turns. There was no apparent difference in the numbers or locations of GFP-positive Deiters cells or pillar cells between control and experimentally treated animals. The threshold of noise necessary to induce GFP within cochlear cells following noise exposure was approximately 92 dB SPL, with the locations of positive cells remaining the same for the range of intensities employed. However, with increasing noise intensities the numbers of GFP positive cells increased. Figure 2, left column, shows a representative case that was exposed to 100 dB noise and then allowed to survive 24 hours. Type I fibrocytes (open arrowheads) were the predominant cell type that showed NF-κB activation. A few fibrocytes in the spiral limbus (open arrow) were activated, as were solitary cells in the type II fibrocyte region (closed arrowheads). There was sporadic activation of epithelial cells along the basilar membrane (closed arrow), which showed no apparent relationship to the noise spectrum or damage to the organ of Corti, which is highly predicable with this noise exposure (Wang et al., 2002). Following noise exposures the distribution of activated cells within the spiral ligament was generally symmetric in the two ears. As a control for the possibility that the cells were activated by some systemic stress associated with the noise exposure, mice with unilateral removal of the malleus were exposed to noise. Activation of type I fibrocytes in these animals was always asymmetric, with few if any activated cells being present on the side which had the malleus removed (Figure 4A, 4B). This asymmetry showed that NF-κB activation in type I fibrocytes following noise exposures was due to local effects within the noise exposed ear and not due to systemic stress.
Figure 2.
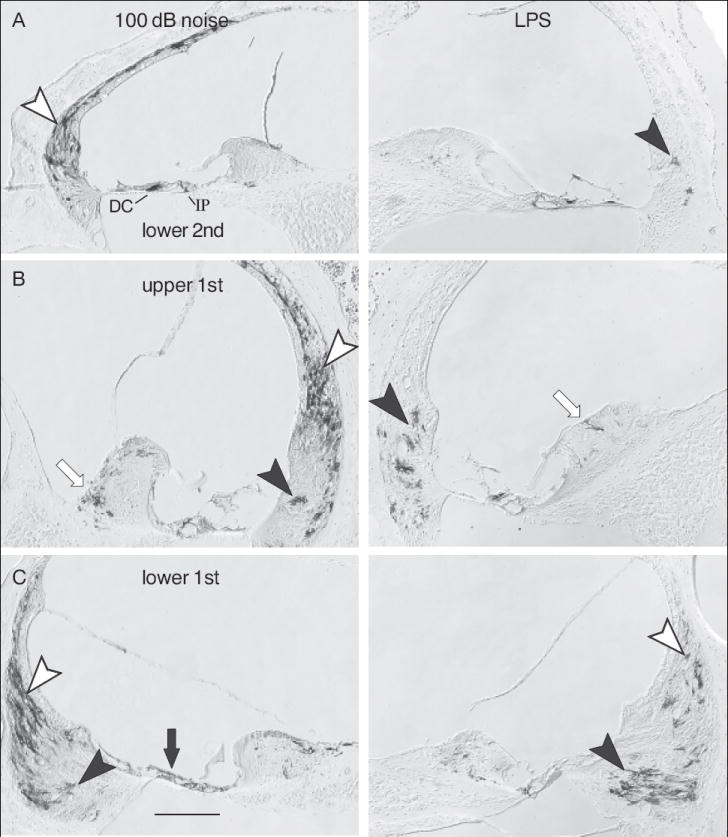
Immunostaining for GFP positive cells in GRP reporter mice following noise exposure and LPS. Each column shows the basal-most three half turns of an ear from a mouse that was exposed to a 100 dB noise (left column) or to intraperitoneal LPS injection (right column). Open arrowheads indicate type I fibrocytes, closed arrowheads indicate type II fibrocytes. Open arrows indicate fibrocytes in the spiral limbus. Closed arrows indicate epithelial cells on the basilar membrane. In panel A, IP indicates inner pillar cell; DC indicates Deiters cell. Calibration bar in C is 100 μm.
Figure 4.

NF-κB activation following noise exposure. A. Immunostaining for GFP in reporter a mouse that was perfused 24 hours following exposure to a 100 dB 8-16 kHz noise for two hours. B. Same case as shown in A except this ear had a conductive hearing loss. Calibration bar in A applies to A and B and indicates 40 microns. C. Immunostaining for p65 in the spiral ligament of a normal CBA mouse that was exposed to 100 dB 8-16 kHz noise for 1 hour and immediately sacrificed. The positive cells within the ellipse are type I fibrocytes with their nuclei stained. In contrast, root cells show their characteristic cytoplasmic staining. Insets show higher magnification of the indicated regions showing exclusive nuclear staining of type I fibrocytes (above) and lack of nuclear staining in roots cells, indicated by asterisks (below). The calibration bar indicates 10 microns and applies to both higher magnification images.
