

Abstract
The mechanism by which the enzyme pyruvate decarboxylase from two yeast species is activated allosterically has been elucidated. A total of seven three-dimensional structures of the enzyme, of enzyme variants, or of enzyme complexes from two yeast species, three of them reported here for the first time, provide detailed atomic resolution snapshots along the activation coordinate. The prime event is the covalent binding of the substrate pyruvate to the side chain of cysteine 221, thus forming a thiohemiketal. This reaction causes the shift of a neighboring amino acid, which eventually leads to the rigidification of two otherwise flexible loops, one of which provides two histidine residues necessary to complete the enzymatically competent active site architecture. The structural data are complemented and supported by kinetic investigations and binding studies, providing a consistent picture of the structural changes occurring upon enzyme activation.
Pyruvate decarboxylases (EC 4.1.1.1) catalyze the non-oxidative decarboxylation of pyruvate, yielding acetaldehyde and carbon dioxide. Together with the enzyme alcohol dehydrogenase (EC 1.1.1.1), which reduces the acetaldehyde to ethanol with the help of the co-substrate NADH, it represents the metabolic pathway of alcoholic fermentation. PDC3 is localized in the cytosol of cells from yeasts, plant seeds, and a few bacteria. The catalytic activity of PDC depends on the presence of the cofactor thiamine diphosphate (ThDP), which is bound mainly via a divalent metal ion (magnesium in most cases) to the protein moiety. Many detailed kinetic studies have been published on yeast PDC wild types (1–9). A number of ScPDC variants were analyzed, too (1–9). Some active site variants (E51A, D28A, E477Q) proved to be almost catalytically inactive. PDCs are multisubunit enzymes. The typical molecular mass of one subunit is 59–61 kDa. The tetramer is the catalytically active state of most PDCs. Higher oligomers (octamers) have been described for PDCs from plant seeds (10, 11) or some fungi (12). However, studies on structure function relationships of yeast PDCs showed that the dimer is the minimum functional unit of the enzyme displaying considerable catalytic activity (13, 14). The two closely related pyruvate decarboxylases from Saccharomyces cerevisiae (ScPDC) and Kluyveromyces lactis (KlPDC) are well characterized ThDP-dependent enzymes, which share 86.3% identical amino acid residues. They have been studied in great detail by means of kinetic investigations and spectroscopic studies. Both enzymes are allosterically regulated as reflected by sigmoid steady state kinetics and lag phases in their progress curves. The substrate PYR activates the initially inactive yeast PDCs in a time-dependent manner. Kinetic studies reveal a slow isomerization as triggered by substrate binding to a separate regulatory site (15). A number of substrate surrogates have been identified, which are able to activate PDC as well. The effects of pyruvamide (PA; for the chemical structure, see Scheme 1) on the activation kinetics have been studied in detail for ScPDC (15) and for KlPDC (16). Phosphonate analogues (among them methyl acetylphosphonate, MAP, Scheme 1) of pyruvate have been applied to elucidate the catalytic cycle (17–21) or to trap reaction intermediates in crystal structures (22–24). Chemical modification of PDCs with group-specific reagents pointed to an important role of cysteine residues (25). Site-directed mutagenesis of cysteine residues to alanine or serine demonstrated that residue Cys-221 might be the decisive one for enzyme activation (1, 4, 26, 27). Consequently, it was postulated that the region around Cys-221 is the regulatory site of PDC, and formation of a thiohemiketal at this side chain was proposed. However, a number of questions remained elusive. (i) How is the activator fixed at the regulatory site? (ii) What are the prime structural properties of the active state as compared with the inactive state? (iii) How is the signal transmitted from the regulatory to the active site? (iv) Which are the decisive features of the active site in the activated state that render efficient catalysis possible? To answer these questions, we present here the crystal structures of KlPDC with the bound substrate surrogate MAP and of the ScPDC variants D28E and E477Q with bound substrate PYR along with kinetic studies on the activating effect of both activators and binding studies using the small angle x-ray solution scattering (SAXS) method.
SCHEME 1.

Chemical structures of the substrate pyruvate, the activators pyruvamide and methyl acetylphosphonate, and the thiohemiketal from pyruvate and cysteine, respectively.
EXPERIMENTAL PROCEDURES
Enzyme Purification—The recombinant wild type and the variants D28A and E477Q have been purified according to the procedures of Killenberg-Jabs et al. (3), KlPDC according to Kutter et al. (28).
Protein Crystallization—KlPDC was dissolved in 20 mm citrate buffer, pH 6.1, 1 mm dithiothreitol, 5 mm ThDP, 5 mm MgSO4, 80 mm MAP. The same solution without MAP, but with 7–23% (w/v) PEG 2000/PEG 6000 (1:1 ratio) as precipitant, was used as reservoir. Well diffracting crystals were obtained after 10 days equilibration at 8 °C at 20% PEG and 1 mg of KlPDC per ml. Stored stock solutions of ScPDC variants were diluted into 12 mm citrate/1.3 mm MES, pH 6.3, 1.3 mm dithiothreitol, 1.3 mm ThDP, 1.3 mm MgSO4, 630 mm PYR, 5 μg of yeast alcohol dehydrogenase per ml, 2 mm NADH+H+. A buffer containing 18 mm citrate/2 mm MES, pH 6.3, 2 mm dithiothreitol, 2 mm ThDP, 2 mm MgSO4 together with 7–24% (w/v) PEG 2000/PEG 6000 (1:1 ratio) was used as reservoir solution. Well diffracting crystals resulted from these batches after 14 days of incubation on ice at PEG concentration of 22.5% (w/v) and 1 mg of enzyme per ml.
