

Abstract
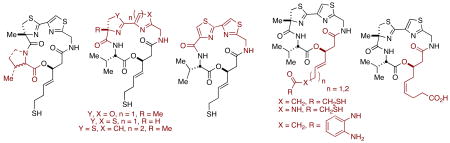
Fourteen analogs of the marine natural product largazole have been prepared and assayed against HDACs 1,2, 3, and 6. Olefin cross-metathesis was used to efficiently access six variants of the side-chain zinc-binding domain, while adaptation of our previously reported modular synthesis allowed probing of the macrocyclic cap group
Histone deacetylases (HDACs) control gene transcription via regulation of lysine acetylation and their selective inhibition is currently a major area of research in cancer chemotherapy.1 The eighteen known HDACs are divided into four classes based on sequence homology to yeast counterparts.2 The significance of individual classes and isoforms to cancer growth and inhibition is not well understood, yet the majority of data suggest that class I HDACs are the most relevant, tractible cancer targets.3
The pharmacophore model for HDAC inhibition consists of three elements: (1) a surface recognition unit which interacts with the rim of the binding pocket, (2) a metal-binding domain which coordinates to the active site zinc ion, and (3) a linker that connects the surface recognition site to the zinc-binding domain.1b Numerous HDAC inhibitors (HDACi), both natural and synthetic, are known and variations in all three features have variably contributed to potency and selectivity in new HDACi.4,5
To date, the most potent and selective HDACi known is largazole (1a, Figure 1), a densely functionalized macrocyclic depsipeptide isolated from the cyanobacterium Symploca sp. by Luesch and co-workers.6 We have recently disclosed a concise, modular, and scalable total synthesis of largazole and demonstrated its picomolar activity against HDACs 1, 2, and 3, as well as low nanomolar cytotoxicity against a number of chemoresistant cancer cell lines.7,8 Additionally, we have disclosed a detailed conformation-activity relationship model for largazole, FK228, and their corresponding amide isosteres with insights into the key contacts and associated spatial determinants that provide this remarkable level of activity.9 Herein we describe our continuing efforts to modify the structural scaffold of largazole with the goal of further defining and expanding structure-activity relationships within the family of macrocyclic HDACi’s. Our over-arching aim in this context, is to perturb the class- and isoform-selectivity as initially observed in robust biochemical assays such that differences in specific enzymatic activity might later be correlated with phenotypic responses in cell-based assays.
Figure 1.

Selected largazole analogs and azumamide E.
Our reported route to largazole proved highly reproducible and we were able to rapidly adapt it to simple variants of the macrocyclic core.7 Thus we were able to easily access milligram quantities of the C-2 epimer (3) and the enantiomer (2) of largazole. Additionally, we sought to perturb the conformation of the macrocycle by imparting greater rigidity; our initial foray in this capacity, replaced the valine residue with proline (4). Compound 4 could be obtained in only slightly diminished overall yield via the same synthetic route we deployed in the total synthesis of largazole (see Supporting Information).
Two methods were employed to alter side chain functionality and access a series of largazole chimeras. For the largazole-azumamide hybrid (9), the cis-geometry of the alkene residue necessitated its early introduction. Thus, aldol condensation of aldehyde 510 with thiazolidine-2-thione 6 provided the necessary β-hydroxy acid building block (7, Scheme 1 & Supporting Information). For other variants investigated, late-stage introduction of the zinc-binding side arms via cross metathesis proved expedient. Cross metathesis had previously been used by Luesch, Phillips, and Cramer to attach the natural side-chain in their syntheses of largazole itself and was investigated independently in our laboratories.8a–d
Scheme 1.

Synthesis of largazole-azumamide Hybrid.
Cramer et al. have demonstrated that, at least where largazole is concerned, the four-atom linker length relative to the thiol is optimal for maximum HDAC inhibition.8d However, literature precedent has shown that a four- to five- atom chain is optimal in small molecules bearing alternative zinc-binding functionality. Therefore, in the series of analogs prepared via metathesis both the four- and the five- atom tethers were synthesized.