In contrast to results following noise exposure, following LPS administration a very different pattern of NF-κB activation was present. Figure 2 (right column) shows a representative case following LPS administration. As with noise exposures, following LPS administration, predominantly connective tissue cells within the spiral ligament showed NF-κB activation. In contrast to results with noise exposures, following LPS administration most activated cells were present in the type II fibrocyte region (closed arrowheads), which was largely free of activated cells in the noise experiments. There were a few activated cells within the spiral limbus (open arrow), within the type I fibrocyte region (open arrowhead), and on the basilar membrane (closed arrow), but most of the activated cells could be identified as type II fibrocytes by their location inferior to the spiral prominence and their expansive cytoplasm, which contrasts with the fusiform appearance of type I fibrocytes.
An additional difference between the distribution of activated cells in the noise experiments and the LPS experiments was that following LPS administration, the activated cells were strikingly asymmetric between the two ears. Positive cells in every tenth section from 12 LPS injected mice were counted and the ratio of the cell numbers on the side with greater numbers divided by those with the lesser numbers. These ratios were compared to a similar number of mice exposed to noise binaurally. For noise exposed mice, the mean ratio was 1.3. For LPS injected mice the mean ratio was 19.4. The total number of LPS-activated cells in given mice varied as much as a factor of 8. The lowest left-right ratio was in a case with 91 positive cells in the left ear, and 169 in the right. Cell activation following LPS showed no apparent preference for the left or right ear. The number of GFP positive spiral limbus cells was also asymmetrical and here the asymmetry always matched that of the spiral ligament cells. There was no sign of infiltrating cells in the cochleas of either the LPS or the noise stimulated animals, as assessed in the representative cases that were screened for using immunostaining for F4/80. Similar findings were obtained with male and female mice.
To confirm the identity of cells that showed NF-κB activation following different stresses reporter mice were immunostained both for GFP and for Na+,K+-ATPase. The latter protein has been shown to characterize type II fibrocytes (Schulte and Adams, 1989). Figure 3A shows almost no spatial overlap of noise-induced GFP fluorescence of type I fibrocytes (green) with the region of type II fibrocytes (shown in red). In contrast, Figure 3B shows cells that were activated by an LPS injection. In this case almost all activated cells lie within the red type II fibrocyte region.
Figure 3.
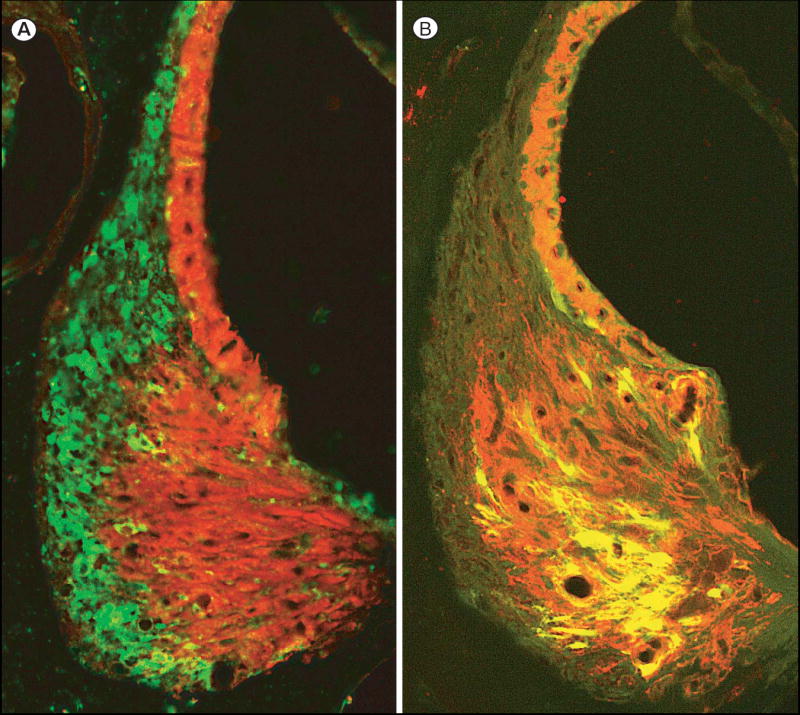
Fluorescent micrographs contrasting the localization of NF- B activation in type I and type II fibrocytes. A. Image of GFP fluorescent type I fibrocytes (green) of a noise exposed mouse merged with red type II fibrocytes and stria vascularis. The latter structures are immunopositive for Na+, K+,-ATPase. (See Fig. 1D for key.) B. Merged image of GFP fluorescent type II fibrocytes in a mouse injected with 50 μg LPS. The type II fibrocytes appear yellow because they are also positive for Na+, K+,-ATPase, which is red as in panel C.