Data Collection, Structure Determination, and Refinement—For cryoprotection, crystals were incubated in a 1:1 mixture of reservoir and an aqueous solution of 32–42% (w/v) PEG 400 and 5% (v/v) glycerol for 1 min (in the case of MAP-KlPDC, 10–15 s). Diffraction data were collected at the beamlines X12 (European Molecular Biology Laboratory (EMBL) Outstation Hamburg c/o DESY) and ID14-2 (European Synchrotron Radiation Facility (ESRF), Grenoble, France). For indexing, integration, and scaling, the programs Denzo and Scalepack (29) were used. Intensities were converted to structure factor amplitudes using the program Truncate (30). KlPDC (PDB ID code 2VK4) was used as a search model for the MAP-KlPDC complex, and PA-ScPDC (PDB ID code 1QPB) was used as a search model for the PYR-ScPDCD28A complex and for the PYR-ScPDCE477Q complex. Refinement was realized with the program Refmac5 (30).
Determination of Protein Concentration—Whenever possible, the protein concentration was determined spectrophotometrically from the UV-spectra at 280 nm (Jasco V-560 UV/VIS spectrophotometer) using a molar extinction coefficient of 60,000 m-1·cm-1 for one PDC subunit. In all other cases, the Bradford method (31) was applied.
Kinetic Measurements—All measurements were carried out at 10 °C and pH 6.0, the optimum of catalytic activity, according to the coupled optical assay of Holzer et al. (32). In this assay, acetaldehyde, the product of the PDC-catalyzed reaction, is reduced with NADH to ethanol by the auxiliary enzyme alcohol dehydrogenase. A protein concentration of 120 μg/ml yeast alcohol dehydrogenase (catalytic activity 300 units/mg) guarantees the suppression of artifactual lag phases. 0.05 m MES buffer, pH 6.0, 0.15 m ammonium sulfate and an observation wavelength of 355 nm were applied. The corresponding extinction coefficient for NADH at this wavelength was determined to be 4764 m-1·cm-1. Preincubation experiments using MAP have been carried out in two different manners. First, PDC (1 mg/ml) was incubated for 30 min in the presence of varying MAP concentrations. The test reaction was started by a 1:500 dilution of the preincubation mixture into the test assay. Subsequently, full progress curves were recorded to determine the initial (v0) and the steady state velocity (vSS). In this case, the MAP concentrations in the preincubation mixture and the assay mixture were identical.
Secondly, the enzyme (6.7 mg/ml) was incubated with several MAP concentrations (15–500 mm) at 25 °C in a time-dependent manner. For each MAP concentration, a series of measurements were taken after different preincubation times t. To determine v0(t) after each individual preincubation run, an aliquot from the preincubation mix was diluted (1:500) into the test assay. The Holzer assay was carried out at 10 °C. In this type of experiment, the test did not contain any MAP except the amount introduced by the aliquot taken from the preincubation mixture. The empirical v0(t) values were fitted according to
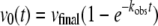 |
(Eq. 1) |
SAXS with Synchrotron Radiation—Measurements were performed at beamline X33 at the EMBL Hamburg outstation c/o DESY (camera length 2.7 m, MAR345 image plate detector, vacuum sample cell) at 16 °C and at protein concentrations of ∼2.5 mg/ml. The buffer system was the same as used for kinetic measurements, but 2 mm dithiothreitol was added. The momentum transfer axis s (s = 4πsinθ/λ, where 2θ is the scattering angle, and λ = 0.15 nm, the x-ray wavelength) was calibrated using collagen or tripalmitin as standards. The scattering patterns were collected for 120 s. MAR image files were extracted during data collection for intensity normalization (transmitted flux, detector response, scaling of the s axis) by the data reduction program Automar (33). Buffer scattering was subtracted using the program Primus-mar (34). The molecular masses were obtained from the ratio of the forward scattering intensity I(0) of the samples and that of the molecular mass standard bovine serum albumin.
RESULTS AND DISCUSSION
Kinetic Effects of MAP
As MAP was used as a substrate surrogate in crystallographic studies on KlPDC, it was of paramount importance to demonstrate that this analogue does indeed act as an activator of this enzyme. In the absence of any effectors, KlPDC displays typical sigmoidal steady state kinetics (16). After incubation of the enzyme with MAP, the sigmoidicity of the v versus [S] plot becomes gradually suppressed with increasing analogue concentrations (Fig. 1A). At a MAP concentration of 75 mm, the steady state kinetics of the enzyme is essentially hyperbolic. Moreover, at PYR concentrations below 1 mm, the activating effect of MAP is also documented by the higher absolute values of the steady state rates vSS (Fig. 1A, inset). This effect is even more apparent from the corresponding progress curves (Fig. 1B). In the absence of MAP, the progress curves of KlPDC show lag phases, which reflect the conversion of the initial inactive enzyme state into the activated enzyme state (16) according to the substrate-driven branch in Scheme 2. Empirical progress curves can be fitted to Equation 2, which conforms to the mechanism in Scheme 2 (16, 35).