Compounds 11 and 12 bear the well-known α-aminobenzamide group present in MS-275.11 Meanwhile, compounds 13, 14 and 15, 16 contain α-thioamides and α-thioketones, respectively. These two motifs were identified as potential candidates in a computational study done by Vanommeslaeghe and have demonstrated promise in subsequent medicinal efforts.12 Of note, our yields in the metathesis reaction conform to the results reported by Cramer.8d Our initial yields, employing Grubb’s second-generation ruthenium catalyst were low with poor conversion. The Hoveyda-Grubbs second-generation catalyst proved much more efficient. All Boc- and Trityl- protecting groups were removed prior to biological assay (see Supporting Information).
We also sought to probe the significance of the methyl substituent on the thiazoline ring. Condensation of nitrile 17 with L-cysteine proved remarkably facile, proceeding in near quantitative yield (Scheme 3). Initial efforts at coupling to ester 19 provided poor yields of the desired acyclic precursor 20. The major product was thiazole-thiazole 21, resulting from in situ oxidation. Optimization of conditions for this coupling eventually allowed for up to 62% yield of the desired product. Oxidation could be the cause of the somewhat diminished yields in the cyclization of 20. Compound 23 could not be detected in NMR spectra of the crude reaction mixtures from cyclization of 20. Moreover, 23 could not be prepared directly from its acyclic precursor 21. Instead, oxidation of 22 under standard conditions provided 23. This macrocycle clearly contains some added constraint as demonstrated by presence of rotamers in the 1H NMR spectrum in CDCl3. Both substrate 22a and 23a could be deprotected in good yield using our standard conditions.
Scheme 3.

Synthesis of cysteine & thiazole-thiazole analogs.
Replacement of the thiazole moiety with a pyridine residue in the heterocyclic backbone was readily ammenable to our synthetic strategy (Scheme 4). Known chloro-nitrile 24 could be Boc-protected and then condensed with α-methyl cysteine to provide acid 27. Subsequent deprotection, coupling, and cyclization provided analog 29.
Scheme 4.

Synthesis of thiazole to pyridine substitution.
We also sought to perform additional single-atom replacements within the largazole macrocyclic scaffold to interrogate very small structural and attendant conformational changes manifest. Due to the inherent acid instability of oxazolines, additional protecting group manipulations were required for synthesis of oxazoline-oxazole substrate 39 (Scheme 5). Thus, oxazole 31 could be saponified and coupled to α-methyl serine.13 Switching the nitrogen protecting group then allowed for cyclization and deprotection/acylation with thiazolidine-2-thione 35 to obtain alcohol 36. Coupling to Fmoc-L-valine then provided the acyclic precursor 38.
Scheme 5.

Synthesis of the oxazoline-oxazole analog.
Finally, deprotection under basic conditions and cyclization gave macrocycle 39a. In this case, removal of the trityl group was performed with iodine in methanol, yielding exclusively the disulfide homodimer (39c) resulting from spontaneous autoxidation of the incipient thiol (39b). The dimer is reduced to the active thiol (39b) under the reducing conditions (TCEP) of our biochemical assay. We next performed comparative profiling of largazole thiol and the thiol synthetic derivatives described herein, for inhibitory potency and selectivity against HDAC1, HDAC2, HDAC3/NCoR and HDAC6. A complete description of the assay has been provided previously7 and is described in detail in Supporting Information.