To confirm that the reporter mice were reporting NF-κB activation, in some cases, the ears of noise treated non-transgenic mice were immunostained for the RelA (p65) isoform of NF-κB to seek evidence of nuclear localization of RelA, the cytologic sign of NF-κB activation. In normal cells, cytoplasmic localization of RelA is present in type I fibrocytes (Adams, 2002). Figure 4C shows nuclear localization within type I fibrocytes in normal CBA mice that were fixed immediately following a one hour exposure to 100 dB SPL noise. Type I but not type II fibrocytes show nuclear localization of RelA. Root cells (Fig. 4C lower inset), which are epithelial cells, show high levels of immunoreactivity for RelA but they never showed nuclear localization for RelA. In the reporter mice they never showed GFP localization following noise exposure or following inflammatory stress. The case shown in Fig. 4C had a control ear with a unilateral conductive hearing loss. The cochlea with the hearing loss contained no cells with nuclear staining for RelA. Thus, the identity of cells that showed NF-κB activation in response to noise exposure was the same when assayed by immunostaining for NF-κB nuclear localization and by the GFP reporter. In a previous report we showed that LPS administration leads to nuclear localization of P65 (Adams, 2002). Collectively these studies demonstrate that type 1 fibrocytes activate NF-κB in response to noise stimulation and type II fibrocytes activate NF-κB in response to LPS challenge.
Perhaps the most intriguing part of these findings is that epithelial cells (including sensory cells) showed no consistent NF-κB activation following the noise exposure, even though it is known that the noise levels employed in this study damage hair cells and result in substantial hearing loss (Wang et al., 2002). Neither were spiral ganglion cells found to activate NF-KB following any of the stress challenges.
Other systemic inflammatory stresses induce NF-κB activation in type II fibrocytes
One possible explanation for LPS-induced NF-κB activation of type II fibrocytes is that those cells express the LPS receptor TLR4. This idea was tested by immunostaining the cochlea for TLR4. No staining for TLR4 was found in type II fibrocytes. Hashimoto et al. (2005) have reported that systemic LPS administration can induce TLR4 expression in outer sulcus cells (cells which were never found to be activated in the present experiments), but they did not find TLR4 positive type II fibrocytes. Another explanation of LPS-induced NF-κB activation is that peritoneal cells responded to the intraperitoneal LPS by secreting TNF, additional cytokines and/or Hsp90 and circulating products from this reaction(s) induced NF-KB activation in the cochlea. Preliminary tests of this hypothesis were done by injecting other agents that are known to induce TNF secretion, anti-CD3 (Magness et al., 2004) and taxol (Byrd-Leifer, 2001). In response to both agents, asymmetric activation of type II fibrocytes was induced in a pattern that was indistinguishable from the activation that was seen following LPS administration. These findings do not support the idea that LPS was acting directly upon the cochlea. What may have stimulated the type II fibrocytes remains to be determined. When dexamethasone was injected prior to LPS injection, no activated cells were found. Animals injected with saline prior to LPS were indistinguishable from those given LPS alone (not shown).
Discussion
The reporter mouse used in the present experiments permitted identification of individual cells in which NF-κB was activated, regardless of which the five forms of NF-κB was involved. The results show that the ability to determine which cells in complex tissues employ NF-κB in response to different stressors offers opportunities to get a more realistic picture of the circumstances under which it operates. Remarkably few cochlear cell classes showed activation in response to noise trauma or systemic inflammatory challenge. Although both classes of challenge were found to activate NF-κB, each stimulus activated predominantly one cochlear cell type with a characteristic and unexpected anatomical pattern. The levels of noise used here are known to induce sensory cell damage and hearing loss, but not sensory cell death (Wang et al., 2002), but neither sensory cells nor the ganglion cells that innervate them showed any evidence of NF-κB activation in response to noise exposures. Somewhat unexpectedly, only fibrocytes were activated by noise exposures. The nature of the stress that provoked this activation is not clear because little is known about these cells’ functions. The sensory epithelium moves only nanometers in response to acoustic stimulation (Harris, 1968), so it is unlikely that type I fibrocytes located remotely from the basilar membrane would be mechanically stressed by sound. Further, the present results would not have been predicted by previous work on acoustic trauma or cochlear inflammation, much of which implicates reactive oxygen species in cochlear epithelial cells’ responses to acoustic trauma (Henderson et al., 2006). The finding of activation exclusively in connective tissue cells casts a new light upon the inner ear’s responses to acoustic stress and to systemic inflammatory stresses. It also raises questions about what stress pathways may be involved in sensory cell responses to noise-induced stress. NF-κB is generally assumed to be present in all cells and is known to directly or indirectly affect expression of hundreds of genes (Pahl, 1999; Ahn and Aggarwal, 2005; Ghosh et al., 1998), so its importance in biology is difficult to overstate. Although it is known to play a central role in many stress responses (Mercurio and Manning, 1999), it is clear that it does not participate in all stress responses. The lack of NF-κB activation in cochlear epithelial cells by traumatic noise exposure may reflect the fact that noise trauma seldom occurs in nature and was therefore most likely not a significant source of stress while the mammalian ear was evolving. The negative results with epithelial cells and ganglion cells emphasizes how little is known about the nature of the stress responsive pathways in various inner ear cell classes. On the other hand, the findings concerning connective tissue cells’ involvement in noise-induced and inflammatory responses in the cochlea affords opportunities for extending this research to learn more about these poorly understood cells and their functional roles in hearing.