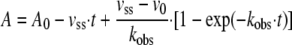 |
(Eq. 2) |
with A, absorbance at time t, A0, initial absorbance, kobs, the observed first order rate constant for the substrate activation process, v0, the initial velocity, and vSS, the steady state velocity. Upon preincubation of KlPDC with MAP, the initial reaction rate (v0) is increased as the concentration of potentially active enzyme (LEa, LEaL in Scheme 2) generated by the activator analogue increases. Eventually, at a MAP concentration of 75 mm, the progress curve appears to be a straight line in accordance with the hyperbolic v versus [S] plots obtained under the very same conditions (Fig. 1, A and B). Initial rates (v0) and steady state rates (vSS) can be evaluated from empirical progress curves (Equation 2, for details, see Krieger et al. (16)). The plot of the ratio v0/vSS versus MAP concentration (Fig. 1C) clearly demonstrates that MAP is able to completely activate KlPDC. For MAP, an effective half-saturation value of 35 mm can be extracted from the sigmoidal fit in Fig. 1C. Time-dependent preincubation studies revealed that the MAP-triggered activation of KlPDC is a rather slow process as compared with the activation by its native substrate PYR (Fig. 1D). Taking the chemical structures of both compounds into account (Scheme 1), the additional methyl residue and the bulky phosphonate moiety of MAP, as compared with the carboxylate of PYR, may cause a steric hindrance for the binding at the enzyme molecule. In the presence of MAP substrate, saturation is reached at slightly higher substrate concentrations (Fig. 1A), pointing to a weak competitive inhibition by this effector. In summary, the kinetic data presented here demonstrate that MAP is an activator for yeast PDCs, thus justifying its application in crystallographic studies. The preliminary kinetic model in Scheme 2 essentially illustrates the competition of substrate and activator molecules for the regulatory site in accordance to the crystallographic findings detailed below. Binding of either substrate or activating ligands at the regulatory site triggers a slow transition of the initially inactive enzyme form into the activated one. The rate-limiting step of this transition is the protein isomerization associated with the rate constants kiso and kilo, respectively. All other steps are assumed to be fast by comparison. In terms of the model, the dependence of kobs on the concentration of PYR in Fig. 1D relates to the substrate-driven branch, whereas the dependence of kobs on the concentration of MAP is reflected in the ligand-driven branch. Additionally, the mechanism allows for the competition effects for the active site in the activated state being present in the steady state data.
FIGURE 1.

MAP kinetics. A, influence of MAP on the shape of the v versus [S] plot (circles, 0 mm MAP, squares, 20 mm MAP, triangles, 40 mm MAP, and inverse triangles, 75 mm MAP). The lines represent sigmoid fits according to equation v([S]) = Vmax · [S]2/(A + B · [S] + [S]2) with the following values for the parameters for Vmax (units/mg), A (mm2), and B (mm), respectively, 0 mm MAP, 8.77, 2.52, 0.27; 20 mm MAP, 9.16, 0.72, 1.88; 40 mm MAP, 9.30, 10-9, 2.83; 75 mm MAP, 9.39, 10-9, 3.26. The uncertainties of these values are less than 20% throughout, except for the A values in the presence of 40–75 mm MAP, which are essentially zero, demonstrating Michaelis-Menten behavior of the activated enzyme with B as apparent Km. Inset, enlarged view at low substrate concentrations. B, transients of the KlPDC-catalyzed reaction at 3 mm PYR after preincubation with MAP (increasing concentrations from top to bottom as in A). For better comparison, original data are normalized to the steady state rate and to the same initial absorbance. C, dependence of the ratio of initial rate (v0) and steady state rate (vSS) on MAP concentration. The rates were obtained from transients (see B for primary data) according to Equation 2. The line represents a fit to the equation v0/vSS = K1 [L] + [L]2/(A + K1 · [L] + [L]2) with A = KL · Kilo · K1. The dissociation constants correspond to Scheme 2, and [L] represents the MAP concentration during preincubation. Fit parameters are K1 = 2.5 ± 2 mm, A = 1460 ± 142 mm2. D, activator concentration dependence of the apparent activation rate constant kobs (circles, PYR; squares, MAP). The kobs values were obtained from discontinuous measurements (for details, see the section “Kinetic Measurements”). The error bars represent the fitting errors. In the absence of MAP, the line represents the fit according to the equation kobs = kiso · [S]/([S] + Ka) + k-iso · KmS/(KmS + [S]) (16); in the presence of MAP, a line is drawn for better visualization only.
SCHEME 2.

Proposed kinetic model for the allosteric activation of yeast pyruvate decarboxylases in simultaneous presence of both the substrate S and the artificial activator L. Ka and KL are the primary dissociation constants at the regulatory site for S and L, respectively. kiso and kilo represent the forward rate constants for the conformation change driven by S and L, respectively. k-iso and k-ilo are the associated rate constants for the reverse reaction. Kiso and Kilo are defined by Kiso = (k-iso/kiso) and Kilo = (k-ilo/kilo), respectively. KmS is the Michaelis-Menten constant for the substrate-activated enzyme, whereas Kml refers to the ligand-activated enzyme. K1 is the dissociation constant of the artificial activator for the ligand-activated enzyme. Ligands/substrates written on the left of the enzyme species are bound to the regulatory site of the enzyme, and those written on the right are bound to the active site. In the absence of L, this activation scheme reduces to the established model of reference (16).