In brief, small-molecule inhibitors are arrayed at twelve-point dose-response (3-fold increments) in 384-well library plates and transferred by robotic pin device to replicate assay plates containing assay buffer under reducing conditions (TCEP 200 μM). A liquid handling device then transfers a tripeptide substrate terminating in acetyl-lysine and amide conjugated to 4-methyl-7-aminocoumarin (AMC), recombinant human histone deacetylase (BPS Bioscience, San Diego, CA), and recombinant human trypsin (Sigma-Aldrich, St. Louis, MO). Following deacetylase hydrolysis of acetyl-lysine, trypsin cleavage liberates the AMC fluorophore. Kinetic (fluorescence per unit time) and end-point (total fluorescence) data are captured by a multilabel plate reader. Replicate data are analyzed by curve-fit using logistic regression (Spotfire Decision-Site). A summary of assay data are in Table 1.
Table 1.
Biochemical inhibiton of human HDACs (IC50, μM).
| compound | HDAC1 | HDAC2 | HDAC3 | HDAC6 |
|---|---|---|---|---|
| largazole thiol (1b) | 0.0012 | 0.0035 | 0.0034 | 0.049 |
| enantiomer (2) | 1.2 | 3.1 | 1.9 | 2.2 |
| C-2 epimer (3) | 0.030 | 0.082 | 0.084 | 0.68 |
| proline substitution (4) | 0.11 | 0.80 | 0.58 | 13 |
| largazole-azumamide hybrid (9b) | > 30 | > 30 | > 30 | > 30 |
| benzamide (11b) | 0.27 | 4.1 | 4.1 | > 30 |
| benzamide (12b) | 23 | 29 | 14 | > 30 |
| thioamide (13b) | 0.67 | 1.6 | 0.96 | 0.7 |
| thioamide (14b) | 1 | 1.9 | 1.5 | 0.24 |
| cysteine substitution 22b | 0.0019 | 0.0048 | 0.0038 | 0.13 |
| thiazole-thiazole (23b) | 0.077 | 0.12 | 0.085 | > 30 |
| thiazole-pyridine substitution (29b) | 0.00032 | 0.00086 | 0.0011 | 0.029 |
| oxazoline-oxazole (39c) | 0.00069 | 0.0017 | 0.0015 | 0.045 |
| SAHA | 0.010 | 0.026 | 0.017 | 0.013 |
| MS-275 | 0.045 | 0.13 | 0.17 | > 30 |
Several striking observations emerge from this dataset. The largazole enantiomer (2) exhibits a decrease in potency by almost exactly three orders of magnitude for all isoforms tested, underscoring the obligate, stereochemical and conformation-activity relationship between the natural product and its protein targets. This is further substantiated by the intermediate potency of the C-2 epimer (3), the valine-to-proline substitution (4) and the oxdized and strained thiazole-thiazole derivative (23b). It is significant to note that the single-atom substitutions of the sulfur atoms for oxygen atoms in the oxazoline-oxazole derivative (29b) provided a compound equipotent to largazole itself. Our synthetic approach has allowed rapid diversification of the zinc-binding arm, modulating both potency and specificity (as for 11b, 12b, 13b and 14b). While none of the zinc-binding replacements for the natural 3-hydroxy-7-mercaptohept-4-enoic acid moiety present in largazole, FK228 and spiruchostatin yielded more potent analogs, many additional zinc-binding replacements are evident and will be evaluated in due course. We were pleased to observe a significant increase in potency with pyridine substitution of the thiazole; this compound (29b) possesses sub-nanomolar activity against Class I HDACs, and will be studied further in translational models of cancer. Compound 29b now constitutes the most biochemically potent Class I HDAC inhibitor known being between three~four times more potent than largazole itself against HDACs 1, 2 and 3. Most notably, we have demonstrated that the methyl substituent of the thiazoline ring is non-essential for the dramatic potency of the natural product (cf. 22b). The commercial availability of relatively inexpensive cysteine, compared with that of the α-methylcysteine residue of natural largazole, permitted for a reduction in the overall synthetic approach to 22b by four steps, establishing a high-yielding, scalable, five-step synthesis of this agent. This highly efficient synthesis is compatible with further derivitization and potential for practical scale-up endeavors. The present study provides additional insight into the structural, functional, stereochemical and conformational aspects of the largazole molecular scaffold that constitutes the basis for the further design and synthesis of extraordinarily potent HDAC inhibitors with potential therapeutic significance. Studies along these lines are under intensive investigation in our laboratories.