In the present report an NF-κB reporter mouse is employed to visualize which cochlear cells showed activation of NF-κB following exposure to traumatizing noise or following administration of inflammatory agents. Immunolocalization of nuclear NF-κB confirmed the results of noise-induced activation in the reporter mice. Likewise, the present finding of systemic LPS administration inducing NF-κB activation in type II fibrocytes was confirmed by a previous report of immunostaining of nuclear p65 (a form of NF-κB) in those cells following LPS administration (Adams, 2002). The power of the present reporter mouse approach is that activation of all forms of NF-κB are reported and that the reporter is retained within activated cells for at least one day post activation so that it is not necessary to do an exhaustive time series in order to capture the brief period during which NF-κB is in the nucleus. Given the breath of the NF-κB forms reported by the assay and the relative lack of temporal constraints on the post stress time for detecting activation, the limited number of cell types that were found to be activated was remarkable.
There have been a number of reports of NF- B activation in the cochlea following stresses, including noise exposure (Ramkumar et al., 2004; Masuda et al., 2006; Tahera et al., 2006a; Tahera et al., 2006b; Nagashima et al., 2007; Selivanova et al., 2007; Miyao et al., 2008), administration of ototoxic drugs (Watanabe et al., 2002; Jiang et al., 2005;So et al., 2008; Chung et al., 2008), and inflammatory challenges (Moon et al., 2007; Miyao et al., 2008). Some of these did not include reports of which cell classes showed NF- B activation. Of those that did, agreements with the present results are mixed. Masuda et al., (2006) used immunostaining for P65 and P50 to identify NF- B activated cells in the cochlea following noise exposure. As in the present study, they found no noise-induced nuclear translocation indicative of NF- B activation in ganglion cells or in sensory cells. Rather, activated cells were exclusively within the spiral ligament and stria vascularis. The principal difference between those results and the present findings appears to be that they found noise-induced activation of type II fibrocytes (their Figure 7). This apparent difference from the present results may be due the fact that they used a noise intensity that was more than two orders of magnitude greater than was used in the present study, and/or due to their use of C57/Bl6J mice, which are more vulnerable to noise damage than CBA/CaJ mice used in the present study.
In contrast with Masuda et al., 2006, and with the present results, Tahera et al. (2006a; 2006b) found abundant nuclear NF- B localization of ganglion cells, even in mice with no noise stress. This unlikely finding may have resulted from the use of a non-specific NF- B antibody or some other error in immunostaining. Likewise, the report that NF- B was still present in the nuclei of ganglion cells and epithelial cells 8 days following stress (Miyao et al., 2008) needs confirmation and an explanation. Although we never found NF- B activation in sensory cells or ganglion cells, we cannot rule out the possibility that such activation could occur if much higher intensity noise had been utilized. We limited noise exposure levels to those that have been shown to permanently damage hair cells without destroying them so as to exclude effects that may be associated with tissue repair that would be induced by physical disruption of the tissue, such as occurs when extreme noise levels are employed. Consequently, our results can not be taken as strong evidence that sensory cells and ganglion cells never activate NF- B following noise trauma, but if it occurs, it appears to do so it happens at extreme levels of trauma. In any case, the relatively low levels of noise required to activate connective tissue cells shows that these cells either have much lower thresholds for noise-induced stress or that they utilize different tactics for dealing with the stresses.