Activator Binding Studies Using SAXS
It had been demonstrated earlier (36) that addition of PA to ScPDC resulted in a significant increase of the radius of gyration RG, the scattering parameter describing the maximum distance of two points within a particle in solution. These changes in RG had been interpreted in terms of a global rearrangement within the protein molecule without alteration of the oligomerization state of the enzyme (because of the unchanged forward scattering intensity I(0), which correlates with the molecular mass of the particle). Here we illustrate, to the best of our knowledge for the first time, the activator concentration dependence of RG for the binding of PA and MAP to KlPDC and of the substrate PYR to the catalytically inactive variant ScPDCE477Q. All plots exhibit a clear saturation of the ligand binding (Fig. 2). The increase of the RG values in the presence of the ligands reflects the formation of activated enzyme species (LEa and LEaL). As shown in Fig. 2, MAP and PA have similar half-saturation values for KlPDC (21 and 38 mm, respectively) and for ScPDC (27 and 35 mm, respectively, data not shown). However, by far the lowest half-saturation value is found for the native substrate PYR with a value of 5.7 mm. This finding corresponds to the striking difference between Ka and KL (Scheme 2) as illustrated in Fig. 1D. The increase of RG, in turn, is similar for MAP and PYR binding (0.31 and 0.29 nm). The lowest shift of RG was found with PA (0.21 nm). Notably, the half-saturation values of MAP drawn from spectroscopic activation kinetics and SAXS binding studies, respectively, conform closely to each other (36 mm from Fig. 1C and 21 mm from Fig. 2). This indicates that both methods monitor the same process.
FIGURE 2.

Dependence of the scattering parameter RG of PDC on the concentration of the added activator. In the case of PA and MAP, KlPDC was used, and in the case of PYR, ScPDCE477Q was used (triangles, PA; squares, MAP; inverse triangles, PYR; lines, hyperbolic fits for KlPDC, sigmoid fit for ScPDCE477Q). The following values for half-saturation were obtained: for PYR, 5.7 ± 0.23 mm (n = 1.85); for MAP, 20.7 ± 2.39 mm; for PA, 37.8 ± 1.36 mm).
So far, only two types of overall tetramer crystal structures of yeast PDCs are known, the open (37) and half-side closed (28, 38, 39) conformation (Fig. 3). The described increase of RG by activator binding can be interpreted as a conformation change from the half-side closed to the open tetramer form. Moreover, theoretical scattering patterns can be calculated from both crystal structure models by using the program Crysol (40). On this basis, volume fractions of the corresponding tetramer forms can be calculated from the experimental scattering patterns (34) for all the applied activator concentrations. Surprisingly, the resulting plot is superimposable to the plot of RG versus activator concentration illustrated in Fig. 2.
FIGURE 3.

The overall crystal structures of PDC tetramers representing MAP-KlPDC and PYR-ScPDC variants in A and of native KlPDC (28), PA-ScPDC (38), and ketomalonate-activated ScPDC (39) in B. The subunits are shown with different colors in space-filling mode.
Structural Implications
Overall Structures—The crystal structure of KlPDC in complex with MAP was determined to 2.3 Å resolution, and those of the ScPDC variants in complex with PYR were determined to 1.7 Å (D28A) and 1.4 Å resolution (E477Q), respectively (for details, see Table 1). The final models comprise four times 562 amino acid residues, each of the subunits harboring one cofactor molecule ThDP, one Mg2+, and two molecules of activator. The asymmetric units contain the PDC tetramer. The overall folds of the subunits within one dimer (Fig. 3) are almost identical to that of native species (KlPDC, r.m.s.d. 2.28 Å for 1116 superimposed Cα-atoms, ScPDCE477Q, r.m.s.d. 0.97 Å for 1074 superimposed Cα-atoms, ScPDCD28A, r.m.s.d. 0.96 Å for 1074 superimposed Cα-atoms). Differences were found at the surface of the tetramer, i.e. at the middle domains and the C-terminal α-helices. The MAP molecules are located at the active sites of KlPDC with distances of 2.22 Å between the Cα-atoms of MAP and the C2 atoms of ThDP within one tetramer and at the regulatory sites, covalently bound to Cys-221 (bond length 1.82 Å). PYR molecules are found with distances of 1.69 Å (Cα–C2) at the active sites and 2.12 Å (Cα–S) at the regulatory sites in ScPDCD28A and with distances of 2.04 Å to Cys-221 of ScPDCE477Q but not in the active sites of the latter variant. The absence of PYR in the active site of ScPDCE477Q might be associated with the disrupted thiazolium ring of the cofactor molecule in this species. The tetramers of MAP-KlPDC, PYR-ScPDCD28A, and PYR-ScPDCE477Q are superimposable without significant differences (r.m.s.d. of 0.5–0.6 Å for 2248 Cα-atoms) All three crystal structures represent the open and planar tetramer conformation (Fig. 3A), which is fixed by the rigidification of the middle domain together with two loop regions (104–113 and 288–304).
TABLE 1.