Supplementary Material
Scheme 2.

Metathesis route to largazole hybrids.
Acknowledgments
We thank the CSU Cancer Supercluster and the NIH for financial support and for a postdoctoral fellowship for AAB (NCI Grant CA136283). Mass Spectra were obtained on instruments supported by the NIH Shared Instrument Grant GM49631. We thank Ralph Mazitschek for the provision of acetylated substrate. JEB acknowledges support by grants from the National Cancer Institute (1K08CA128972) and the Burroughs-Wellcome Foundation (CAMS).
Footnotes
Supporting Information Available. Spectroscopic data and experimental details for the preparation of all new compounds as well as procedures for the biochemical HDAC assay used in these experiments are provided. This material is available free of charge via the internet at http://pubs.acs.org.
References
- 1.Somech R, Israeli S, Simon A. Cancer Treat Rev. 2004;30:461. doi: 10.1016/j.ctrv.2004.04.006.Miller TA, Witter DJ, Belvedere S. J Med Chem. 2003;46:5097. doi: 10.1021/jm0303094.Moradei O, Maroun CR, Paquin I, Vaisburg A. Curr Med Chem; Anti-Cancer Agents. 2005;5:529. doi: 10.2174/1568011054866946.Bolden JE, Peart MJ, Johnstone RW. Nat Rev Drug Discovery. 2006;5:769. doi: 10.1038/nrd2133.
- 2.Taunton J, Hassig CA, Schreiber SL. Science. 1996;272:408–11. doi: 10.1126/science.272.5260.408.Grozinger CM, Hassig CA, Schreiber SL. Proc Nat Acad Sci USA. 1999;96:4868. doi: 10.1073/pnas.96.9.4868.Johnstone RW. Nature Rev Drug Disc. 2002;1:287. doi: 10.1038/nrd772.
- 3.Karagiannis TC, El-Osta A. Leukemia. 2007;21:61–5. doi: 10.1038/sj.leu.2404464. and references therein. [DOI] [PubMed] [Google Scholar]
- 4.Richon VM, Emiliani S, Verdin E, Webb Y, Breslow R, Rifkind RA, Marks PA. Proc Natl Acad Sci U S A. 1998;95:3003–7. doi: 10.1073/pnas.95.6.3003.Yoshida M, Kijima M, Akita M, Beppu T. J Biol Chem. 1990;265:17174–9.Darkin-Rattray SJ, Gurnett AM, Myers RW, Dulski PM, Crumley TM, Allocco JJ, Cannova C, Meinke PT, Colletti SL, Bednarek MA, Singh SB, Goetz MA, Dombrowski AW, Polishook JD, Schmatz DM. Proc Natl Acad Sci U S A. 1996;93:13143–7. doi: 10.1073/pnas.93.23.13143.Kijima M, Yoshida M, Sugita K, Horinouchi S, Beppu T. J Biol Chem. 1993;268:22429–35.Nakao Y, Yoshida S, Matsunaga S, Shindoh N, Terada Y, Nagai K, Yamashita JK, Ganesan A, van Soest RW, Fusetani N. Angew Chem Int Ed Engl. 2006;45:7553–7. doi: 10.1002/anie.200602047.Izzo I, Maulucci N, Bifulco G, De Riccardis F. Angew Chem Int Ed Engl. 2006;45:7557–60. doi: 10.1002/anie.200602033.Wen S, Carey KL, Nakao Y, Fusetani N, Packham G, Ganesan A. Org Lett. 2007;9:1105–8. doi: 10.1021/ol070046y.Ueda H, Nakajima H, Hori Y, Fujita T, Nishimura M, Goto T, Okuhara M. J Antibiot (Tokyo) 1994;47:301–10. doi: 10.7164/antibiotics.47.301.Shigematsu N, Ueda H, Takase S, Tanaka H, Yamamoto K, Tada T. J Antibiot (Tokyo) 1994;47:311–4. doi: 10.7164/antibiotics.47.311.Ueda H, Manda T, Matsumoto S, Mukumoto S, Nishigaki F, Kawamura I, Shimomura K. J Antibiot (Tokyo) 1994;47:315–23. doi: 10.7164/antibiotics.47.315.Yurek-George A, Habens F, Brimmell M, Packham G, Ganesan A. J Am Chem Soc. 2004;126:1030–1. doi: 10.1021/ja039258q.