Exposure of mice to low level noise has been shown to protect the ears from subsequent traumatizing noise exposure (Yoshida and Liberman, 2000). Activation of type I fibrocytes by noise stimulation suggest that these cells may play a role in protection of the ear from noise damage. How this might be achieved is not immediately clear, but two possibilities suggest themselves. Activation of NF- B in type I fibrocytes could result in up-regulation of inflammatory cytokines and/or nitric oxide (Ichimiya et al., 2000; Hashimoto et al., 2005), both of which are known to regulate gap junctional permeability. A combination of TNF and a purinergic agonist has been shown to modulate gap junctional connectivity and connexin 26 immunostaining in the trigeminal ganglion (Damodaram et al., 2009) and in immortalized mouse hepatocytes (Temme et al., 1998). Increased nitric oxide synthase has been reported to be correlated with decreased connexin 26 levels (Pitre et al., 2001). Decreasing gap junctional permeability in type I fibrocytes would be expected to deprive the stria vascularis of K+ ions, which should lead to decreasing endolymphatic potential. Decreasing endolymphatic potential would produce a transient decrease in cochlear sensitivity to sound and thereby decrease its vulnerability to noise-induced trauma. The obvious limitation of this hypothesis is that it remains to be demonstrated that nitric oxide or inflammatory cytokines actually affect permeability of gap junctions within the cochlea. Another possibility is that NF- B activation could result in type I fibrocytes secreting signaling compounds such as inflammatory cytokines that affect cochlear epithelial cells in a paracrine fashion. Such signaling of nearby root cells, for example, could ultimately result in signals being transmitted to sensory cells or to their adjacent epithelial cells via gap junctions (Kikuchi et al., 1995) or by purinergic signaling (Gale et al., 2004). How such signals might lead to protection of sensory cells from acoustic trauma remains a matter of conjecture. Both of these possibilities are based on the premise that type I fibrocytes are sensitive to excessive acoustic stimulation, perhaps by sensing K+ flux and/or other associated ion changes through the tissue. Their location between the primary site of K+ uptake from perilymph (type II fibrocytes) and the stria vascularis has them situated in a key site for controlling ion input to the stria (Kikuchi et al., 2000; Wangemann, 2006). Clearly, much work will be needed to test these and other possibilities.
Loss of function of type II fibrocytes would likely have a drastic impact upon hearing due to their critical roles in K+ ion uptake from perilymph. Transgenic mice with degenerated type II fibrocytes have hearing threshold elevations (Delprat et al., 2005). The finding that these cells may be selectively stressed by systemic inflammation raises the possibility that their vulnerability to systemically administered inflammatory stress may underlie two poorly understood forms of hearing loss, sudden hearing loss and immune-mediated hearing loss. Both types of hearing loss are usually unilateral, like the response of type II fibrocytes to systemic inflammatory challenge reported here. Both types of hearing loss are responsive to treatment with anti-inflammatory steroids (Chen et al., 2003; Ruchenstein, 2004). The fact that steroids are potent blockers of NF-κB activation suggests that, if the activation of NF-κB in type II fibrocytes by systemic inflammatory agents can result in a loss of function of these cells, this activation could be blocked or reversed by steroid treatment (Auphan et al., 1995; Scheinman et al., 1995). It remains to be determined what effects inflammatory stresses may have upon type II fibrocytes but the present results show that among cochlear cells these cells are selectively vulnerable to such stresses and that this effect can be blocked by steroid administration. Clearly systemic inflammations do not routinely cause unilateral hearing loss, but the present findings suggest that if a second stress were compounded with the largely unilateral stress upon type II fibrocytes like that shown in the present results, the result could compromise hearing in that ear.
The present results provide important insight into previously unrecognized signaling pathways activated in response to excessive noise, a health hazard of increasing clinical importance (Nelson et al., 2005). Understanding how the cochlea protects itself from acoustic trauma is of paramount importance, given the epidemic proportions of noise-induced hearing loss (Nelson et al., 2005; Catlin, 1986; Kurmis and Apps, 2007). Uncovering two previously unrecognized cell specific stress-related responses within the ear by use of the NF-κB reporter mouse demonstrates the power of the present approach for identifying individual cells that are stress responsive in vivo. There is every reason to believe that similar insights could be gained by this approach in a wide variety of other tissues.
Acknowledgments
This work is supported by grants from the National Institutes of Health DC03929 to JA AI062773, DK043351 to RJX and AI27849, AI46731 to BS. We thank Drs Charles Liberman, Saumil Merchant, Ian Rosenberg and Joe Avruch for critical reading of the manuscript. We also thank Sarah McCaffrey for expert technical assistance.