Data collection and refinement statistics for MAP-KlPDC, PYR-ScPDCD28A, and PYR-ScPDCE477Q

where Ii(hkl) is the intensity of the observation i of the reflection hkl and N is the redundancy (41)
Loop Structuring—The most salient feature common to all activated structures described herein is the well defined electron density for two loop regions (residues 104–113 and 288–304, respectively), which is virtually absent in the crystal structure of native ScPDC (37) or poorly defined in the structure of native KlPDC (28). It may therefore be concluded that loop structuring and loop translocation are the decisive events in the activation process. The gained rigidity of both loop regions at all subunits within the tetramer enforces the planar and symmetric dimer arrangement within the tetramer (Fig. 3A). Both loops are located in the neighborhood of the active site at the core region of the tetramer (see Figs. 5 and 6). It should be noted that these loops, although cooperating in the completion of one active site, belong to different subunits (38). In the frozen state, these loop regions are stabilized internally by a number of H-bonds (see Fig. 6C and supplemental materials). Loop 104–113 additionally develops a short α-helix and closes partially over the active site. His-114 and His-115, the next upstream neighbors of loop 104–113, are part of the active site. For His-114, an essential function in PDC catalysis has been proposed from kinetic studies with accordant variants from yeast and bacteria (7, 8, 42, 43). Tittmann et al. (44) postulated a specific role for His-114 (together with Asp-28) during release of the reaction product acetaldehyde. Because of the location of both loop regions in the tetramer core, i.e. the inner interface between two dimers of the tetramer, crystal contacts can clearly be excluded as reason for loop structuring.
FIGURE 5.

View from the surface of KlPDC molecule down to one active site. A and B, the location of the histidine residues 114 and 115 in native KlPDC (A)- and MAP-activated KlPDC (B). The cofactor ThDP and the histidine residues are presented in stick mode (individually colored atoms), and all other residues are shown as a tube of the Cα line, with loop 104–113 in blue, loop 288–304 in red, and the C-terminal helix in green.
FIGURE 6.

Snapshots of the signal transduction pathway from the regulatory to the active site in yeast PDCs. The different subunits are shown as Cα trace in different colors, the labeled residues are shown as sticks, and the color of the label is that of the subunit the residue belongs to. A, overlay of one regulatory site before (carbon atoms in green) and after covalent binding of the activator MAP at Cys-221 (carbon atoms in beige). The 4 Å shift of the thiohemiketal is clearly to be seen. B, the shift of the thiohemiketal causes direct interactions to residues Ala-286 and Gly-287 and restructures the whole loop region 288–304 (beige tube; loop conformation before activation is shown in green). C, the structured loop 288–304 induces direct interactions to the other loop region 104–113, and the H-bonds between both loops are illustrated as blue dashed lines. The new conformation of this loop (beige, loop conformation before activation in green) reorients the adjacent residues His-114 and His-115, which now can directly interact with the activator molecule bound at the C2 atom of ThDP.
Regulatory Site—The most striking result of this study is the evidence for a covalent C–S bond at the side chain of Cys-221 in the activated structures, confirming results of studies on the activation mechanism by chemical modification (25) and mutation of cysteine residues (1, 4, 26, 27) in ScPDC. The covalent character of this bond is well documented by the crystallographic C–S distance (1.82 Å) as well as by the tetrahedral configuration at the former carbonyl carbon of both MAP (Fig. 4A) and PYR (Fig. 4B). Notably, the sp3 character of the Cα-atom of PYR in both activated ScPDC variants is somewhat less developed than that of the equivalent carbon atom in the MAP-activated KlPDC. Together with the longer distances between the Cα-atom of PYR and the S-atom of Cys-221, this could be indicative of a dynamic equilibrium of non-covalently and covalently bound PYR at the regulatory site. Apart from Cys-221, the following residues are located in vicinity (within a 5 Å distance) to the bound activator (for MAP-KlPDC): His-92, Arg-161, His-225, Gly-286, Ala-287, Leu-288, His-310, Ser-311, Tyr-313, Met-326, and five waters. Thus, the regulatory site is predominantly lined by positively charged side chains, which can interact electrostatically with the phosphonate moiety of MAP or the carboxylate moiety of PYR, respectively. This electrostatic interaction might guide the negatively charged activator molecule to the regulatory site. Surprisingly, H-bonds are only formed from main chain carbonyl oxygen atoms of Gly-286 and Ala-287 to the α-hydroxyl group (the former carbonyl group) of the covalently bound MAP.
FIGURE 4.

View of the regulatory sites with the activators bound covalently to residue Cys-221 for MAP-KlPDC (A) and PYR-ScPDCE477Q (B). Electron density is shown at a σ-level of 2.0 in the 2Fo - Fc map (blue) and at a σ-level of 4.0 in the omit map (green). Amino acid residues are illustrated in stick mode. The labeled residues can directly interact with the thiohemiketal. Labels in A correspond also to B.
Effects on the Active Site—Interestingly, all side chains constituting the active site apart from Asp-28, His-114, and His-115 remain unaffected by the binding of the activators at Cys-221. Furthermore, no significant distortion of the vital V-conformation of the cofactor is seen in the activated state. However, a comparison of the native and the MAP-activated crystal structures of KlPDC revealed that the two amino acids His-114 and His-115 undergo marked structural reorientations upon activator binding (Fig. 5). The histidine side chains are now directed toward the substrate-binding site, i.e. the distances of their ring nitrogens to the C2 atom of the cofactor ThDP are diminished from 12–15 to 6–7 Å. Concomitantly, the main chain of residue Asp-28/Ala-28 is rotated by ∼35°, and the side chain is orientated toward His-115. This reorientation basically restructures the active site into its enzymatically competent architecture, triggered by activator binding at Cys-221. A survey of all Cα-atom shifts recorded upon activation is given in the supplemental information.