- 5.For seminal work on HDACi hybrids: Furumai R, Komatsu Y, Nishino N, Khochbin S, Yoshida M, Horinouchi S. Proc Nat Acad Sci USA. 2000;98:87–92. doi: 10.1073/pnas.011405598.Nishino N, Jose B, Okamura S, Ebisusaki S, Kato T, Sumida Y, Yoshida M. Org Lett. 2003;5:5079–82. doi: 10.1021/ol036098e.
- 6.Taori K, Paul VJ, Luesch H. J Am Chem Soc. 2008;130:13506. doi: 10.1021/ja7110064. [DOI] [PubMed] [Google Scholar]
- 7.Bowers AA, West N, Taunton J, Schreiber SL, Bradner JE, Williams RM. J Am Chem Soc. 2008;130:11219–22. doi: 10.1021/ja8033763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.For additional syntheses of Largazole, see: Nasveschuk CG, Ungermannova D, Liu X, Phillips AJ. Org Lett. 2008;10:3595–98. doi: 10.1021/ol8013478.Ghosh AK, Kulkarni S. Org Lett. 2008;10:3907–9. doi: 10.1021/ol8014623.Ying Y, Taori K, Kim H, Hong J, Luesch H. J Am Chem Soc. 2008;130:8455–59. doi: 10.1021/ja8013727.Seiser T, Kamena F, Cramer N. Angew Chem Int Ed. 2008;47:6483–85. doi: 10.1002/anie.200802043.Ren Q, Dai L, Zhang H, Tan W, Xu Z, Ye T. Synlett. 2008;15:2379.
- 9.Bowers AA, Greshock TJ, West N, Estiu G, Schreiber SL, Wiest O, Williams RM, Bradner JE. J Am Chem Soc. doi: 10.1021/ja807772w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wen S, Carey KL, Nakao Y, Fusetani N, Packham G, Ganesan A. Org Lett. 2007;9:1105–8. doi: 10.1021/ol070046y. [DOI] [PubMed] [Google Scholar]
- 11.Wang DF, Helquist P, Wiech NL, Wiest O. J Med Chem. 2005;48:6936–47. doi: 10.1021/jm0505011.Ryan QC, Headlee D, Acharya M, Sparreboom A, Trepel JB, Ye J, Figg WD, Hwang K, Chung EJ, Murgo A, Melillo G, Elsayed Y, Monga M, Kalnitskiy M, Zwiebel J, Sausville EA. Journal of Clinical Oncology. 2005;23:3912–22. doi: 10.1200/JCO.2005.02.188.Bouchain G, Delorme D. Curr Med Chem. 2003;10:2359–72. doi: 10.2174/0929867033456585.
- 12.Vanommeslaeghe K, Loverix S, Geerlings P, Tourwe D. Bioorg Med Chem. 2005;13:6070–82. doi: 10.1016/j.bmc.2005.06.009.Vanommeslaeghe K, De Proft F, Loverix S, Tourwe D, Geerlings P. Bioorg Med Chem. 2005;13:3987–92. doi: 10.1016/j.bmc.2005.04.001.
- 13.α-Methylserine provided as a generous gift by Ajinomoto Co., Japan.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.