Abbreviations
- ABC
avidin-biotin-(horseradish peroxidase) complex
- dB
decibel
- EDTA
ethylenediaminetetraacetic acid
- GFP
green fluorescent protein
- gm
gram
- IKB
I kappa B
- ICCUA
Institutional Committee for the Care and Use of Animals
- i.p
intraperitoneally
- LPS
lipopolysaccharide
- kHz
kilohertz
- μg
microgram
- NF-κB
nuclear factor kappa B
- ng
nanogram
- SPL
sound pressure level
- TLR
toll-like receptor
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abe S, Katagiri T, Saito-Hisaminato A, Usami S, Inoue Y, Tsunoda T, Nakamura Y. Identification of CRYM as a candidate responsible for nonsyndromic deafness, through cDNA microarray analysis of human cochlear and vestibular tissues. Am J Hum Genet. 2003;72:73–82. doi: 10.1086/345398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams JC. Clinical implications of inflammatory cytokines in the cochlea: a technical note. Otol Neurotol. 2002;23:316–322. doi: 10.1097/00129492-200205000-00015. [DOI] [PubMed] [Google Scholar]
- Ahn KS, Aggarwal BB. Transcription factor NF-kappaB: a sensor for smoke and stress signals. Ann N Y Acad Sci. 2005;1056:218–233. doi: 10.1196/annals.1352.026. [DOI] [PubMed] [Google Scholar]
- Auphan N, DiDonato JA, Rosette C, Helmberg A, Karin M. Immunosuppression by glucocorticoids: inhibition of NF-kappa B activity through induction of I kappa B synthesis. Science. 1995;270:286–290. doi: 10.1126/science.270.5234.286. [DOI] [PubMed] [Google Scholar]
- Byrd-Leifer CA, Block EF, Takeda K, Akira S, Ding A. The role of MyD88 and TLR4 in the LPS-mimetic activity of Taxol. Eur J Immunol. 2001;31:2448–2457. doi: 10.1002/1521-4141(200108)31:8<2448::aid-immu2448>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- Catlin FI. Noise-induced hearing loss. Am J Otol. 1986;7:141–149. [PubMed] [Google Scholar]
- Chen CY, Halpin C, Rauch SD. Oral steroid treatment of sudden sensorineural hearing loss: a ten year retrospective analysis. Otol Neuroto. 2003;24:728–733. doi: 10.1097/00129492-200309000-00006. [DOI] [PubMed] [Google Scholar]
- Delprat B, Ruel J, Guitton MJ, Hamard G, Lenoir M, Pujol R, Puel JL, Brabet P, Hamel CP. Deafness and cochlear fibrocyte alterations in mice deficient for the inner ear protein otospiralin. Mol Cell Biol. 2005;25:847–853. doi: 10.1128/MCB.25.2.847-853.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gale JE, Piazza V, Ciubotaru CD, Mammano F. A mechanism for sensing noise damage in the inner ear. Curr Biol. 2004;14:526–529. doi: 10.1016/j.cub.2004.03.002. [DOI] [PubMed] [Google Scholar]
- Ghosh S, May MJ, Kopp EB. NF-kappa B and Rel proteins: evolutionarily conserved mediators of immune responses. Annu Rev Immunol. 1998;16:225–260. doi: 10.1146/annurev.immunol.16.1.225. [DOI] [PubMed] [Google Scholar]
- Hacker H, Karin M. Regulation and function of IKK and IKK-related kinases. Sci STKE. 2006;2006:re13. doi: 10.1126/stke.3572006re13. [DOI] [PubMed] [Google Scholar]
- Harris GG. Brownian motion in the cochlear partition. J Acoust Soc Am. 1968;44:176–186. doi: 10.1121/1.1911052. [DOI] [PubMed] [Google Scholar]
- Hashimoto S, Billings P, Harris JP, Firestein GS, Keithley EM. Innate immunity contributes to cochlear adaptive immune responses. Audiol Neurootol. 2005;10:35–43. doi: 10.1159/000082306. [DOI] [PubMed] [Google Scholar]
- Hayden MS, Ghosh S. Signaling to NF-kappaB. Genes Dev. 2004;18:2195–2224. doi: 10.1101/gad.1228704. [DOI] [PubMed] [Google Scholar]
- Henderson D, Bielefeld EC, Harris KC, Hu BH. The role of oxidative stress in noise-induced hearing loss. Ear Hear. 2006;27:1–19. doi: 10.1097/01.aud.0000191942.36672.f3. [DOI] [PubMed] [Google Scholar]
- Israel A. The IKK complex: an integrator of all signals that activate NF-kappaB? Trends Cell Biol. 2000;10:129–133. doi: 10.1016/s0962-8924(00)01729-3. [DOI] [PubMed] [Google Scholar]