Signal Transfer from the Thiohemiketal at Cys-221 to the Active Site—The question of how the signal of activation is transmitted from Cys-221 to the active site is a matter of debate that has not been decided to this very day. What can be drawn about signal transduction from the new structures? Firstly, the formation of the thiohemiketal at Cys-221, which itself is not part of either loop, shifts the side chain of this amino acid about 4 Å from its original position in the native structures (Fig. 6A). Subsequently, this conformation change enforces the reorientation of a number of amino acid residues in the interaction radius of the thiohemiketal, such as His-92, His-225, Gly-286, Ala-287, His-310, and Ser-311 (Fig. 6B). The arising interaction network fixes the complete R-domain. Loop 288–304 is part of that domain and is completely restructured. This loop forms a number of interactions with another, originally flexible loop 104–113 of the PYR domain of the adjacent subunit, especially residues Leu-289, Ser-290, and Asp-291, with residues Leu-112, Leu-113 and becomes structured itself (Fig. 6C). Due to this interaction between both loops, the signal is transmitted to the other subunit within the functional dimer. Further stabilization of the loop 104–113 arises from interactions with the C-terminal helix, which in the activated state closes over the active site (Fig. 5B). The position of the latter is now identical to those in the three-dimensional structures of the non-activated species ZmPDC (pyruvate decarboxylase from Zymomonas mobilis) (45) and EcIPDC (indolepyruvate decarboxylase from Enterobacter cloacae) (46). Eventually, the signal is transmitted to His-114 and His-115, which adopt a new orientation (Figs. 5B and 6C). These conformation changes in the vicinity of the cofactor ThDP make the active site now competent for effective catalysis and binding of the substrate.
Molecular Causes of Cofactor Activation—Phenomenologically, the activation of ScPDC and KlPDC is reflected in lag phases of their respective progress curves as well as in the sigmoidal shape of the steady state kinetics (15, 16). Both enzyme species are potentially inactive at the start of the reaction (15, 16, 47). A decade ago, Kern et al. (48) have shown that the prime molecular target of activation in ScPDC is the deprotonation at the C2 atom of ThDP, which requires a tremendous shift of the pKa value of the C2 proton to become catalytically competent (49). The structural basis of this kinetic effect remained, however, largely elusive. Evidently, some structural differences between the active sites in the native and the activated state must account for the observed acceleration of the H/D exchange at the C2 atom of the cofactor as triggered by activation in both yeast PDCs. The results of the current crystallographic study allow some preliminary insight. First and foremost, the active site as such is complete only in the activated state. A vital role has been ascribed to His-113 in ZmPDC, as part of a catalytic dyad supporting aldehyde release (44, 50). Its position is equivalent to that of His-114 in ScPDC and KlPDC, respectively, which reorients upon activation. Both histidines, His-114 and His-115, are required for efficient catalysis. Moreover, the H114F/H115F variant, although being almost inactive, shows perfect Michaelis-Menten behavior, pointing to abolished activation (7). These observations alone, however, do not yet explain the accelerated deprotonation at the C2 atom of the cofactor, as neither His-114 nor His-115 is specifically involved in this process. Secondly, the structured loops, particularly the region 104–113, shield the active site against the solvent. Thus, the microenvironment of the cofactor is probably less polar in the activated than in the native state. A non-polar environment dramatically promotes deprotonation at the C2 atom of the cofactor as the neutral ylide species is generated in this process (51). Thus, the partial closure of the active site might well be the decisive molecular cause of the activation of the cofactor. Thirdly, solvent shielding might likewise contribute to the stabilization of non-polar reaction intermediates, as e.g. the enamine and, additionally, promote the general sequestration of substrates and intermediates. On the other hand, substrate molecules must have access to the active site during catalysis. Therefore, the active site cannot be totally shielded from the solvent. It should be noted that even the rigidified loop structures retain a considerable measure of mobility, which allows substrate access. It remains to be clarified whether a quasi-periodic closure and reopening of the active site in the activated state is coupled to particular stages of the catalytic cycle as advocated by Kluger and Smyth (52) or is even synchronized with a dynamic alternation between covalently and non-covalently associated PYR at Cys-221 as proposed by Alvarez et al. (53) and defended recently by Schowen (54). Finally, the question arises as to why nature chose thiohemiketal formation, a rare mode of covalent modification, which is, to the best of our knowledge, unique as a principle of enzyme regulation. Probably, thiohemiketal formation was favored as PYR lacks an extended hydrophobic moiety that could function as a partner in non-covalent ligand-protein interaction.
Supplementary Material
Acknowledgments
We thank the Jordan group at Rutgers for the variants D28A and E477Q, Birgitta Seliger for preparation of the enzyme species from S. cerevisiae, and Anja Seidel and Robert Eckensthaler for crystallization of MAP-KlPDC. Access to EMBL beamlines X12 and X33 at the DORIS storage ring, DESY, Hamburg and to beamline ID14-2 at the ESRF in Grenoble is acknowledged.
The atomic coordinates and structure factors (codes 2VJY, 2VK1 and 2VK8) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
The on-line version of this article (available at http://www.jbc.org) contains two supplemental tables and two supplemental figures.
Footnotes
The abbreviations used are: PDC, pyruvate decarboxylase; ScPDC, pyruvate decarboxylases from Saccharomyces cerevisiae; KlPDC, pyruvate decarboxylases from Kluyveromyces lactis; ThDP, thiamine diphosphate; MAP, methyl acetylphosphonate; PYR, pyruvate; PA, pyruvamide; SAXS, small angle x-ray solution scattering; PEG, polyethylenglycol; MES, 4-morpholineethanesulfonic acid; r.m.s.d., root mean square deviation.