- Jiang H, Sha SH, Schacht J. NF-kappaB pathway protects cochlear hair cells from aminoglycoside-induced ototoxicity. J Neurosci Res. 2005;79:644–651. doi: 10.1002/jnr.20392. [DOI] [PubMed] [Google Scholar]
- Kikuchi T, Kimura RS, Paul DL, Adams JC. Gap junctions in the rat cochlea: immunohistochemical and ultrastructural analysis. Anat Embryol (Berl) 1995;191:101–118. doi: 10.1007/BF00186783. [DOI] [PubMed] [Google Scholar]
- Kikuchi T, Adams JC, Miyabe Y, So E, Kobayashi T. Potassium ion recycling pathway via gap junction systems in the mammalian cochlea and its interruption in hereditary nonsyndromic deafness. Med Electron Microsc. 2000;33:51–56. doi: 10.1007/s007950070001. [DOI] [PubMed] [Google Scholar]
- Kurmis AP, Apps SA. Occupationally-acquired noise-induced hearing loss: a senseless workplace hazard. Int J Occup Med Environ Health. 2007;20:127–136. doi: 10.2478/v10001-007-0016-2. [DOI] [PubMed] [Google Scholar]
- Kwok OT, Nadol JB., Jr Correlation of otosclerotic foci and degenerative changes in the organ of Corti and spiral ganglion. Am J Otolaryngol. 1989;10:1–12. doi: 10.1016/0196-0709(89)90086-0. [DOI] [PubMed] [Google Scholar]
- Magness ST, Jijon H, Van Houten Fisher N, Sharpless NE, Brenner DA, Jobin C. In vivo pattern of lipopolysaccharide and anti-CD3-induced NF-kappa B activation using a novel gene-targeted enhanced GFP reporter gene mouse. J Immunol. 2004;173:1561–1570. doi: 10.4049/jimmunol.173.3.1561. [DOI] [PubMed] [Google Scholar]
- Masuda M, Nagashima R, Kanzaki S, Fujioka M, Ogita K, et al. Nuclear factor-kappa B nuclear translocation in the cochlea of mice following acoustic overstimulation. Brain Res. 2006;1068:237–247. doi: 10.1016/j.brainres.2005.11.020. [DOI] [PubMed] [Google Scholar]
- Merchant SN, Linthicum FH, Nadol JB., Jr Histopathology of the inner ear in DFNA9. Adv Otorhinolaryngol. 2000;56:212–217. doi: 10.1159/000059105. [DOI] [PubMed] [Google Scholar]
- Mercurio F, Manning AM. NF-kappaB as a primary regulator of the stress response. Oncogene. 1999;18:6163–6171. doi: 10.1038/sj.onc.1203174. [DOI] [PubMed] [Google Scholar]
- Minowa O, Ikeda K, Sugitani Y, Oshima T, Nakai S, Katori Y, Suzuki M, Furukawa M, Kawase T, Zheng Y, et al. Altered cochlear fibrocytes in a mouse model of DFN3 nonsyndromic deafness. Science. 1999;285:1408–1411. doi: 10.1126/science.285.5432.1408. [DOI] [PubMed] [Google Scholar]
- Miyao M, Firestein GS, Keithley EM. Acoustic trauma augments the cochlear immune response to antigen. Laryngoscope. 2008;118:1801–1808. doi: 10.1097/MLG.0b013e31817e2c27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon SK, Woo JI, Lee HY, Park R, Shimada J, et al. Toll-like receptor 2-dependent NF-kappaB activation is involved in nontypeable Haemophilus influenzae-induced monocyte chemotactic protein 1 up-regulation in the spiral ligament fibrocytes of the inner ear. Infect Immun. 2007;75:3361–3372. doi: 10.1128/IAI.01886-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagashima R, Sugiyama C, Yoneyama M, Kuramoto N, Kawada K, et al. Acoustic overstimulation facilitates the expression of glutamate-cysteine ligase catalytic subunit probably through enhanced DNA binding of activator protein-1 and/or NF-kappaB in the murine cochlea. Neurochem Int. 2007;51:209–215. doi: 10.1016/j.neuint.2007.04.023. [DOI] [PubMed] [Google Scholar]
- Nelson DI, Nelson RY, Concha-Barrientos M, Fingerhut M. The global burden of occupational noise-induced hearing loss. Am J Ind Med. 2005;48:446–458. doi: 10.1002/ajim.20223. [DOI] [PubMed] [Google Scholar]
- Pahl HL. Activators and target genes of Rel/NF-kappaB transcription factors. Oncogene. 1999;18:6853–6866. doi: 10.1038/sj.onc.1203239. [DOI] [PubMed] [Google Scholar]
- Parahy C, Linthicum FH., Jr Otosclerosis: relationship of spiral ligament hyalinization to sensorineural hearing loss. Laryngoscope. 1983;93:717–720. doi: 10.1288/00005537-198306000-00003. [DOI] [PubMed] [Google Scholar]