References
- 1.Baburina, I., Gao, Y., Hu, Z., Jordan, F., Hohmann, S., and Furey, W. (1994) Biochemistry 33 5630-5635 [DOI] [PubMed] [Google Scholar]
- 2.Killenberg-Jabs, M., König, S., Hohmann, S., and Hübner, G. (1996) Biol. Chem. Hoppe-Seyler 377 313-317 [DOI] [PubMed] [Google Scholar]
- 3.Killenberg-Jabs, M., König, S., Eberhardt, I., Hohmann, S., and Hübner, G. (1997) Biochemistry 36 1900-1905 [DOI] [PubMed] [Google Scholar]
- 4.Baburina, I., Li, H., Bennion, B., Furey, W., and Jordan, F. (1998) Biochemistry 37 1235-1244 [DOI] [PubMed] [Google Scholar]
- 5.Li, H., Furey, W., and Jordan, F. (1999) Biochemistry 38 9992-10003 [DOI] [PubMed] [Google Scholar]
- 6.Li, H., and Jordan, F. (1999) Biochemistry 38 10004-10012 [DOI] [PubMed] [Google Scholar]
- 7.Liu, M., Sergienko, E. A., Guo, F. S., Wang, J., Tittmann, K., Hübner, G., Furey, W., and Jordan, F. (2001) Biochemistry 40 7355-7368 [DOI] [PubMed] [Google Scholar]
- 8.Sergienko, E. A., and Jordan, F. (2001) Biochemistry 40 7369-7381 [DOI] [PubMed] [Google Scholar]
- 9.Joseph, E., Wei, W., Tittmann, K., and Jordan, F. (2006) Biochemistry 45 13517-13527 [DOI] [PubMed] [Google Scholar]
- 10.Mücke, U., König, S., and Hübner, G. (1995) Biol. Chem. Hoppe-Seyler 376 111-117 [DOI] [PubMed] [Google Scholar]
- 11.Zehender, H., Trescher, D., and Ullrich, J. (1987) Eur. J. Biochem. 167 149-154 [DOI] [PubMed] [Google Scholar]
- 12.Alvarez, M. E., Rosa, A. L., Temporini, E. D., Wolstenholme, A., Panzetta, G., Patrito, L., and Maccioni, H. J. F. (1993) Gene (Amst.) 130 253-258 [DOI] [PubMed] [Google Scholar]
- 13.Killenberg-Jabs, M., Jabs, A., Lilie, H., Golbik, R., and Hübner, G. (2001) Eur. J. Biochem. 268 1698-1704 [PubMed] [Google Scholar]
- 14.Kutter, S., Spinka, M., Koch, M. H. J., and König, S. (2007) Prot. J. 26 585-591 [DOI] [PubMed] [Google Scholar]
- 15.Hübner, G., Weidhase, R., and Schellenberger, A. (1978) Eur. J. Biochem. 92 175-181 [DOI] [PubMed] [Google Scholar]
- 16.Krieger, F., Spinka, M., Golbik, R., Hübner, G., and König, S. (2002) Eur. J. Biochem. 269 3256-3263 [DOI] [PubMed] [Google Scholar]
- 17.Biryukov, A. I., Bunik, V. I., Zhukov, Y. N., Khurs, E. N., and Khomutov, R. M. (1996) FEBS Lett. 382 167-170 [DOI] [PubMed] [Google Scholar]
- 18.Bunik, V. I., Biryukov, A. I., and Zhukov, Y. N. (1992) FEBS Lett. 303 197-201 [DOI] [PubMed] [Google Scholar]
- 19.Kluger, R., and Nakaoka, K. (1974) Biochemistry 13 910-914 [DOI] [PubMed] [Google Scholar]
- 20.Kluger, R., and Pike, D. C. (1977) J. Am. Chem. Soc. 99 4504-4506 [DOI] [PubMed] [Google Scholar]
- 21.O'Brien, T. A., Kluger, R., Pike, D. C., and Gennis, R. B. (1980) Biochim. Biophys. Acta 613 10-17 [DOI] [PubMed] [Google Scholar]
- 22.Arjunan, P., Sax, M., Brunskill, A., Chandrasekhar, K., Nemeria, N., Zhang, S., Jordan, F., and Furey, W. (2006) J. Biol. Chem. 281 15296-15303 [DOI] [PubMed] [Google Scholar]
- 23.Bera, A. K., Polovnikova, L. S., Roestamadji, J., Widlanski, T. S., Kenyon, G. L., McLeish, M. J., and Hasson, M. S. (2007) J. Am. Chem. Soc. 129 4120-4121 [DOI] [PubMed] [Google Scholar]
- 24.Wille, G., Meyer, D., Steinmetz, A., Hinze, E., Golbik, R., and Tittmann, K. (2006) Nat. Chem. Biol. 2 324-328 [DOI] [PubMed] [Google Scholar]
- 25.Hübner, G., König, S., and Schellenberger, A. (1988) Biomed. Biochim. Acta 47 9-18 [PubMed] [Google Scholar]
- 26.Zeng, X. P., Farrenkopf, B., Hohmann, S., Dyda, F., Furey, W., and Jordan, F. (1993) Biochemistry 32 2704-2709 [DOI] [PubMed] [Google Scholar]
- 27.Wang, J., Golbik, R., Seliger, B., Spinka, M., Tittmann, K., Hübner, G., and Jordan, F. (2001) Biochemistry 40 1755-1763 [DOI] [PubMed] [Google Scholar]
- 28.Kutter, S., Wille, G., Relle, S., Weiss, M. S., Hübner, G., and König, S. (2006) FEBS. J. 273 4199-4209 [DOI] [PubMed] [Google Scholar]
- 29.Otwinowski, Z., and Minor, W. (1997) Methods Enzymol. 276 307-326 [DOI] [PubMed] [Google Scholar]
- 30.Collaborative Computational Project Number 4 (1994) Acta Crystallogr. Sect. D Biol. Crystallogr. 50 760-76315299374 [Google Scholar]
- 31.Bradford, M. (1976) Anal. Biochem. 72 248-254 [DOI] [PubMed] [Google Scholar]
- 32.Holzer, H., Schultz, G., Villar-Palasi, C., and Jüntgen-Sell, J. (1956) Biochem. Z. 327 331-344 [PubMed] [Google Scholar]
- 33.Petoukhov, M. V., Konarev, P. V., Kikhney, A. G., and Svergun, D. I. (2007) J. Appl. Crystallogr. 40 223-228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Konarev, P. V., Volkov, V. V., Sokolova, A. V., Koch, M. H. J., and Svergun, D. I. (2003) J. Appl. Crystallogr. 36 1277-1282 [Google Scholar]
- 35.Werther, T., Spinka, M., Tittmann, K., Schütz, A., Golbik, R., Mrestani-Klaus, C., Hübner, G., and König, S. (2008) J. Biol. Chem. 283 5344-5354 [DOI] [PubMed] [Google Scholar]
- 36.Hübner, G., König, S., Schellenberger, A., and Koch, M. H. J. (1990) FEBS Lett. 266 17-20 [DOI] [PubMed] [Google Scholar]
- 37.Arjunan, P., Umland, T., Dyda, F., Swaminathan, S., Furey, W., Sax, M., Farrenkopf, B., Gao, Y., Zhang, D., and Jordan, F. (1996) J. Mol. Biol. 256 590-600 [DOI] [PubMed] [Google Scholar]
- 38.Lu, G., Dobritzsch, D., Baumann, S., Schneider, G., and König, S. (2000) Eur. J. Biochem. 267 861-868 [DOI] [PubMed] [Google Scholar]
- 39.Furey, W., Arjunan, P., Chen, L., Sax, M., Guo, F., and Jordan, F. (1998) Biochim. Biophys. Acta 1385 253-270 [DOI] [PubMed] [Google Scholar]
- 40.Svergun, D., Barberato, C., and Koch, M. H. J. (1995) J. Appl. Cryst. 28 768-773 [Google Scholar]
- 41.Weiss, M. S. (2001) J. Appl. Cryst. 34 130-135 [Google Scholar]
- 42.Candy, J. M., Koga, J., Nixon, P. F., and Duggleby, R. G. (1996) Biochem. J. 315 745-751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Huang, C. Y., Chang, A. K., Nixon, P. F., and Duggleby, R. G. (2001) Eur. J. Biochem. 268 3558-3565 [DOI] [PubMed] [Google Scholar]
- 44.Tittmann, K., Golbik, R., Uhlemann, K., Khailova, L., Schneider, G., Patel, M., Jordan, F., Chipman, D. M., Duggleby, R. G., and Hübner, G. (2003) Biochemistry 42 7885-7891 [DOI] [PubMed] [Google Scholar]
- 45.Dobritzsch, D., König, S., Schneider, G., and Lu, G. (1998) J. Biol. Chem. 273 20196-20204 [DOI] [PubMed] [Google Scholar]
- 46.Schütz, A., Sandalova, T., Ricagno, S., Hübner, G., König, S., and Schneider, G. (2003) Eur. J. Biochem. 270 2312-2321 [DOI] [PubMed] [Google Scholar]
- 47.Hübner, G., and Schellenberger, A. (1986) Biochem. Int. 13 767-772 [PubMed] [Google Scholar]
- 48.Kern, D., Kern, G., Neef, H., Tittmann, K., Killenberg-Jabs, M., Wikner, C., Schneider, G., and Hübner, G. (1997) Science 275 67-70 [DOI] [PubMed] [Google Scholar]
- 49.Washabaugh, M. W., and Jencks, W. P. (1988) Biochemistry 27 5044-5053 [DOI] [PubMed] [Google Scholar]
- 50.Schenk, G., Leeper, F. J., England, P., and Duggleby, R. G. (1997) Eur. J. Biochem. 248 63-71 [DOI] [PubMed] [Google Scholar]
- 51.Crosby, J., and Lienhard, G. E. (1970) J. Am. Chem. Soc. 92 5707-5716 [DOI] [PubMed] [Google Scholar]
- 52.Kluger, R., and Smyth, T. (1981) J. Am. Chem. Soc. 103 1214-1216 [Google Scholar]
- 53.Alvarez, F. J., Ermer, J., Hübner, G., Schellenberger, A., and Schowen, R. L. (1995) J. Am. Chem. Soc. 117 1678-1683 [Google Scholar]
- 54.Schowen, R. L. (2007) Isot. Environ. Health Stud. 43 1-16 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.