- Pitre DA, Seifert JL, Bauer JA. Perineurium inflammation and altered connexin isoform expression in a rat model of diabetes related peripheral neuropathy. Neurosci Lett. 2001;303:67–71. doi: 10.1016/s0304-3940(01)01696-2. [DOI] [PubMed] [Google Scholar]
- Ramkumar V, Whitworth CA, Pingle SC, Hughes LF, Rybak LP. Noise induces A1 adenosine receptor expression in the chinchilla cochlea. Hear Res. 2004;188:47–56. doi: 10.1016/S0378-5955(03)00344-7. [DOI] [PubMed] [Google Scholar]
- Ruckenstein MJ. Autoimmune inner ear disease. Curr Opin Otolaryngol Head Neck Surg. 2004;12:426–430. doi: 10.1097/01.moo.0000136101.95662.aa. [DOI] [PubMed] [Google Scholar]
- Selivanova O, Brieger J, Heinrich UR, Mann W. Akt and c-Jun N-terminal kinase are regulated in response to moderate noise exposure in the cochlea of guinea pigs. ORL J Otorhinolaryngol Relat Spec. 2007;69:277–282. doi: 10.1159/000103871. [DOI] [PubMed] [Google Scholar]
- Scheinman RI, Gualberto A, Jewell CM, Cidlowski JA, Baldwin AS., Jr Characterization of mechanisms involved in transrepression of NF-kappa B by activated glucocorticoid receptors. Mol Cell Biol. 1995;15:943–953. doi: 10.1128/mcb.15.2.943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulte BA, Adams JC. Distribution of immunoreactive Na+,K+-ATPase in gerbil cochlea. J Histochem Cytochem. 1989;37:127–134. doi: 10.1177/37.2.2536055. [DOI] [PubMed] [Google Scholar]
- Spicer SS, Schulte BA. Differentiation of inner ear fibrocytes according to their ion transport related activity. Hear Res. 1991;56:53–64. doi: 10.1016/0378-5955(91)90153-z. [DOI] [PubMed] [Google Scholar]
- Tahera Y, Meltser I, Johansson P, Hansson AC, Canlon B. Glucocorticoid receptor and nuclear factor-kappa B interactions in restraint stress-mediated protection against acoustic trauma. Endocrinology. 2006a;147:4430–4437. doi: 10.1210/en.2006-0260. [DOI] [PubMed] [Google Scholar]
- Tahera Y, Meltser I, Johansson P, Bian Z, Stierna P, et al. NF-kappaB mediated glucocorticoid response in the inner ear after acoustic trauma. J Neurosci Res. 2006b;83:1066–1076. doi: 10.1002/jnr.20795. [DOI] [PubMed] [Google Scholar]
- Temme A, Traub O, Willecke K. Downregulation of connexin32 protein and gap-junctional intercellular communication by cytokine-mediated acute-phase response in immortalized mouse hepatocytes. Cell Tissue Res. 1998;294:345–350. doi: 10.1007/s004410051184. [DOI] [PubMed] [Google Scholar]
- Teufert KB, Linthicum F., Jr Paget disease and sensorineural hearing loss associated with spiral ligament degeneration. Otol Neurotol. 2005;26:387–391. doi: 10.1097/01.mao.0000169773.23668.3f. [DOI] [PubMed] [Google Scholar]
- Trowe MO, Maier H, Schweizer M, Kispert A. Deafness in mice lacking the T-box transcription factor Tbx18 in otic fibrocytes. Development. 2008;135:1725–1734. doi: 10.1242/dev.014043. [DOI] [PubMed] [Google Scholar]
- Wang Y, Hirose K, Liberman MC. Dynamics of noise-induced cellular injury and repair in the mouse cochlea. JARO. 2002;3:248–268. doi: 10.1007/s101620020028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wangemann P. Supporting sensory transduction: cochlear fluid homeostasis and the endocochlear potential. J Physiol. 2006;576:11–21. doi: 10.1113/jphysiol.2006.112888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe K, Inai S, Jinnouchi K, Bada S, Hess A, et al. Nuclear-factor kappa B (NF-kappa B)-inducible nitric oxide synthase (iNOS/NOS II) pathway damages the stria vascularis in cisplatin-treated mice. Anticancer Res. 2002;22:4081–4085. [PubMed] [Google Scholar]
- Yoshida N, Liberman MC. Sound conditioning reduces noise-induced permanent threshold shift in mice. Hear Res. 2000;148:213–219. doi: 10.1016/s0378-5955(00)00161-1. [DOI] [PubMed] [Google Scholar]